
Abstract
Mitochondria are key cellular signaling platforms, affecting fundamental processes such as cell proliferation, differentiation and death. However, it remains unclear how mitochondrial signaling affects other organelles, particularly lysosomes. Here, we demonstrate that mitochondrial respiratory chain (RC) impairments elicit a stress signaling pathway that regulates lysosomal biogenesis via the microphtalmia transcription factor family. Interestingly, the effect of mitochondrial stress over lysosomal biogenesis depends on the timeframe of the stress elicited: while RC inhibition with rotenone or uncoupling with CCCP initially triggers lysosomal biogenesis, the effect peaks after few hours and returns to baseline. Long-term RC inhibition by long-term treatment with rotenone, or patient mutations in fibroblasts and in a mouse model result in repression of lysosomal biogenesis. The induction of lysosomal biogenesis by short-term mitochondrial stress is dependent on TFEB and MITF, requires AMPK signaling and is independent of calcineurin signaling. These results reveal an integrated view of how mitochondrial signaling affects lysosomes, which is essential to fully comprehend the consequences of mitochondrial malfunction, particularly in the context of mitochondrial diseases.
Similar content being viewed by others


Introduction
Mitochondria are fundamental organelles in metabolism and cell biology1. In addition to the long characterized metabolic roles, including energy metabolism and citrate cycle, iron and calcium metabolism and regulation of apoptosis, mitochondria are increasingly recognized as key signaling platforms affecting not only major cellular processes but also far-reaching systemic regulatory mechanisms2. Furthermore, it is now clear that mitochondria interact with other organelles, both through physical contact sites and through signaling pathways3,4. The interaction between mitochondria and the endoplasmic reticulum (ER) provides pivotal examples of both: the mitochondrial-ER contact sites have been extensively studied and are known to be key players in cellular calcium homeostasis5, while mitochondrial dysfunction has also been shown to trigger ER stress6.
Recently, it was also reported that acute mitochondrial malfunction results in impairment of lysosomes and induction of lysosomal biogenesis7.
The lysosomes are part of the endolysosomal system, and have traditionally been associated with degradation of cellular components8,9. In the recent years, the lysosomes have been shown to be much more than an acid recycling bag, as their role in amino acid sensing and autophagy regulation has been elucidated8,9. Furthermore, a lysosomal stress response that increases lysosomal biogenesis was also identified10,11. This stress response is mediated by the transcription factors of the microphtalmia family, TFEB, MITF, TFEC and TFE310,11,12. These transcription factors are associated to the lysosome during basal conditions, where they become phosphorylated by the mTORC1 complex. Upon lysosomal stress or amino acid starvation, mTORC1 is inactivated and Ca2+ is released from the lysosomes, resulting in the activation of calcineurin which can dephosphorylate TFEB and related molecules13. Dephosphorylated TFEB can then relocate to the nucleus and drive its transcriptional program. TFE3, TFEC and MITF seem to have similar regulatory mechanisms.
Under acute mitochondrial stress, lysosomes become dysfunctional and TFEB relocates to the nucleus14,15,16. The mechanisms linking these processes remain however not completely understood7. In this study, we show how acute and chronic respiratory chain defects have opposite effects on lysosomal biogenesis, both in cultured cells and in vivo in a mouse model of respiratory chain dysfunction. While acute mitochondrial stress triggers a AMPK-TFEB/MITF pathway that leads to increased lysosomal biogenesis, chronic mitochondrial stress results in repression of lysosomal biogenesis.
Results
Lysosomal biogenesis is differentially regulated by acute and chronic mitochondrial dysfunction
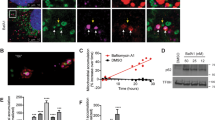
To assess how lysosomal biogenesis is affected by mitochondrial malfunction, we first subjected wild-type mouse embryonic fibroblasts (MEF) to a mitochondrial respiratory chain (RC) complex I inhibitor, rotenone (250 nM). We observed that the transcript levels of several lysosomal genes are rapidly increased upon RC complex I inhibition, and eventually return to baseline levels after 12 h treatment (Fig. 1A). This result suggests that the effect of mitochondrial malfunction on lysosomal biogenesis is dependent on the duration of the mitochondrial perturbation. We then performed a similar experiment, but treating the MEF with a mitochondrial uncoupler, carbonyl cyanide 3-chlorophenylhydrazone (CCCP, 10 μM), which is widely used to induce mitophagy. Upon CCCP treatment, the expression of lysosomal genes increases rapidly and returns to baseline, albeit later than the rotenone treatment (Fig. 1B). Overall, the pattern resulting from perturbation of mitochondrial respiratory chain and oxidative phosphorylation is the same: rapid up-regulation of lysosomal genes, followed by a return to the baseline after few hours. The longer curve observed in CCCP treatment may be related to the relative effect of the two treatments on mitochondrial inner membrane potential (ΔΨm): rotenone has a small effect on ΔΨm while CCCP strongly affects ΔΨm (Supplementary Figure 1A). The effects of rotenone and CCCP on the overall activity of the respiratory chain, as measured by O2 consumption, show that the treatments are working as expected (Supplementary Figure 1B). Furthermore, these results show that the effect of acute and chronic mitochondrial respiratory chain deficiency in lysosomal biogenesis may be different. To test this, we treated MEFs with rotenone (250 nM) for 5 days, and observed a strong reduction in the transcript levels of three of the lysosomal genes (cathepsin D was not significantly changed) (Fig. 1C). We then tested other chronic models of respiratory chain deficiency, to further validate this result. First, we used patient fibroblasts with complex I deficiency. These cells are permanently deficient in complex I activity, and thus represent a model of chronic respiratory chain deficiency17,18. Accordingly, the transcript levels of the four tested lysosomal genes are significantly repressed in these cells (Fig. 1D). Then, we tested MEFs obtained from a mouse model of mitochondrial malfunction (Coq9R239X, knock-in of a patient mutation in Coq9). These mice lack a functional COQ9 protein and present instability of mitochondrial supercomplex I-III19. The transcript levels of the four lysosomal genes tested were robustly decreased in Coq9R239X MEFs (Fig. 1E). Finally, we tested the heart of the Coq9R239X mice, in which the transcript levels of the four lysosomal genes were significantly decreased (Fig. 1F). These results, obtained by long-term chemical inhibition of the respiratory chain and genetic impairment of the respiratory chain in fibroblasts, as well as by genetic impairment of the respiratory chain in vivo, support a model in which acute mitochondrial respiratory chain deficiency triggers lysosomal biogenesis, but chronic mitochondrial respiratory chain deficiency represses it.

(A) Relative transcript levels of LAMP1, GAA, CTSD and CTSF in MEFs treated with the respiratory chain complex I inhibitor rotenone (250 nM), collected at 0, 4, 8 and 12 hours of treatment, normalized to GAPDH. The bars represent average and standard error of the mean (S.E.M.) (N = 4) with p-values indicated (*p < 0.05; **p < 0.005; ***p < 0.001). (B) Relative transcript levels of LAMP1, GAA, CTSD and CTSF in MEFs treated with the uncoupler carbonyl cyanide 3-chlorophenylhydrazone (CCCP) (10 μM), collected at 0, 4, 8, 12 and 24 hours of treatment, normalized to GAPDH. The bars represent average and S.E.M. (N = 4) with p-values indicated (*p < 0.05; **p < 0.005; ***p < 0.001). (C) Relative transcript levels of LAMP1, GAA, CTSD and CTSF in MEFs treated with the respiratory chain complex I inhibitor rotenone (250 nM) for 5 days, normalized to GAPDH. The white bar corresponds to the control MEFs (treated with DMSO) and the red bars to the MEFs treated with rotenone. The bars represent average and S.E.M. (N = 4); t test p-values (*p < 0.05; **p < 0.005; ***p < 0.001). (D) Relative transcript levels of LAMP1, GAA, CTSD and CTSF in fibroblasts from patients with mutations in respiratory chain complex I subunits (red bars) and control individuals (white), normalized to GAPDH. Patient 1 presents a mutation in the NDUFV1 subunit26 and patient two has a mutation in the NDUFV2 subunit27. The bars represent average and S.E.M. (N = 4) with p-values indicated (*p < 0.05; **p < 0.005; ***p < 0.001). (E) Relative transcript levels of LAMP1, GAA, CTSD and CTSF in MEFs from Coq9R239X mice, normalized to HPRT. The white bar corresponds to the wild-type mice MEFs and the blue bars to the Coq9R239X MEFs. The bars represent average and S.E.M. (N = 4) with t test p values indicated (*p < 0.05; **p < 0.005; ***p < 0.001). (F) Relative transcript levels of LAMP1, GAA, CTSD and CTSF in heart from Coq9R239X mice, normalized to GAPDH. The white bars corresponds to the wild-type mice, the blue bars to the Coq9R239X. The bars represent average and S.E.M. (N = 5) with p-values indicated (*p < 0.05; **p < 0.005; ***p < 0.001).
However, given that we observed a limited number of genes, we could not guarantee that the hundreds of genes that encode for lysosomal proteins have a similar behavior. Therefore, we took advantage of two publicly-available transcriptional datasets of dopaminergic neuroblastoma cells subject to rotenone treatment (GSE35642 and GSE4773). We used a database of lysosomal proteins20 to define a list of “lysosomal transcripts” (Supplementary Table 1). This “lysosomal transcripts” list allows us to study the overall effect of mitochondrial impairment over the expression of lysosomal genes, by monitoring how many of those genes had significantly changed expression upon rotenone treatment, and in which direction (up-regulated or down-regulated) – the experimental strategy is illustrated in Fig. 2A. In the first study21 (GSE35642), after one week of treatment with two different concentrations of rotenone (5 nM and 50 nM), there was a higher number of up-regulated lysosomal genes, suggesting an overall increase in lysosomal biogenesis (Supplementary Figure 2A). However, after four weeks of treatment the effect was completely reversed, with a much higher number of lysosomal genes being down-regulated (Supplementary Figure 2A). In the second study22 (GSE4773), the number of up- and down-regulated genes was similar after one week of treatment, but the amount of down-regulated genes increased progressively at 2- and 4-weeks of treatment (Supplementary Figure 2B).

(A) Illustration of the experimental strategy employed to determine lysosomal-related genes that are significantly affected by inhibition of the mitochondrial respiratory chain. (B) Relative transcript levels of LAMP1 in SK-N-MC neuroblastoma cells treated with the respiratory chain complex I inhibitor rotenone (50 nM) collected at 1 and 4 weeks of treatment, after RMA normalization. The white bar corresponds to the control MEFs and the red bars to the MEFs treated with rotenone with t test p-values indicated (*p < 0.05; **p < 0.005; ***p < 0.001). (C) Relative transcript levels of GAA in SK-N-MC neuroblastoma cells treated with the respiratory chain complex I inhibitor rotenone (50 nM) collected at 1 and 4 weeks of treatment, after RMA normalization. The white bar corresponds to the control MEFs and the red bars to the MEFs treated with rotenone with t test p-values indicated (*p < 0.05; **p < 0.005; ***p < 0.001). (D) Relative transcript levels of CTSD in SK-N-MC neuroblastoma cells treated with the respiratory chain complex I inhibitor rotenone (50 nM) collected at 1 and 4 weeks of treatment, after RMA normalization. The white bar corresponds to the control MEFs and the red bars to the MEFs treated with rotenone with t test p-values indicated (*p < 0.05; **p < 0.005; ***p < 0.001). (F) Relative transcript levels of CTSF in SK-N-MC neuroblastoma cells treated with the respiratory chain complex I inhibitor rotenone (50 nM) collected at 1 and 4 weeks of treatment, after RMA normalization. The white bar corresponds to the control MEFs and the red bars to the MEFs treated with rotenone with t test p-values indicated (*p < 0.05; **p < 0.005; ***p < 0.001).
We then tested how the four genes that we have used as proxy for lysosomal biogenesis were affected. We focused on the study which employed 50 nM rotenone treatment, which is closer to our conditions (GSE35642). None of the four genes was down-regulated after 1 week of 50 nM rotenone treatment, but all four genes tested showed a significant down-regulation after 4 weeks of treatment (Fig. 2B–E). These results are in agreement with the long-term effects of respiratory chain inhibition that we described in Fig. 1. Furthermore, they show that these four genes constitute a good proxy for the rest of the lysosomal genes.
We further used the GSE35642 dataset to determine how the genes that are changed after 1 week of 50 nM rotenone treatment behave at four weeks of rotenone treatment in neuroblastoma cells. We filtered the genes whose expression was significantly changed both at 1-week and 4-week treatments, and followed what happens to the direction of the change. Of the 32 transcripts meeting these criteria, 23 were increased after 1 week, and 9 decreased. Remarkably, 22 out of the 23 transcripts increased at 1 week were significantly down-regulated after 4 weeks of 50 nM rotenone treatment (Supplementary Figure 2C and Supplementary Table 2). Furthermore, the 9 transcripts that were down-regulated at week 1, were all up-regulated at week 4 (Supplementary Figure 2C, and Supplementary Table 2).
Altogether, these results show that lysosomal biogenesis undergoes a biphasic response to mitochondrial malfunction, with up-regulation of lysosomal biogenesis after the onset of mitochondrial respiratory chain deficiency, followed by repression after persistence of the mitochondrial defect.
Induction of lysosomal biogenesis by acute mitochondrial stress is TFEB/MITF-dependent
Given the known role of TFEB in the regulation of a gene expression program of lysosomal biogenesis, we addressed the role of TFEB in the response to mitochondrial respiratory chain deficiency. We first observed that the transcript levels of TFEB and other related microphtalmia transcription factors (MITF, TFE3) were following a pattern very similar to the lysosomal transcripts: rapid up-regulation in response to mitochondrial stress followed by return to the baseline, both under rotenone or CCCP treatment (Fig. 3A). The transcript levels of TFEC, another microphtalmia transcription factor, were very low in MEF and its quantification wasn’t sufficiently reliable. To directly assess if TFEB was involved in the lysosomal biogenesis induced by acute mitochondrial stress, we decided to move to another cell type more amenable to genetic manipulation than primary MEF. We employed HeLa cells, which are easier to transfect and not limited by early passage number. We first verified that the treatment of HeLa cells with rotenone and CCCP affects TFEB in a manner similar to what we observed in MEF, which was the case (Fig. 3B). The levels of TFEB protein follow a similar pattern (Fig. 3C). Accordingly, a 4-hour treatment of HeLa cells with CCCP results in an increase in the number of lysosomes (Fig. 3D).

(A) Relative transcript levels of TFEB, MITF and TFE3 in MEFs treated with DMSO (controls, white bars), rotenone (250 nM, red) and CCCP (10 μM, blue), collected at 0, 4, 8 and 12 hours of treatment, normalized to GAPDH mRNA transcript level. The bars represent average and S.E.M. an (N = 4) with p-values indicated (*p < 0.05; **p < 0.005; ***p < 0.001). (B) Relative transcript levels of TFEB, MITF and TFE3 in HeLa cells treated with DMSO (controls, white bars) rotenone (250 nM, red) and CCCP (10 μM, blue), collected at 0, 4, 8, 12, 16, 20 and 24 hours of treatment, normalized to GAPDH mRNA transcript level. The bars represent average and S.E.M. (N = 6) with p-values indicated (*p < 0.05; **p < 0.005; ***p < 0.001). (C) Western blot analysis of whole-cells extracts for TFEB in HeLa cells treated with CCCP (10 μM), collected at 0, 1, 4, 8, 12, 16, 20 and 24 hours of treatment, and corresponding quantification (normalized to GAPDH). The white bar corresponds to the control HeLa cells and the blue bars to the HeLa cells treated with CCCP. The bars represent average and S.E.M. (N > 4) with p-values indicated (*p < 0.05; **p < 0.005; ***p < 0.001). (D) Representative spinning-disk microscopy images of HeLa cells treated 4 hours with CCCP 10 μM. The cells were immunostained with LAMP1-488 and DAPI. The number of the lysosomes was quantified using ImageJ. The average number of lysosomes and respective standard error of the mean are represented in the graph below. At least 15 cells of each group were used for the quantification. The statistics was done using t test, and the p-value is indicated (*p < 0.05; **p < 0.005; ***p < 0.001). (E) Transcript levels of LAMP1, GAA, CTSD and CTSF mRNA transcript levels in TFEB/MITFkd HeLa cells treated 4 hours with DMSO or CCCP (10 μM) normalized to GAPDH. The bars represent average and S.E.M. (N = 4) with t test p-values indicated (*p < 0.05; **p < 0.005; ***p < 0.001).
We then prepared a stable knock-down of TFEB in HeLa cells, and verified the efficiency of the silencing by western blotting (Supplementary Figure 3A). We tested five different constructs, independently, and picked the two clones with the most efficient silencing for further studies. Since MITF also has the ability to trigger lysosomal biogenesis, we decided to also knock-down MITF both independently and together with TFEB knock-down. We used siRNA against MITF in the scrambled and stable TFEB knock-down lines. We assessed the efficiency of MITF knock-down by qPCR (Supplementary Figure 3B), and also verified that the knock-down of MITF does not affect TFEB levels and vice-versa (Supplementary Figure 3B). We then treated the scrambled, TFEB knock-down, MITF knock-down and double TFEB/MITF knock-down with CCCP and determined how the transcript levels of lysosomal genes are affected. As expected, CCCP treatment increased the levels of LAMP1 and GAA in scrambled cells, but was unable to do so in TFEB knock-down and double TFEB/MITF knock-down cells (Fig. 3E). MITF knock-down was sufficient to inhibit the induction of LAMP1 but not of GAA (Fig. 3E). These results show that lysosomal biogenesis induced by acute mitochondrial stress is TFEB/MITF dependent.
AMPK signaling is necessary for lysosomal biogenesis induced by acute mitochondrial stress
We then set to determine what are the mechanisms linking acute mitochondrial stress with activation of TFEB/MITF signaling. We reasoned that given the known role of AMPK as a major responder to mitochondrial stress, and an inhibitor of mTORC1, which in turn inhibits TFEB activity, AMPK might be a key mediator between mitochondrial stress and TFEB activity (illustrated in Fig. 4A). Accordingly, AMPK phosphorylation is increased after 4 h treatment with 10 μM CCCP (Fig. 4B). Thus, we subjected HeLa cells to a 4-hour 10 μM CCCP treatment and simultaneously treated one group of cells with dorsomorphine (also referred to as “compound c”), a commonly used inhibitor of AMPK23. The treatment with CCCP expectedly increased the transcript levels of lysosomal genes LAMP1, GAA, CTSD and CTSF, but this increase was blocked or strongly ablated in the presence of dorsomorphin (Fig. 4C). These results show that AMPK signaling is needed for the activation of TFEB by acute mitochondrial stress. We next tested if activation of AMPK, in the absence of mitochondrial stress, was enough to trigger TFEB-dependent transcription. For this we treated HeLa cells with A769662, a commonly used AMPK activator24, for 4-hours and determined the transcript levels of lysosomal genes. No significant increase was observed (Fig. 4D), despite A769662 was able to activate AMPK (Supplementary Figure 4). This implies that AMPK activation per se is not sufficient to trigger TFEB-dependent transcription in the absence of mitochondrial stress.

(A) Mitochondrial stress leads in lysosomal biogenesis through TFEB. The damage mitochondria release signalling that affects AMPK, which is involved in lysosomal biogenesis through TFEB family pathway. (B) Western blot analysis of whole-cells extracts showing increased AMPK activation, by measuring AMPK and AMPK-P in HeLa cells treated 4 hours with CCCP (10 μM). The graph shows the ratio of AMPK-P/AMPK protein normalized to HPRT. The white bar corresponds to the control HeLa cells and the red bar to the HeLa cells treated with CCCP. The bars represent average and S.E.M. (N = 4) with t test p-value indicated (*p < 0.05; **p < 0.005; ***p < 0.001). (C) Transcript levels of LAMP1, GAA, CTSD and CTSF mRNA transcript levels in HeLa cells treated 4 hours with CCCP 10 μM and dorsomorphin (compound C) (10 μM), normalized to GAPDH mRNA transcript level. The white bar corresponds to the control HeLa cells, the bars with pattern to HeLa cells treated with CCCP and the blue bars corresponds to HeLa cells treated with compound C. The bars represent average and S.E.M. (N = 6) with p-values indicated (*p < 0.05; **p < 0.005; ***p < 0.001). (D) Transcript levels of of LAMP1, GAA, CTSD and CTSF mRNA transcript levels in HeLa cells treated 4 hours with A769962, normalized to GAPDH mRNA transcript level. The white bar corresponds to the control HeLa cells and the red bars to the HeLa cells treated with A769662 (100 μM). The bars represent average and S.E.M. (N = 4). No statistically significant changes were observed. (E) Transcript levels of LAMP1, GAA, CTSD and CTSF mRNA transcript levels in HeLa cells treated 4 hours with FK506 (5 μM) and CCCP (10 μM), normalized to GAPDH mRNA transcript level. The white bar corresponds to the control HeLa cells, the bars with pattern to HeLa cells treated with CCCP and the orange bars corresponds to HeLa cells treated with FK506. The bars represent average and S.E.M. (N = 6) with p-values indicated (*p < 0.05; **p < 0.005; ***p < 0.001).
In addition to the AMPK-mTORC1 axis, TFEB activity was recently shown to be also regulated by calcineurin. To assess the role of calcineurin in our experimental paradigm, we treated cells simultaneously with 10 μM CCCP and calcineurin inhibitor13 FK506, and found no effect on the transcript levels of TFEB targets (Fig. 4E). This result implies that the regulation of TFEB in response to acute mitochondrial stress occurs mostly through the AMPK-mTORC1 axis and is independent of calcineurin.
Discussion
This study addresses the mechanisms by which mitochondrial malfunction, and particularly respiratory chain deficiency, impact lysosomal biogenesis. We show here that acute and chronic perturbations of the mitochondrial respiratory chain have opposite effects in lysosomal biogenesis: while acute mitochondrial respiratory chain stress triggers AMPK- and TFEB/MITF-dependent lysosomal biogenesis, chronic mitochondrial respiratory chain dysfunction actually results in a repression of TFEB/MITF transcriptional activity and lysosomal biogenesis.
The impact of mitochondrial defects in cellular function has been addressed by numerous studies in recent years. Several mechanisms of mitochondrial stress were identified, which culminate in the modulation of cellular signaling pathways and of gene expression programs2. The impact of mitochondrial defects in other organelles has been subject to less attention. Nevertheless, it was shown that mitochondrial malfunction can trigger endoplasmic reticulum stress and the unfolded protein response6. Recently, three studies addressed the effect of mitochondrial malfunction and of mitophagy on lysosomal function and biogenesis. In cultured cells, short-term mitophagy bursts affect the regulation of the microphtalmia transcription factor family15,16, which includes TFEB, MITF, TFEC and TFE3, while mtDNA depletion, and consequent loss of respiratory chain function, perturbs lysosomal function and triggers a program of incomplete lysosomal biogenesis in mouse T cells upon their activation14. However, the mechanisms underlying the impact of mitochondrial malfunction on lysosomes and lysosomal biogenesis remain unaddressed7. Here, we show that mitochondrial malfunction affects lysosomal biogenesis in a biphasic manner. Under acute mitochondrial stress, an AMPK-dependent activation of TFEB/MITF transcriptional response results in the induction of a transcriptional program of lysosomal biogenesis. This response fades after few hours of mitochondrial stress, and returns to basal levels. This biphasic effect is independent of the cell type, since we observed it in mouse embryonic fibroblasts and HeLa cells, and in transcriptional data from neuroblastoma cells. Rather, this biphasic effect seems to be a common response to different mitochondrial stresses employed in our study, such as inhibition of respiratory chain or uncoupling the respiratory chain from the oxidative phosphorylation machinery. Interestingly, in chronic models of mitochondrial malfunction, we find the transcriptional program of lysosomal biogenesis to be repressed. This is observed in vivo in a mouse model of mitochondrial respiratory chain deficiency, in the MEFs obtained from the same mouse, as well as in patient fibroblasts with complex I deficiency. Therefore, the effect is valid in post-mitotic tissues as well as primary cells in culture. Our model of lysosomal biogenesis activation by acute mitochondrial stress is in agreement with other models of acute mitochondrial stress, such as mitophagy bursts and T-cell activation, which also induce TFEB and lysosomal biogenesis14,15,16. The exact kinetics of the biphasic effect may vary depending on the cell type employed, the treatment (compound, concentration) and the genetic environment. For example, Ivankovic et al. show that TFEB mRNA peaks after 18 hours 10 μM CCCP treatment of SH-SY5Y neuroblastoma cells15. In our MEFs and HeLa the same concentration of CCCP induces TFEB much earlier, and by that time point TFEB is already down. It is possible that differences regarding the metabolic functions of MEF and neuroblastoma cells contribute to the different kinetics of the biphasic effect. The 250 nM rotenone treatment in our MEFs also seems to have faster kinetics than a 50 nM rotenone treatment in neuroblastoma cells21,22, but in this case the increased rotenone concentration is likely a factor. Nevertheless, the rotenone treatments in neuroblastoma cells concur with our results in that lysosomal genes eventually become repressed after a long-term treatment. This is in agreement with our long-term treatment with rotenone, but also with the MEFs and tissues from Coq9 mice, and patient fibroblasts with deficiency in complex I, all of which are models of chronic mitochondrial respiratory chain deficiency, and all of which show a robust down-regulation of the lysosomal genes that we were monitoring. The study by Baixauli et al. shows that in cytoplasmic hybrid L929 mouse cells with defects in complex I subunit ND6, TFEB transcript levels are up but LAMP1 transcript levels are not changed14. These results show a different pattern than what we observe in chronic mitochondrial malfunction. It is unlikely that the difference is due to different control genes in the qPCR reactions (beta-actin and beta-microglobulin in Baixauli and colleagues, HPRT and GAPDH in this manuscript), since when we use beta-actin as a control gene we observe the same results as with GAPDH and HPRT (data not shown). It is possible that the long ethidium bromide treatment that the original L929 cells underwent to completely lose their mtDNA has impacted the TFEB signaling pathway – certainly the near future will shed light on how different cell types and different stresses impact how mitochondria and lysosomes communicate. Our result showing down-regulation of lysosomal genes upon long-term mitochondrial stress is consistent across several models (rotenone-treated MEFs, patient fibroblasts, MEFs with genetic inhibition of the RC, tissues from mice with mitochondrial RC defects).
The initial increase in lysosomal biogenesis in response to mitochondrial stress is likely to allow the cells to cope with the increase in autophagy flux and to increase the capacity to remove damaged mitochondria. At some point, after about 12–24 h of mitochondrial stress, the cells start deactivating the transcriptional mechanisms leading to the generation of more lysosomal proteins, possibly as a feed-back mechanism designed to avoid excessive accumulation of lysosomes in the cells. The lysosomes are involved not only in the autophagy pathway but also in endocytic pathway and in exocytosis of cellular components, and thus accumulation of lysosomes might have detrimental effects to other cellular functions25. The importance of avoiding excessive lysosomal biogenesis is underscored by the repression of the transcript levels of lysosomal genes under chronic mitochondrial stress. Interestingly, mitochondrial malfunction seems to be associated with lysosomal impairments, both in acute (TFAM−/− T cells upon activation)14 and chronic stress (e.g., OPA1−/−, AIF−/− MEFs)26, characterized by enlarged dysfunctional lysosomes akin to the saturated organelles observed in lysosomal storage diseases.
Interestingly, AMPK signaling is needed to activate lysosomal biogenesis upon acute mitochondrial malfunction. AMPK is a known responder to mitochondrial stress2, and while the exact mechanism leading to AMPK activation in acute mitochondrial stress remains to be determined, it is likely that several of its known activators, such as ROS23, Ca2+ and lower energy charge27,28, are signals of acute mitochondrial stress. AMPK has broad roles in cellular signaling28, namely in the regulation of compensatory mitochondrial biogenesis27, activation of autophagy and the regulation of mTORC1 activity28. Furthermore, AMPK was recently shown to have the ability to regulate TFEB activity in embryonic stem cells24. The known roles of AMPK and its requirement for lysosomal biogenesis upon acute mitochondrial stress suggest a coordinated cellular response: the increase in AMPK signaling will likely result in ULK1/2 activation and increased formation of autophagosomes29, which can then be used to remove damaged mitochondria from the cytoplasm. However, such a burst of autophagosomes might saturate the lysosomal capacity, and thus the simultaneous induction of lysosomal biogenesis allows for the generation of new lysosomes, enabling them to cope with the increased autophagic flow.
The regulation of TFEB under acute mitochondrial stress seems to be independent of calcineurin. The mechanisms regulating TFEB activity only recently started to be identified, and additional pathways have been proposed that are independent of the initially described roles of mTORC1 and calcineurin30. As already suggested by Nezich and colleagues, the regulation of TFEB intracellular localization and activity seems more complex than initially thought and requires further investigation16. Our data on the role of TFEB in response to acute mitochondrial stress is in agreement with the works by Nezich and colleagues16 as well as Ivankovic and colleagues15, which both demonstrate that TFEB and its related transcription factors are necessary for the increase in lysosomal biogenesis upon mitophagy.
This study unveils a mechanism for the communication between mitochondria and lysosomes under acute mitochondrial stress, and highlights the importance of distinguishing between acute and chronic mitochondrial stresses and the respective cellular responses, in particular regarding the impact in other organelles. While this closely concerns mitochondrial diseases, the impact of dysfunctional mitochondria on other organelles is likely to be relevant for the understanding of neurodegenerative syndromes that involve chronically defective mitochondria (e.g., Parkinson’s disease triggered by mutations in mitochondrial genes such as POLG and PARK2, as well as rotenone-induced Parkisonism).
Methods
Drugs and cellular treatments
The following drugs were used: Carbonyl cyanide 3-chlorophenylhydrazone (CCCP) 10 μM (Fluka), Rotenone 250 nM (Sigma-Aldrich), dorsomorphin (compound C) 10 μM (InvivoGen), A769662 100 μM (Invivogen), FK506 (Tacrolimus) 5 μM (InvivoGen). The experiments were performed in the University Medical Center Göttingen according to the appropriate guidelines.
Cell culture, stable and transient transfection
HeLa cells were grown in DMEM high glucose medium (Gibco) supplemented with 10% fetal bovine serum and 1% Penicillin/Streptomycin at 37 °C and 5% CO2, unless otherwise stated. Patient fibroblasts were collected and maintained according to the ethical guidelines of the Hospital Robert Depré Paris. RNA from patient fibroblasts was extracted using a Qiagen kit according to manufacturer instructions.
Western Blotting
Whole-cell extracts of cultured human cells were prepared in 1,5% n-dodecylmaltoside in PBS as described (Raimundo et al., 2009). Gels were loaded with 50 μg of total protein per well, proteins were separated in 12% SDS-PAGE gels and transferred to polyvinylidene fluoride (PVDF) membranes (Amersham, Life Technologies). The following antibodies were used for immunoblotting: GAPDH (Sigma-Aldrich, G9545), HPRT (Abcam, ab10479), LAMP1 (Abcam, ab24170), TFEB (Novus, NBP1-67872), AMPKα1 (Cell Signaling, 2795), AMPKα1-P (T172) (Cell Signaling, 2535). Band density traces and quantification were determined using ImageJ.
Quantitative RT-PCR
RNA extraction and purification were performed using Crystal RNA mini Kit (Biolab). RNA quantification and quality control were done using Nanodrop (PeqLab) and cDNA was synthesized with High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to the manufacturer’s instructions. cDNA was diluted 1:100, and each 8 μl reaction contained 4 μl diluted cDNA, 0,2 μl dilutions of each primer (from 25 μM stock), and 3,6 μl iTaq Universal SYBR Green Supermix (Bio-Rad).
Lysosomal transcript list and data analysis
Using the Human Lysosome Gene Database (http://lysosome.unipg.it/)16, we defined a list of 435 genes encoding proteins with confirmed lysosomal localization. Then, using the gene symbol, we translated that list using the matrix file of the Affymetrix HG U133 Plus 2.0 microarray platform, which was used in the studies GSE35642 and GSE4773 were conducted. Of the 435 lysosomal genes, 418 had at least one probeset in HG U133 Plus 2.0. The list of these 418 genes, with their gene symbol and the AffyID is presented in Supplementary Table 1 (“lysosomal transcripts”). To determine how many of these genes had significant changes in expression upon rotenone treatments, we downloaded the CEL files of the GSE35642 and GSE4773 datasets. Each experiment was analyzed individually. For both, we normalized the samples by RMA and obtained a differentially-expressed gene (DEG) list as described19. Then, we determined how many of the “lysosomal transcripts” were included in the DEG for each condition. The result is presented in Fig. 2A and B.
Flow Cytometry
Measurement of mitochondrial membrane potential with JC-1 20 μM (ThermoFisher Scientific) was performed according to the manufacturer instructions.
Microscopy
Cells were plated using coverslips on 12 well-plates, treated with CCCP 10 μM (Fluka) and fixed with 4% PFA. The immunostaining was performed using the primary antibody H4A3 (DSHB) according to the manufacturer instructions.
Mice Coq9R239X sample preparation
RNA was extracted from heart of 3- (pre-symptomatic) and 5-month old (post-symptomatic) Coq9R239X mice (C57BL/6 genetic background). Animal experiments were performed according to a protocol approved by the Institutional Animal Care and Use Committee of the University of Granada (procedures 62-CEEA-OH-2014) and were in accordance with the Directive 2010/63/EU on the protection of animals used for scientific purposes and the Spanish law (R.D. 53/2013).
TFEB/MITF silencing
The HeLa cells with stable TFEB knock-down were prepared using lentiviral shRNA delivery. The shRNAs were purchased from Open Biosystems (TermoFisher Scientific). Lentiviral generation was done by growing HEK293T packaging cells in DMEM high glucose (Gibco) supplemented with 10% FBS and after 24 h were transfected with shRNA against target genes and viral components using Lipofectamine 2000, grown and concentrated using Lenti-X Concentrator (Clontech) according to the manufacturer’s instructions. HeLa cells were then seeded at 0.12 × 105 and grown overnight to 70–80% confluence. These cells were then transfected with the lentiviral particles using Polybrene (8 ug/ul). Puromycin (6 mg/m)-resistant transfected were selected as positive for knockdown of interest.
Transient MITF knockdowns were done using siRNAs and Fugene 6 Transfection Reagent (Promega) according to the manufacturer’s instructions. MITF knockdowns were done with RNAi Duplex Oligos from TriFECTa RNAi Kit (IDT) according to the manufacturer’s instructions.
Statistical Analysis
For Western blotting, flow cytometry, quantitative RT-PCR, all data points represent the mean of at least three independent biological replicates, unless otherwise indicated in the respective figures. Error bars represent SEM. P-values were determined using Student’s t test for two group comparisons or ANOVA for multi-group comparisons. *p < 0.05 **p < 0.005 ***p < 0.001.
Primers for qPCR
The primers used for the qPCR reactions are detailed in Supplementary Table 3.
Additional Information
How to cite this article: Fernández-Mosquera, L. et al. Acute and chronic mitochondrial respiratory chain deficiency differentially regulate lysosomal biogenesis. Sci. Rep. 7, 45076; doi: 10.1038/srep45076 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Nunnari, J. & Suomalainen, A. Mitochondria: in sickness and in health. Cell 148, 1145–1159, doi: 10.1016/j.cell.2012.02.035 (2012).
Raimundo, N. Mitochondrial pathology: stress signals from the energy factory. Trends Mol Med 20, 282–292, doi: 10.1016/j.molmed.2014.01.005 (2014).
Giacomello, M. & Pellegrini, L. The coming of age of the mitochondria-ER contact: a matter of thickness. Cell Death Differ 23, 1417–1427, doi: 10.1038/cdd.2016.52 (2016).
Murley, A. & Nunnari, J. The Emerging Network of Mitochondria-Organelle Contacts. Mol Cell 61, 648–653, doi: 10.1016/j.molcel.2016.01.031 (2016).
Raffaello, A., Mammucari, C., Gherardi, G. & Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem Sci, doi: 10.1016/j.tibs.2016.09.001 (2016).
Haynes, C. M., Fiorese, C. J. & Lin, Y. F. Evaluating and responding to mitochondrial dysfunction: the mitochondrial unfolded-protein response and beyond. Trends Cell Biol 23, 311–318, doi: 10.1016/j.tcb.2013.02.002 (2013).
Raimundo, N., Fernandez-Mosquera, L., Yambire, K. F. & Diogo, C. V. Mechanisms of communication between mitochondria and lysosomes. Int J Biochem Cell Biol 79, 345–349, doi: 10.1016/j.biocel.2016.08.020 (2016).
Lim, C. Y. & Zoncu, R. The lysosome as a command-and-control center for cellular metabolism. J Cell Biol 214, 653–664, doi: 10.1083/jcb.201607005 (2016).
Perera, R. M. & Zoncu, R. The Lysosome as a Regulatory Hub. Annu Rev Cell Dev Biol 32, 223–253, doi: 10.1146/annurev-cellbio-111315-125125 (2016).
Roczniak-Ferguson, A. et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Science signaling 5, ra42, doi: 10.1126/scisignal.2002790 (2012).
Settembre, C. et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. Embo J 31, 1095–1108, doi: 10.1038/emboj.2012.32 (2012).
Martina, J. A. et al. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Science signaling 7, ra9, doi: 10.1126/scisignal.2004754 (2014).
Medina, D. L. et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 17, 288–299, doi: 10.1038/ncb3114 (2015).
Baixauli, F. et al. Mitochondrial Respiration Controls Lysosomal Function during Inflammatory T Cell Responses. Cell Metab 22, 485–498, doi: 10.1016/j.cmet.2015.07.020 (2015).
Ivankovic, D., Chau, K. Y., Schapira, A. H. & Gegg, M. E. Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J Neurochem, doi: 10.1111/jnc.13412 (2015).
Nezich, C. L., Wang, C., Fogel, A. I. & Youle, R. J. MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. J Cell Biol 210, 435–450, doi: 10.1083/jcb.201501002 (2015).
Benit, P. et al. Mutant NDUFV2 subunit of mitochondrial complex I causes early onset hypertrophic cardiomyopathy and encephalopathy. Hum Mutat 21, 582–586, doi: 10.1002/humu.10225 (2003).
Benit, P. et al. Large-scale deletion and point mutations of the nuclear NDUFV1 and NDUFS1 genes in mitochondrial complex I deficiency. Am J Hum Genet 68, 1344–1352, doi: 10.1086/320603 (2001).
Garcia-Corzo, L. et al. Dysfunctional Coq9 protein causes predominant encephalomyopathy associated with CoQ deficiency. Hum Mol Genet 22, 1233–1248, doi: 10.1093/hmg/dds530 (2013).
Brozzi, A., Urbanelli, L., Germain, P. L., Magini, A. & Emiliani, C. hLGDB: a database of human lysosomal genes and their regulation. Database: the journal of biological databases and curation 2013, bat024, doi: 10.1093/database/bat024 (2013).
Cabeza-Arvelaiz, Y. & Schiestl, R. H. Transcriptome analysis of a rotenone model of parkinsonism reveals complex I-tied and -untied toxicity mechanisms common to neurodegenerative diseases. PLoS One 7, e44700, doi: 10.1371/journal.pone.0044700 (2012).
Greene, J. G., Greenamyre, J. T. & Dingledine, R. Sequential and concerted gene expression changes in a chronic in vitro model of parkinsonism. Neuroscience 152, 198–207, doi: 10.1016/j.neuroscience.2007.11.029 (2008).
Raimundo, N. et al. Mitochondrial Stress Engages E2F1 Apoptotic Signaling to Cause Deafness. Cell 148, 716–726, doi: 10.1016/j.cell.2011.12.027 (2012).
Young, N. P. et al. AMPK governs lineage specification through Tfeb-dependent regulation of lysosomes. Genes Dev 30, 535–552, doi: 10.1101/gad.274142.115 (2016).
Ballabio, A. & Gieselmann, V. Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta 1793, 684–696, doi: 10.1016/j.bbamcr.2008.12.001 (2009).
Demers-Lamarche, J. et al. Loss of Mitochondrial Function Impairs Lysosomes. J Biol Chem 291, 10263–10276, doi: 10.1074/jbc.M115.695825 (2016).
Bergeron, R. et al. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab 281, E1340–1346 (2001).
Hardie, D. G. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev 25, 1895–1908, doi: 10.1101/gad.17420111 (2011).
Egan, D. F. et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461, doi: 10.1126/science.1196371 (2011).
Li, Y. et al. Protein kinase C controls lysosome biogenesis independently of mTORC1. Nat Cell Biol 18, 1065–1077, doi: 10.1038/ncb3407 (2016).
Acknowledgements
This work was supported by ERC grant 337327 (NR), Rückkehrerförderung-Startförderung from the University Medical Center Goettingen (NR), Emmy Noether Award from the Deutsche Forschungsgemeinschaft (IM), and Peter und Traudl Engelhorn Stiftung (CVD). Daniela Gerke is thanked for technical assistance. PB and PR were supported by grants from Ammi (Association contre les Maladies Mitochondriales), AJII (Association Aide aux Jeunes Handicapés), OLY (Ouvrir les Yeux) and ANR (Agence Nationale de la Recherche). LCL was supported by grants from the Ministerio de Economía y Competitividad, Spain, the ERDF (SAF2015-65786-R) and from the foundation “todos somos raros, todos somos únicos”.
Author information
Authors and Affiliations
Contributions
L.F.M. planned experiments, performed experiments, analyzed data and prepared figures. C.V.D., K.F.Y. and G.L.S. performed experiments. M.L.S. and L.C.L.G. provided samples of mouse tissues. P.B. and P.R. provided RNA samples of patient and control fibroblasts. I.M. planned and performed experiments, analyzed data. NR designed the study, planned experiments, performed experiments, analyzed data. L.F.M. and N.R. wrote the manuscript, which was then commented by all co-authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Fernández-Mosquera, L., Diogo, C., Yambire, K. et al. Acute and chronic mitochondrial respiratory chain deficiency differentially regulate lysosomal biogenesis. Sci Rep 7, 45076 (2017). https://doi.org/10.1038/srep45076
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep45076
This article is cited by
-
Autophagy modulators influence the content of important signalling molecules in PS-positive extracellular vesicles
Cell Communication and Signaling (2023)
-
Crosstalk of organelles in Parkinson’s disease – MiT family transcription factors as central players in signaling pathways connecting mitochondria and lysosomes
Molecular Neurodegeneration (2022)
-
Lysosomal perturbations in human dopaminergic neurons derived from induced pluripotent stem cells with PARK2 mutation
Scientific Reports (2020)
-
Autophagy Induction by Bexarotene Promotes Mitophagy in Presenilin 1 Familial Alzheimer’s Disease iPSC-Derived Neural Stem Cells
Molecular Neurobiology (2019)
-
Emerging roles and regulation of MiT/TFE transcriptional factors
Cell Communication and Signaling (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
