
Abstract
More and more new natural products have been found in Streptomyces species, which become the significant resource for antibiotics production. Among them, Streptomyces lydicus has been known as its ability of streptolydigin biosynthesis. Herein, we present the genome analysis of S. lydicus based on the complete genome sequencing. The circular chromosome of S. lydicus 103 comprises 8,201,357 base pairs with average GC content 72.22%. With the aid of KEGG analysis, we found that S. lydicus 103 can transfer propanoate to succinate, glutamine or glutamate to 2-oxoglutarate, CO2 and L-glutamate to ammonia, which are conducive to the the supply of amino acids. S. lydicus 103 encodes acyl-CoA thioesterase II that takes part in biosynthesis of unsaturated fatty acids, and harbors the complete biosynthesis pathways of lysine, valine, leucine, phenylalanine, tyrosine and isoleucine. Furthermore, a total of 27 putative gene clusters have been predicted to be involved in secondary metabolism, including biosynthesis of streptolydigin, erythromycin, mannopeptimycin, ectoine and desferrioxamine B. Comparative genome analysis of S. lydicus 103 will help us deeply understand its metabolic pathways, which is essential for enhancing the antibiotic production through metabolic engineering.
Similar content being viewed by others

Introduction
Streptomyces species are high-GC Gram-positive bacteria found predominantly in soil1. Through a complex process of morphological and physiological differentiation, Streptomyces species could produce many specialized metabolites used for agricultural antibiotics2. Some fungi can degrade difficult decomposition by lipase and cellulase, which play an important role in soil ecology3. Besides, the resistance genes of insecticide and herbicide in Streptomyces are widely used in transgenic plants4. These secondary metabolites are not essential for bacterial growth but have important roles in microbe-microbe communication5. As a root-colonizing actinomycete, Streptomyces lydicus can produce antibiotics or siderophore for suppressing fungal growth6. The elucidation of the related antimicrobial mechanism will facilitate the finding of novel antibiotics.
With the development of genome sequencing technology, more and more complete genomes of Streptomyces species have been announced. S. lydicus could produce streptolydigin which acts on catalytic function of RNA polymerase and inhibits RNA synthesis7. Our previous studies have identified its biosynthesis pathways of fatty acids8, type II thioesterase9 and nitrogen metabolism10 which are responsible for streptolydigin biosynthesis. Besides, proteomics and metabolomics approaches have been demonstrated in our previous studies on the responses of S. lydicus to pitching ratios during streptolydigin production11,12. However, only one complete genome sequence of S. lydicus, i.e., S. lydicus A02 (accession number CP007699.1), was available in GenBank. Therefore, we have carried out the complete genome sequencing of S. lydicus 103 and constructed its metabolic pathways of antibiotic biosynthesis, including primary metabolism and secondary metabolism. Previous work has shown that heterologously expression of chit42 gene from Trichoderma harzianum P1 in S. lydicus A01 could enhance the chitinase activity and natamycin production13. Further functional characterization of the gene cluster will advance our understanding of the related pathways of antibiotic biosynthesis, and provide insight into the further analysis of the metabolism and gene targets for strain improvement.
Results
Genomic characteristics of S. lydicus 103
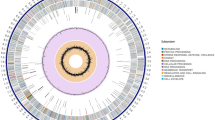
S. lydicus 103 has a chromosome of 8.20 Mb with 72.22% GC content, which contains 6,872 annotated protein-coding genes (Fig. S1 and Table S1). Mostly, the chromosomes of Streptomyces species are linear14. However, the chromosome of S. lydicus 103 in this study is circular, which may lead to more genetic stability. Phylogenetic analysis of S. lydicus 103 with other Streptomyces species has been carried out using CVTree (Fig. S2). BLASTP searches have been performed based on the whole amino acid sequences of S. lydicus 103 against those of other Streptomyces genomes listed in Fig. S2 with E-values less than 10−5. The protein-coding genes with the percent of identity and coverage larger than 80% in all Streptomyces genomes listed in Fig. S2 are defined as core genes (Figs 1 and 2). The protein-coding genes that have no hits within the other Streptomyces genomes listed in Fig. S2 are defined as unique genes. Based on the BLASTP results, 641 core genes and 59 unique genes in S. lydicus 103 are predicted by this definition. With the aid of the distribution of core genes and unique genes, two large genomic islands have been detected (GI-I: from 3812417 to 4085811 bp and GI-II: from 4171990 to 4395111 bp) in S. lydicus 103 based on GC-Profile (Fig. 1 and Table S2). It is thought that Streptomyces achieves the productivity of a wide variety of secondary metabolites by acquiring foreign biosynthetic enzyme genes through horizontal gene transfer15. In GI-I, we found a thiopeptide-lantipeptide biosynthesis pathway, which has the 38% similarity with the cyclothiazomycin biosynthesis pathway (Table 1). Although S. lydicus strain 103 and A02 belong to the same species, S. lydicus A02 has a larger chromosome (9,300,149 bp) than strain 103 (8,201,357 bp). Besides, the genome size of S. lydicus 103 is smaller than two other neighbor species, Streptomyces bingchenggensis BCW-1 and Streptomyces albus J1074 (Fig. S2). By the distribution of COG classification, we can see that in S. lydicus 103 genome, the number of genes related to transcription (K), amino acid transport and metabolism (E), carbohydrate transport and metabolism (G) and the signal transduction mechanisms (T) are more than the other function related genes (Table S3).

With the aid of the distribution of core genes and unique genes, two large genomic islands (red lines) have been detected (GI-I: from 3812417 to 4085811 bp and GI-II: from 4171990 to 4395111 bp) in S. lydicus 103 based on GC-Profile. And the purple and yellow stars present core genes and unique genes of S. lydicus 103, respectively.

BioVenn, a web application for the comparison and visualization of biological lists, has been used for Venn diagrams drawing.
Bacterial toxin-antitoxin (TA) system has been identified in Streptomyces species, such as 22 putative TA loci in Streptomyces coelicolor A3, 27 in Streptomyces avermitilis MA-4680 and 14 in Streptomyces griseus NBRC 13350. Twenty-eight putative type II TA locus have been predicted in S. lydicus 103 genome by TADB, including DUF397, Xre, COG3832 and PIN families (Table S4). Besides, we found the relBE locus in S. lydicus 103, which rarely existed in Streptomyces species. It was reported that over-expression of S. cattleya toxin RelE2sca was lethal in E. coli and S. lividans16. The regulatory mechanism of TA loci in S. lydicus 103 may be necessary to the environmental stress responses and complex secondary metabolisms.
The related metabolism of streptolydigin synthesis in S. lydicus 103
Primary metabolism significantly influences secondary metabolism and serves as building precursors for antibiotic biosynthesis, including acetyl-CoA, glucose-6-phosphate, glyceraldehyde-3-phosphate, and oxaloacetate17. With the aid of KEGG analysis, metabolic network was obtained, including the central carbon metabolism, nitrogen, amino acids and fatty acids metabolism. Among all the KEGG pathways, carbohydrate and amino acid metabolism accounted for the largest proportion.
In the central carbon metabolism, S. lydicus 103 has the complete glycolysis, citrate cycle and pentose phosphate pathway. Acyl-CoA is the important precursor of acetyl-CoA, malonyl-CoA, methylmalonyl-CoA, and ethylmalonyl-CoA (Fig. 3). Cutting phosphofructokinase would transfer carbon metabolic flux of glycolytic pathway to the pentose phosphate pathway, and acetyl-CoA could be significant accumulated and further converted to antibiotics and pyruvate18. In the carbohydrate metabolism, S. lydicus 103 harbors the complete pathway that transfers xylitol to D-ribulose-5P, involving pentose phosphate pathway, and contains endoglucanase and beta-glucosidase, which transfer cellulose to glucose. Furthermore, S. lydicus 103 contains the PTS system and sugar-specific component, thus utilizing the extracellular trehalose and maltose. In the propanoate metabolism, S. lydicus 103 harbors the complete pathway that transfers propanoate to succinate, involving pyruvate metabolism. In the nitrogen metabolism, we found two cycle pathways to transfer CO2 and L-glutamate to ammonia, respectively.

The related metabolism of streptolydigin synthesis in S. lydicus 103.
In the fatty acids biosynthesis, S. lydicus 103 lacks the 3-hydroxyacyl-[acyl-carrier-protein] dehydratase, which is responsible for the dehydration step of the dissociated (type II) fatty-acid biosynthesis system19. Moderate control of lipids biosynthesis may distribute more coenzyme A to the streptolydigin biosynthesis. In the fatty acids degradation, S. lydicus 103 lacks the O-palmitoyltransferase, which is responsible for the hexadecanoyl-CoA degradation. Besides, S. lydicus 103 contains the tesB gene that encodes acyl-CoA thioesterase II [EC:3.1.2.-], taking part in biosynthesis of unsaturated fatty acids, e.g. palmitic acid, stearic acid and oleic acid.
Among the amino acids, glutamic acid was the most favorable as the nitrogen source to form streptolydigin20. In the glutamine and glutamate metabolism, S. lydicus 103 contains the complete pathway to transfer glutamine or glutamate to 2-oxoglutarate, supplying the citrate cycle (Fig. 3). In the cysteine and methionine metabolism, S. lydicus 103 harbors the complete pathways to transformation among the L-cysteine, pyruvate, L-homocysteine and L-methionine. L-methionine was not the direct precursor for streptolydigin biosynthesis, but it provided N-methyl of streptolydigin through S-adenosylmethionine, which was catalyzed by S-adanosylmethionine synthase. In the lysine degradation, S. lydicus 103 lacks lots of related genes, thus restricting the supplement of acetyl-CoA. In the valine, leucine and isoleucine degradation, S. lydicus 103 lacks the 2-oxoisovalerate dehydrogenase E1 component alpha subunit [EC:1.2.4.4] and 2-oxoisovalerate dehydrogenase E2 component (dihydrolipoyl transacylase) [EC:2.3.1.168], thus influencing the biosynthesis of branched chain fatty acid and terpenoid backbone. S. lydicus 103 harbors the complete pathways of lysine, valine, leucine, phenylalanine, tyrosine and isoleucine biosynthesis. In addition to the acyl-CoA, L-valine contributes to the biosynthesis for methylmalonyl-CoA and ethylmalonyl-CoA, and L-methionine contributes to the biosynthesis for chloroethylmalonyl-CoA. Proteomics and metabolomics analyses showed that the pitching ratio influenced the activity of glutamate and proline pathways (both precursors of streptolydigin), and exogenously addition can increase the yield of streptolydigin production21. We found that S. lydicus 103 harbors the complete pathways to transform among the arginine, ornithine, glutamate and proline.
Streptolydigin was a polyketide compound synthesized by type I polyketide pathway, which shares same or similar precursors with the type II polyketide pathways22. Complete biosynthetic pathway of streptolydigin has been identified, so that the combined and metabolic processes could be further interpreted23. S. lydicus 103 harbors 67 ORFs covering a region of 111.2 kb, which are putatively assigned as streptolydigin biosynthesis genes encoding amino-acid permease, isocitrate dehydrogenase, lysophospholipase, erythronolide synthase, phenolphthiocerol synthesis polyketide synthase, cadicidin biosynthesis thioesterase, squalene cyclase, cytochrome P450, methylmalonyl-CoA mutase, glucose-1-phosphate thymidylyltransferase, lipopolysaccharide, biosynthesis protein and electron transfer flavoprotein etc.
The analysis of secondary metabolite pathways in S. lydicus 103
S. lydicus can produce a lot of important secondary metabolites, and a total of 27 gene clusters were predicted to be involved in secondary metabolism by antiSMASH. They are mainly focused on polyketide (PKS), nonribosomal peptide (NRPs) and terpene, and most of them have the really low similarity with the known clusters (Table 1).
As the typical PKS I, the biosynthetic pathway of erythromycin has been illuminated, including 6-deoxyerythronolide B (6-dEB) biosynthesis and glycosylation modification24. The 6-dEB was condensed by a molecule propionyl CoA and 6 molecules methyl malonyl CoA. The PKS gene cluster of erythromycin contains eryA I, eryA II and eryA III and encodes acyl wansferase, acyl carrier protein, ketosynthase, ketoreductase, dehydratase and enoyl reductase. The improvements of the erythromycin yield by metabolic engineering has been reported25. The product of 6-dEB and erythromycin A was reported in titers of 10 mg·L−1 26. As the typical NRPs, mannopeptimycin was first found in industrial bacterium Streptomyces hygroscopicus27. Mannopeptimycin comprises two distinct stereoisomers of amino acids, thus conforming glycosylated cyclic hexapeptide. Besides, with the different R groups, it can form diverse secondary metabolites. We identified a biosynthetic cluster showing 81% similarity with known mannopeptimycin biosynthetic cluster (BGC0000388_c1), which consists of polyprenyl mannose synthase MppG, polyprenyl phospho-mannosyltransferase MppHI, mannopeptimycin peptide synthetase MppAB, alpha/beta hydrolase MppK, ABC transporter MppL, isovaleryltransferase MppMN, PLP-dependent aminotransferase MppQ, putative transcriptional regulator MppS, hypothetical protein MppT, two component response regulator MppU, two component sensor kinase MppV, hypothetical lipoprotein MppW, ABC transporter MppX, conserved hypothetical protein MppYZ in S. lydicus 103.
Besides, S. lydicus 103 harbors the ectione biosynthetic pathway that shows 47% similarity with Streptomyces albulus PD-1. As one kind of compatible solute, ectoine can be used for protecting enzymes, membranes and whole cells against stresses28. The formation of hydroxyectoine in the ectoine producer Halomonas elongatawas was improved by the heterologous expression of the ectoine hydroxylase gene from Streptomyces chrysomallus29. We identify two ectoine dioxygenases (EctD), L-ectoine synthase (EctC), diaminobutyrate-pyruvate aminotransferase (EctB) and L-2,4-diaminobutyric acid acetyltransferase (EctA) in S. lydicus 103, which shows 75% similarity with known ectione biosynthetic cluster (BGC0000853_c1). As the family of siderophores, desferrioxamines can form strong hexadentate complexes with ferric iron. Desferrioxamine B has been used for the treatment of iron overload in human30. S. lydicus 103 harbors the desferrioxamine B biosynthetic pathway that shows 80% similarity with known desferrioxamine B biosynthetic cluster (BGC0000941_c1). Previous research has unambiguously identified desferrioxamine E as the major desferrioxamine siderophore produced by S. coelicolor M145 and has identified a cluster of four genes (desA-D) that directs desferrioxamine biosynthesis in this model actinomycete31. We also identify tetratricopeptide (TPR) protein, DesD-A, HTH domain of SpoOJ/ParA/ParB/repB family, 4-nitrophenylphosphatase, desferrioxamine E transporter and ABC-type Fe3+-siderophore transport system in S. lydicus 103.
Discussion
Although streptolydigin produced by S. lydicus has the activities mentioned above, the yield from the original strain is not very high yet. To achieve higher antibiotic streptolydigin productivity through metabolic regulation, propionate was fed during the fermentation of S. lydicus32. The streptolydigin yield, and the carbon fluxes of pentose phosphate pathway and the anaplerotic reaction were significantly increased after propionate feeding. However, it is very difficult to sharply improve the antibiotic production only by the traditional fermentation optimization and mutagenesis treatment. So it is urgent for us to make clear the metabolic network for antibiotic biosynthesis pathways to further improve the production. For example, the cluster slgE1-slgE2-slgE3 is involved in 3-methylaspartate (the precursor of the tetramic acid) supply. SlgE3, a ferredoxin-dependent glutamate synthase, is responsible for the biosynthesis of glutamate from glutamine and 2-oxoglutarate. The expression of slgE3 is increased up to 9-fold at the onset of streptolydigin biosynthesis33. The asparaginyl-tRNA synthetase-like SlgZ and methyltransferase SlgM enzymes are involved in the biosynthesis of the tetramic acid in S. lydicus. Over-expression of slgZ and slgM in S. lydicus led to a considerable increase in streptolydigin production34. SlnM gene overexpression with different promoters can improve the natamycin production in S. lydicus A0235. The biosynthetic genes or regulatory elements of a metabolite must be characterized prior to metabolic engineering36. Furthermore, modifications to the structures of secondary metabolites can often change the biological activity of the compound37. In this study, we presented the complete genome sequence of S. lydicus 103 and identified the pathways related to streptomyces biosynthesis from primary metabolism to secondary metabolite, which would provide more accurate analysis of the metabolic network and a more rational adjustment of metabolic regulation38.
Genomics-based bottom-up approaches have been developed to unveil biosynthetic pathways of new natural products that were undetected under standard fermentation conditions39. Despite being tapped as antibiotic sources for decades, Streptomyces spp. could produce up to 100,000 antimicrobial metabolites, while only a small proportion have been identified40. As an example, a terpene synthase from S. avermitilis was expressed in E. coli, resulting in the synthesis of the novel tricyclic sesquiterpene, avermitilol41. Chu et al.42 used primary sequence from the human microbiome, and thus bioinformatically predicted and chemical synthesized a new antibiotic. Luo et al.43 applied a plug-and-play synthetic biology strategy to activate a cryptic polycyclic tetramate macrolactams (PTMs) biosynthetic gene cluster from S. griseus and discovered three new PTMs. Besides, transcriptome and metabolome can identify the potential biosynthetic genes by correlating the expression of the secondary metabolite related gene44. In S. lydicus 103, we found many new gene clusters that have really low similarity with known clusters (Table 1). Thus, further studies are desirable for optimization, isolation and identification of the new bio-active molecule. The availability of the genome sequence of S. lydicus 103 provides a framework for biotechnological analysis and characterization of new natural products.
Methods
Bacterial culture and genome sequence
S. lydicus 103, an actinomycete, was isolated from soil. One loop of cells was incubated in a 250 mL flask containing 50 mL seed medium for 48 hours at 28 °C with shaking at 220 r·min−1. The seed medium contained (g·L−1): glucose 5, starch 30, yeast extract 2, peptone 4, K2HPO4 1.5, NaCl 0.5, and MgSO4.7H2O 0.5. Isolation of genomic DNA was carried out using SDS method. Total DNA obtained was subjected to quality control by agarose gel electrophoresis and quantified by Qubit. The genome was sequenced by Single Molecule, Real-Time (SMRT) technology. Sequencing was performed at the Beijing Novogene Bioinformatics Technology Co., Ltd. SMRT Analysis 2.3.0 was used to filter low quality reads and the filtered reads were assembled to the chromosome without gaps. The circular skeleton of chromosome was identified by the long fragment across the head and tail.
Genome annotation and bioinformatics analysis
Transfer RNA (tRNA) genes, Ribosome RNA (rRNA) genes, small RNA (sRNA) genes were predicted with tRNAscan-SE45, rRNAmmer46 and Rfam database47, respectively. Gene prediction was performed with the integrated model by NCBI prokaryotic annotation pipeline48, and gene functional prediction was performed by Blast49 against the databases, KEGG50 (Kyoto Encyclopedia of Genes and Genomes), COG51 (Clusters of Orthologous Groups), Swiss-Prot52, and GO53 (Gene Ontology). The origin of replication (oriC) and putative DnaA boxes were identified using Ori-Finder54. GC-Profile was used to compute the GC content variation in genome sequence and predict the genomic islands55. CGView Server56, a comparative genomics tool for circular genomes, was used to obtain a circular graphical representation of chromosome. A whole genome-based, alignment-free composition vector (CV) method was performed for phylogenetic analysis57 and the phylogenetic tree was generated using the MEGA program58. The toxin-antitoxin (TA) systems were predicted by TADB59. Secondary metabolite gene clusters were predicted by antiSMASH60. BioVenn, a web application for the comparison and visualization of biological lists, was used for Venn diagrams drawing61.
GenBank accession number
The sequence of the S. lydicus 103 genome has been deposited at DDBJ/EMBL/GenBank under the GenBank accession number CP017157.
Additional Information
How to cite this article: Jia, N. et al. Complete genome sequencing and antibiotics biosynthesis pathways analysis of Streptomyces lydicus 103. Sci. Rep. 7, 44786; doi: 10.1038/srep44786 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Paulsen, I. T. Carbon metabolism and its regulation in Streptomyces and other high GC Gram-positive bacteria. Res. Microbiol. 147(6), 535–541 (1996).
Ikeda, H. et al. Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis . Nat. Biotechnol. 21(5), 526–531 (2003).
Ramesh, M., Girish, B. & Mahalingeshwara, K. B. Thermophilic Fungi: Their physiology and enzymes. Microbiol. Mol. Biol. Rev. 64(3), 461–488 (2000).
Thompson, C. J. et al. Characterization of the herbicide-resistance gene bar from Streptomyces hygroscopicus . EMBO J. 6(9), 2519 (1987).
Seipke, R. F., Kaltenpoth, M. & Hutchings, M. I. Streptomyces as symbionts: an emerging and widespread theme? FEMS Microbiol. Rev. 36, 862–876 (2012).
Crawford, D. L., Kowalski, M., Roberts, M. A., Merrell, G. & Deobald, L. A. Discovery, development and commercialization of a microbial biocontrol agent, Streptomyces lydicus WYEC108: history of a decade long endeavor. SIM News. 55(3), 88–95 (2005).
Temiakov, D. et al. Structural basis of transcription inhibition by antibiotic streptolydigin. Mol. Cell. 19, 655–666 (2005).
Zhao, G.-R. et al. fabC of Streptomyces lydicus involvement in the biosynthesis of streptolydigin. Appl. Microbiol. Biotechnol. 83, 305–313 (2009).
Yu, F.-M., Qiao, B., Zhu, F., Wu, J.-C. & Yuan, Y.-J. Functional analysis of type II thioesterase of Streptomyces lydicus AS 4.2501. Appl. Biochem. Biotechnol. 135(2), 145–158 (2006).
Li, L.-Z., Qiao, B. & Yuan, Y.-J. Nitrogen sources affect streptolydigin production and related secondary metabolites distribution of Streptomyces lydicus AS 4.2501. Chin. J. Chem. Eng. 15(3), 403–410 (2007).
Cheng, J.-S. et al. Metabolic analysis reveals the amino acid responses of Streptomyces lydicus to pitching ratios during improving streptolydigin production. Appl. Microbiol. Biotechnol. 97(13), 5943–5954 (2013).
Cheng, J.-S., Lv, X.-M. & Yuan, Y.-J. Investigation of proteomic responses of Streptomyces lydicus to pitching ratios for improving streptolydigin production. Biotechnol. Bioprocess Eng. 17(5), 997–1007 (2012).
Wu, Q. et al. Construction of a Streptomyces lydicus A01 transformant with a chit42 gene from Trichoderma harzianum P1 and evaluation of its biocontrol activity against Botrytis cinerea . J. Microbiol. 51(2), 166–173 (2013).
Lin, Y. S., Kieser, H. M., Hopwood, D. A. & Chen, C. W. The chromosomal DNA of Streptomyces lividans 66 is linear. Mol. Microbiol. 10, 923–933 (1993).
Chater, K. F., Biro, S., Lee, K. J., Palmer, T. & Schrempf, H. The complex extracellular biology of Streptomyces . FEMS Microbiol. 34, 171–198 (2010).
Li, P. et al. Identification and characterization of chromosomal relBE toxin-antitoxin locus in Streptomyces cattleya DSM46488. Sci. Rep. 6, 32047 (2016).
Rokem, J. S., Lantz, A. E. & Nielsen, J. Systems biology of antibiotic production by microorganisms. Nat. Prod. Rep. 24, 1262–1287 (2007).
Borodina, I. et al. Antibiotic over production in Streptomyces coelicolor A3(2) mediated by phosphofructokinase deletion. J. Biol. Chem. 283, 25186–25199 (2008).
Heath, R. J. & Rock, C. O. Roles of the FabA and FabZ beta-hydroxyacyl-acyl carrier protein dehydratases in Escherichia coli fatty acid biosynthesis. J. Biol. Chem. 271, 27795–277801 (1996).
Li, L.-Z., Qiao, B. & Yuan, Y.-J. Nitrogen sources affect streptolydigin production and related secondary metabolites distribution of Streptomyces lydicus AS 4.2501. Chin. J. Chen. Eng. 15(3), 403–410 (2007).
Cheng, J.-S., Cui, S.-F., Ding, M.-Z. & Yuan, Y.-J. Insights into the roles of exogenous glutamate and proline in improving streptolydigin production of Streptomyces lydicus with metabolomic analysis. J. Ind. Microbiol. Biotechnol. 40, 1303–1314 (2013).
Chen, H. & Harrison, P. H. Investigation of the origin of C2 units in biosynthesis of streptolydigin. Org. Lett. 6, 4033–4036 (2004).
Olano, C. et al. Deciphering biosynthesis of the RNA polymerase inhibitor streptolydigin and generation of glycosylated derivatives. Chem. Biol. 16, 1031–1044 (2009).
Ward, S. L. et al. Chalcomycin biosynthesis gene cluster from Streptomyces bikiniensis:novel features of anunusual ketolide produced through expression of the chm polyketide synthase in Streptomyces fradiae. Antimicrob Agents Chemother. 48(12), 4703–4712 (2004).
Jiang, M. & Pfeifer, B. A. Metabolic and pathway engineering to influence native and altered erythromycin production through E. coli . Metab. Eng. 19, 42–49 (2013).
Zhang, H., Wang, Y., Wu, J., Skalina, K. & Pfeifer, B. A. Complete biosynthesis of erythromycin A and designed analogs using E. coli as a heterologous host. Chem. Biol. 17, 1232–1240 (2010).
Baker, D. D. & Alvi, K. A. Small-molecule natural products: new structures, new activities. Curr. Opin. Biotechnol. 15(6), 576–583 (2004).
Lippert, K. & Galinski, E. A. Enzyme stabilisation by ectoine-type compatible solutes: protection against heating, freezing and drying. Appl. Microbiol. Biotechnol. 37, 61–65 (1992).
Prabhu, J., Schauwecker, F., Grammel, N., Keller, U. & Bernhard, M. Functional expression of the ectoine hydroxylase gene (thpD) from Streptomyces chrysomallus in Halomonas elongata . Appl. Environ. Microbiol. 70(5), 3130–3132 (2004).
Propper, R. D., Shurin, S. B. & Nathan, D. G. Reassessment of the use of desferrioxamine B in iron overload. New England J. Medicine. 294(26), 1421–1423 (1976).
Barona-Gomez, F., Wong, U., Giannakopulos, A. E., Derrick, P. J. & Challis, G. L. Identification of a cluster of genes that directs desferrioxamine biosynthesis in Streptomyces coelicolor M145. J. Am. Chem. Soc. 126(50), 16282–16283 (2004).
Li, L.-Z., Zheng, H. & Yuan, Y.-J. Effects of propionate on streptolydigin production and carbon flux distributionin Streptomyces lydicus AS 4.2501. Chin. J. Chem. Eng. 15(2), 143–149 (2007).
Cristina, G. et al. Amino acid precursor supply in the biosynthesis of the RNA polymerase inhibitor streptolydigin by Streptomyces lydicus . J. Bacteriol. 193(16), 4214–4223 (2011).
Dina, H. H. et al. Biosynthesis of the RNA polymerase inhibitor streptolydigin in Streptomyces lydicus: tailoring modification of 3-Methyl-aspartate. J. Bacteriol. 193(10), 2647–2651 (2011).
Wu, H. L. et al. SlnM gene overexpression with different promoters on natamycin production in Streptomyces lydicus A02. J. Ind. Microbiol. Biotechnol. 41, 163–172 (2014).
Weber, T. et al. Metabolic engineering of antibiotic factories: new tools for antibiotic production in actinomycetes. Trends in Biotechnol. 33(1), 15–26 (2015).
Cummings, M., Breitling, R. & Takano, E. Steps towards the synthetic biology of polyketide biosynthesis. FEMS Microbiol. Lett. 351, 116–125 (2014).
Parekh, S., Vinci, V. A. & Strobel, R. J. Improvement of microbial strains and fermentation processes. Appl. Microbol. Biotechnol. 54, 287–301 (2000).
Winter, J. M., Behnken, S. & Hertweck, C. Genomics-inspired discovery of natural products. Curr. Opin. Chem. Biol. 15, 22–31 (2011).
Watve, M. G., Tickoo, R., Jog, M. M. & Bhole, B. D. How many antibiotics are produced by the genus Streptomyces? Arch. Microbiol. 176, 386–390 (2001).
Chou, W. K. et al. Genome mining in Streptomyces avermitilis: cloning and characterization of SAV_76, the synthase for a new sesquiterpene, avermitilol. J. Am. Chem. Soc. 132, 8850–8851 (2010).
Chu, J. et al. Discovery of MRSA active antibiotics using primary sequence from the human microbiome. Nat. Chem. Biology. 12(12), 1004–1006 (2016).
Luo, Y.-Z. et al. Activation and characterization of a cryptic polycyclic tetramate macrolactam biosynthetic gene cluster. Nat. Commun. 4, 2894 (2003).
Yonekura-Sakakibara, K. & Saito, K. Transcriptome coexpression analysis using ATTED-II for integrated transcriptomic/metabolomic analysis. Methods Mol. Biol. 1011, 317–326 (2013).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25(5), 0955–0964 (1997).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35(9), 3100–3108 (2007).
Gardner, P. P. et al. Rfam: updates to the RNA families database. Nucleic Acids Res. 37 (suppl 1), D136–D140 (2009).
Tatusova, T. et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44(14), 6614–6624 (2016).
Altschul, S. F. et al. Basic local alignment search tool. J. Mol. Biol. 215(3), 403–410 (1990).
Kanehisa, M. et al. The KEGG resource for deciphering the genome. Nucleic Acids Res. 32 (suppl 1), D277–D280 (2004).
Tatusov, R. L. et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 4(1), 41 (2003).
Magrane, M. UniProt Knowledgebase: a hub of integrated protein data. Database: J. Biological Databases & Curation. bar009 (2011).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat. Genetics. 25(1), 25–29 (2000).
Gao, F. & Zhang, C. T. Ori-Finder: a web-based system for finding oriCs in unannotated bacterial genomes. BMC Bioinformatics. 9, 79 (2008).
Gao, F. & Zhang, C. T. GC-Profile: a web-based tool for visualizing and analyzing the variation of GC content in genomic sequences. Nucleic Acids Res. 34, W686–W691 (2006).
Grant, J. R. & Stothard, P. The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 36 (suppl 2), W181–W184 (2008).
Xu, Z. & Hao, B. L. CVTree update: a newly designed phylogenetic study platform using composition vectors and whole genomes. Nucleic Acids Res. 37, W174–W178 (2009).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Shao, Y. et al. TADB: a web-based resource for Type 2 toxin-antitoxin loci in Bacteria and Archaea. Nucleic Acids Res. 39, D606–D611 (2011).
Blin, K. et al. antiSMASH 2.0-a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. gkt449 (2013).
Hulsen, T., de Vlieg, J. & Alkema, W. BioVenn-a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics. 9(1), 488 (2008).
Acknowledgements
This work was funded by the Ministry of Science and Technology of China (“973” Program: 2014CB745102), and the National Natural Science Foundation of China (21390203, 31571358, 21621004 and 21676190), the China National 863 High-Tech Program (2015AA020101) and the International S&T Cooperation Program of China (2015DFA00960). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
F.G., M.Z.D. and Y.J.Y. designed the project and experiments; N.J. performed the experiments; F.G. and Y.J.Y. contributed reagents/materials/analysis tools; N.J., M.Z.D., H.L. and F.G. analyzed the final data and wrote the manuscript. All the authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Jia, N., Ding, MZ., Luo, H. et al. Complete genome sequencing and antibiotics biosynthesis pathways analysis of Streptomyces lydicus 103. Sci Rep 7, 44786 (2017). https://doi.org/10.1038/srep44786
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep44786
This article is cited by
-
Genome features and secondary metabolite potential of the marine symbiont Streptomyces sp. RS2
Archives of Microbiology (2023)
-
Integrated genomics and proteomics analysis of Paenibacillus peoriae IBSD35 and insights into its antimicrobial characteristics
Scientific Reports (2022)
-
Comparative genomics of Alexander Fleming’s original Penicillium isolate (IMI 15378) reveals sequence divergence of penicillin synthesis genes
Scientific Reports (2020)
-
Antarctic Streptomyces fildesensis So13.3 strain as a promising source for antimicrobials discovery
Scientific Reports (2019)
-
Impacts of horizontal gene transfer on the compact genome of the clavulanic acid-producing Streptomyces strain F613-1
3 Biotech (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
