
Abstract
Alkenyl ether phospholipids are a major sub-class of ethanolamine- and choline-phospholipids in which a long chain fatty alcohol is attached at the sn-1 position through a vinyl ether bond. Biosynthesis of ethanolamine-containing alkenyl ether phospholipids, plasmalogens, is regulated by modulating the stability of fatty acyl-CoA reductase 1 (Far1) in a manner dependent on the level of cellular plasmalogens. However, precise molecular mechanisms underlying the regulation of plasmalogen synthesis remain poorly understood. Here we show that degradation of Far1 is accelerated by inhibiting dynamin-, Src kinase-, or flotillin-1-mediated endocytosis without increasing the cellular level of plasmalogens. By contrast, Far1 is stabilized by sequestering cholesterol with nystatin. Moreover, abrogation of the asymmetric distribution of plasmalogens in the plasma membrane by reducing the expression of CDC50A encoding a β-subunit of flippase elevates the expression level of Far1 and plasmalogen synthesis without reducing the total cellular level of plasmalogens. Together, these results support a model that plasmalogens localised in the inner leaflet of the plasma membranes are sensed for plasmalogen homeostasis in cells, thereby suggesting that plasmalogen synthesis is spatiotemporally regulated by monitoring cellular level of plasmalogens.
Similar content being viewed by others


Introduction
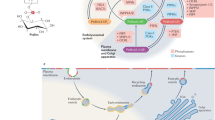
Alkenyl ether phospholipids are a subclass of glycerophospholipids characterized by a vinyl ether linkage of fatty alcohols to the sn-1 position of the glycerol backbone. Synthesis of ethanolamine (Etn)-containing alkenyl ether phospholipids (plasmalogens) is initiated in peroxisomes and completed in the endoplasmic reticulum (ER) through seven-step reactions1. Thereafter, plasmalogens are transported to the post-Golgi compartments mainly via a non-secretory pathway, in a manner dependent on the cellular ATP level2. The initial two steps of plasmalogen synthesis are catalyzed by peroxisomal matrix enzymes, dihydroxyacetonephosphate acyltransferase (Dhapat) and alkylglycerone phosphate synthase (Agps), in which 1-alkyl-dihydroxyacetonephosphate (DHAP) is generated by replacing the acyl chain of 1-acyl-DHAP with a long chain fatty alcohol that is synthesized by fatty acyl-CoA reductase 1 (Far1), a peroxisomal C-tail anchored protein3,4,5. Deficiency of plasmalogen synthesis evoked by dysfunction of peroxisomal enzymes essential for the plasmalogen synthesis causes rhizomelic chondrodysplasia punctata (RCDP), a fatal genetic disease6,7,8. Plasmalogen deficiency is also found in peroxisome biogenesis disorders (PBDs) where plasmalogens level is severely reduced in several organs such as brain, heart, and kidney from patients with PBDs9, implying that de novo synthesis of plasmalogens plays a pivotal role in the tissue plasmalogen homeostasis.
We earlier showed that plasmalogen synthesis is regulated by modulating the stability of Far1 in response to the cellular levels of plasmalogens4,5. Elevation of plasmalogens is found in tumour10,11,12, whereas plasmalogens are reduced in the patients with several diseases including sporadic Alzheimer’s disease and Pelizaeus-Merzbacher disease13,14,15. In addition, the cellular plasmalogen level plays an important role in cholesterol homeostasis at multiple steps including cholesterol synthesis16, esterification of cholesterol17, high-density lipoprotein (HDL)-mediated cholesterol efflux18, internalization of cholesterol19, and distribution of cholesterol20. Given these findings, plasmalogen synthesis must be tightly regulated to maintain the homeostasis of plasmalogens by sensing the cellular level of plasmalogens. However, the mechanisms of regulation in plasmalogen synthesis remain undefined. To address this issue, we investigated how cells sense the cellular level of plasmalogens. The results suggest that plasmalogens localised in the inner leaflet of the plasma membrane are sensed, of which signal is transferred to peroxisomes to modulate the stability of Far1.
Results
Plasmalogen-induced degradation of Far1 in wild-type cells
Plasmalogen biosynthesis is regulated by modulating the stability of Far1 in response to the cellular level of plasmalogens4. We earlier showed that restoration of plasmalogens in the plasmalogen-deficient cell mutants such as agps ZPEG251 or elevation of plasmalogens in wild-type cells enhances the degradation of Far14,5. In addition, the expression level of Far1 in CHO-K1 and HeLa cells was reduced in the presence of cycloheximide (CHX) (Fig. 1), whereas degradation of Far1 is suppressed in ZPEG2514, implying that Far1 is degraded by the turnover rate to maintain the homeostasis of plasmalogens through the constitutive sensing of cellular plasmalogens in wild-type cells (hereafter called plasmalogen-induced degradation of Far1).

CHO-K1 (A) and HeLa (B) cells were cultured in the presence (+) or absence (−) of CHX for the indicated time. Expression level of Far1 at each time point in the absence (diamond) or presence (square) of CHX was assessed. Peroxisomal membrane protein Pex3p and β–actin for a loading control were detected with respective specific antibodies (left panels). Molecular masses were indicated on the right, using the migration of standard mass markers. Relative amounts of Far1 at each time point were represented by taking as 100 those at the time point of CHX addition in respective cells (right panels). *p < 0.05, **p < 0.01; Student’s t test compared with the expression level of Far1 at each time point in the absence of CHX. Data of unprocessed original blots are available in Supplementary Fig. S2.
Dynamin-mediated endocytosis is involved in the regulation of plasmalogen synthesis
Plasmalogens are transported from the ER to the post-Golgi compartments in a manner dependent on ATP, but independent on secretory pathway2. However, plasmalogens are not localised to peroxisomes2,21,22. Therefore, we suspected that plasmalogens in either ER or the post-Golgi compartments including plasma membrane are sensed for the regulation of plasmalogen synthesis. To test if plasmalogens in the plasma membrane are sensed, we attempted to inhibit endocytic pathways with Dynasore, an inhibitor of dynamin23. Dynamin plays a crucial step in the fission of newly formed vesicles in several endocytic pathways, including clathrin-mediated and clathrin-independent endocytic processes such as caveolin- as well as clathrin- and caveolin-independent pathways24. When cells were incubated with 14C-Etn in the presence of Dynasore, plasmalogen synthesis was reduced to about 30% of that in mock-treated cells, whereas synthesis of phosphatidylethanolamine (PE) was elevated (Fig. 2, lanes 2 and 3). These results are similar to the synthesis of plasmalogens and PE in CHO cell mutants defective in early step(s) of plasmalogen synthesis including DHAPAT-defective NRel-425 and AGPS-deficient ZPEG2512 and NZel-126. In contrast, treatment with chlorpromazine (CPZ), an inhibitor of clathrin-dependent endocytosis and macropinocytosis27, lowered the synthesis of both plasmalogens and PE in a similar fashion, indicating that the plasmalogen synthesis was not specifically reduced by CPZ, rather incorporation of 14C-Etn and/or the common pathway(s) for the synthesis of both plasmalogens and PE were suppressed (Fig. 2, lanes 2 and 4). However, expression and peroxisomal localisation of Dhapat and Agps were not altered in the presence of Dynasore (Supplementary Fig. S1A,B). We next assessed the expression of Far1, a rate-limiting enzyme of plasmalogen synthesis4,5. Protein level of Far1 was reduced upon the treatment with Dynasore in CHO-K1 (Fig. 3A, lanes 2 and 3), which was further decreased in the presence of CHX (Fig. 3A, lane 4). In contrast, expression of Far1 was not altered by treatment with Dynasore in agps ZPEG251 (Fig. 3B), where accumulation of transferrin (Tf) in recycling endosomes was inhibited with Dynasore as reported23 (Supplementary Fig. S1C, lower panel). Transcription of FAR1 in CHO-K1 cells was not reduced in the presence of Dynasore (Fig. 3C). Together, these results suggest that plasmalogen-induced degradation of Far1 is augmented by Dynasore. However, total cellular level of plasmalogens was not elevated in the presence of Dynasore (Fig. 3D).

(A) CHO-K1 cells were incubated with DMSO, Dynasore, or CPZ for 30 min and further metabolically labelled with 14C-Etn for 5 h in the absence (−) and presence of each inhibitor. The biosynthesis of PE and plasmalogen (2-acyl-GPE) were detected with a FLA-5000 imaging analyser (upper panel). Total cellular level of phospholipids including PE, PS, phosphatidylinositol (PI), plasmalogens, phosphatidylcholine (PC), and SM were detected with iodine vapor (lower panel). Note that biosynthesis of plasmalogens was specifically reduced in the presence of Dynasore. (B) Biosynthesis of PE (open bar), plasmalogens (dark grey bar), PE plus plasmalogens (light grey bar) was represented by taking the amounts of PE plus plasmalogens as 100 in mock-treated (−) CHO-K1. Statistical analysis was performed by one-way ANOVA with Dunnett’s post hoc test as compared with the synthesis of respective lipids in mock-treated cells. *p < 0.05, **p < 0.01, and ***p < 0.001.

(A) CHO-K1 cells were cultured for 5 h in the presence (+) of CHX (lane 1), Dynasore (lane 3), or CHX plus Dynasore (lane 4) and were assessed for the expression level of Far1 (upper panel). β–actin was used as a loading control. Relative expression levels of Far1 were represented by taking those as 100 in mock-treated cells (lane 2, lower panel). Note that Far1 expression was reduced in the presence of Dynasore in CHO-K1 cells (lane 3). **p < 0.01 and ***p < 0.001; two-way ANOVA with Tukey-Kramer post hoc test. (B) agps ZPEG251 was cultured for 5 h in the presence (+) or absence (−) of Dynasore and were assessed for the expression level of Far1 (upper panel). Relative expression levels of Far1 were represented by taking those as 100 in mock-treated cell (lower panel). n.s., not significant; t test. (C) Expression level of FAR1 relative to β–Actin was determined by real-time PCR using total RNA, and represented by taking as 100 that in CHO-K1 (−). n.s., not significant; t test. (D) Total cellular plasmalogens were analysed by LC-ESI-MS/MS and represented by taking that as 100 in CHO-K1 (−). n.s., not significant; t test. (E) Lipids were extracted from cells cultured in the absence (−) or presence (+) of Dynasore and assessed by TLC (upper panel) as described in Materials and Methods. Cholesterol was detected with iodine vapor and quantified (lower panel) using an image analysis software (Multi Gauge). n.s., not significant; t test. (F) CHO-K1 cells were cultured for 5 h in the presence (+) or absence (−) of MiTMAB and assessed for the expression level of Far1 (upper panel). Relative expression levels of Far1 were represented by taking those as 100 in mock-treated cells (lower panel). **p < 0.01; t test. Data of unprocessed original blots are available in Supplementary Fig. S2.
Dynasore inhibits GTPase activity of dynamin-like protein 1 (Dlp1) both in vitro23 and in vivo28,29. Therefore, we investigated whether Dynasore-mediated inactivation of Dlp1 induces degradation of Far1 using dlp1 CHO mutant ZP12130. Consistent with the partial defect of plasmalogen synthesis in ZP12130, expression level of Far1 in ZP121 was higher than that in CHO-K1 cells (Supplementary Fig. S1D), implying that inactivation of Dlp1 is not the primary cause for the reduction of the expression level of Far1 upon the treatment with Dynasore in CHO-K1 cells. Dynasore also reduces cellular cholesterol and disrupts rafts31. However, we did not observe the reduction of cholesterol when CHO-K1 cells were incubated with Dynasore (Fig. 3E). Finally, we verified the expression level of Far1 with myristyl trimethyl ammonium bromide (MiTMAB), an inhibitor that inhibits GTPase activity of dynamin by disrupting the interaction between pleckstrin homologydomain of dynamin and phospholipids32, and found that the expression level of Far1 was reduced to about 70% of that in mock-treated CHO-K1 cells upon the treatment with MiTMAB (Fig. 3F). Taken together, these results suggest that inhibition of dynamin-mediated endocytic pathways most likely accelerates plasmalogen-induced degradation of Far1.
Knockdown of flotillin-1 enhances plasmalogen-induced degradation of Far1
We further investigated whether inhibition of caveolin- or flotillin-1 (Flot1)-mediated endocytosis enhances plasmalogen-induced degradation of Far1. To this end, plasmalogen-deficient agps ZPEG251 cells were cultured with purified plasmalogens in the presence of PP2, an inhibitor specific for Src-kinase essential for both types of caveolin-dependent and Flot1-mediated endocytosis33,34. Expression of Far1 was lowered in ZPEG251 cells upon culturing with purified plasmalogens as compared to that in untreated ZPEG251, which was further reduced in the presence of Dynasore (Fig. 4A, lanes 1–3), confirming that inhibition of dynamin-mediated endocytosis induces degradation of Far1 (Fig. 3A, lanes 2 and 3). Protein level of Far1 was similarly reduced in ZPEG251 in the presence of PP2 and purified plasmalogens, whereas PP2 alone did not affect the expression level of Far1 (Fig. 4A, lanes 4 and 5). Under these conditions, total amount of cellular plasmalogens was not altered by the treatment with Dynasore and PP2 as compared with those in ZPEG251 cells cultured with purified plasmalogens (Fig. 4B). Furthermore, protein level of Far1 was reduced by nearly 50% upon knocking down FLOT1 using two independent double-stranded RNAs (dsRNAs) (Fig. 5A, lower panel), where Flot1 protein level was lowered to about 20% of that in mock-treated cells (Fig. 5A, upper panel). Consistent with these results, the knockdown of FLOT1 reduced the plasmalogen synthesis to about 50% level of that in HeLa cells (Fig. 5B), whereas transcription of FAR1 and total cellular plasmalogen level were not altered (Fig. 5C). In contrast, protein level of Far1 was not altered under the more than 60% reduction of caveolin1 (Cav1) expression in HeLa cells treated with CAV1 RNAi (Fig. 6A). Next, we examined whether elevation of plasmalogens stimulates degradation of Far1 in HepG2 cells, a cell line that shows no detectable caveolins and caveolae35 but expresses Flot1 (Fig. 6B). When HeLa cells were cultured in the presence of Etn, cellular plasmalogen level was increased about 1.5-fold as compared to that in mock-treated cells16, thereby stimulating the degradation of Far1 by sensing the elevated level of plasmalogens (Fig. 6B,C, lanes 1 and 2)4,5. Similar extent of the degradation of Far1 was observed in HepG2 cells as the plasmalogen level was increased by supplementation of Etn (Fig. 6B,C, lanes 3 and 4), implying that plasmalogen-induced degradation of Far1 is not abrogated under the suppressed caveolin-mediated endocytosis. Together, these results suggest that inhibition of Flot1-mediated endocytosis most likely enhances plasmalogen-induced degradation of Far1.

(A) agps ZPEG251 was cultured in the presence (lanes 2–4) or absence (lanes 1 and 5) of purified plasmalogens (puri. Pls) for 6 h, and further cultured in the presence of Dynasore (lane 3) or PP2 (lanes 4 and 5) for 5 h. Expression levels of Far1 were assessed by immunoblotting (upper panel) and represented (lower panel) by taking as 100 that in ZPEG251 (lane 1). ###p < 0.001; one-way ANOVA with Dunnett’s post hoc test as compared with mock-treated cells. n.s., not significant. *p < 0.05 and **p < 0.01; one-way ANOVA with Dunnett’s post hoc test as compared with ZPEG251 treated with purified plasmalogens (lane 2). (B) Cellular plasmalogens were analysed by LC-ESI-MS/MS and represented by taking as 100 that in ZPEG251 cultured in the presence of purified plasmalogens (lane 2). **p < 0.01 and ***p < 0.001; one-way ANOVA with Tukey post hoc test as compared with ZPEG251. n.s., not significant. Data of unprocessed original blots are available in Supplementary Fig. S2.

(A) Expression levels of Far1 and Flot1 were assessed in HeLa cells transfected with control dsRNA (lane 1) or two different dsRNAs (lanes 3 and 4) against FLOT1. Expression level of Far1 was represented by taking as 100 that in mock-treated cell (lower panel). (B) Plasmalogen synthesis was assessed in mock-treated (−) or FLOT1-knocked down (#68 and #69) HeLa cells by metabolically labelling with 14C-Etn for 2 h (upper panel). Biosynthesis of plasmalogens was represented by taking as 100 that in mock-treated cells (lower panel). *p < 0.05, **p < 0.01, and ***p < 0.001; one-way ANOVA with Dunnett’s post hoc test as compared with mock-treated cells. (C) Left panel, total cellular level of plasmalogens was represented by taking as 100 that in mock-treated cells. Right panel, expression level of FAR1 relative to ribosomal protein 3 (RPL3)63 was determined by real-time PCR using total RNA, and represented by taking as 100 that in HeLa cells (−). n.s., not significant; one-way ANOVA with Dunnett’s post hoc test as compared with mock-treated cells. Data of unprocessed original blots are available in Supplementary Fig. S2.

(A) Expression levels of Far1 (solid bar) and Cav1 (open bar) were assessed in HeLa cells transfected with dsRNAs against CAV1. β–actin was used as a loading control. Expression level of Far1 was represented by taking as 100 that in mock-treated cell (right panel). **p < 0.01; t test versus mock-treated cells. n.s., not significant. (B) HeLa and HepG2 were cultured in the presence of Etn (5 μM) for 48 h and assessed for expression level of Far1, β–actin, Pex3p, Cav1, and Flo1. Note that Cav1 is not expressed in HepG2 cells35. Cellular plasmalogens were analysed by LC-ESI-MS/MS and represented by taking those as 100 in mock-treated respective cells. **p < 0.01; t test versus mock-treated respective cells. (C) Expression level of Far1 was represented by taking those as 100 in mock-treated respective cells. **p < 0.01; t test versus mock-treated respective cells. Data of unprocessed original blots are available in Supplementary Fig. S2.
Sensing of plasmalogens in the inner leaflet of plasma membrane
Given the findings that plasmalogen-induced degradation of Far1 is enhanced by the knockdown of Flot1 without elevating total cellular plasmalogen level (Fig. 5C), we suspected that the degradation of Far1 is accelerated by sensing plasmalogens locally enriched in the plasma membrane upon inhibiting Flot1-mediated endocytosis. In this hypothesis, elevation of plasmalogens in lipid rafts might be a prerequisite for the sensing because plasmalogens are enriched in lipid rafts2,36 and knocking down of FLOT1 inhibits the endocytosis of raft-associated proteins such as CD59, i.e. a glycosylphosphatidylinositol (GPI)-linked protein, and a cholera toxin B subunit in HeLa cells37. Therefore, we verified the amount of plasmalogens and sphingomyelin (SM) in the detergent-resistant membranes (DRMs) in FLOT1-knocked down HeLa cells (Fig. 7). When HeLa cells were solubilized with Triton X-100 on ice and separated into DRMs and Triton X-100 soluble fractions by floatation on a 30% iodixanol gradient, Cav1 was mostly recovered in DRMs, while Tf receptor (Tfr), a non-raft protein, was completely solubilized by Triton X-100 as reported2 (Fig. 7B, lanes 1 and 2). In FLOT1-knocked down HeLa cells, Cav1 and SM, not Tfr, were recovered in DRMs similar to those in mock-treated HeLa cells where the transfection of one of the dsRNAs against FLOT1 induced protein level of Tfr (Fig. 7B,C). However, amount of plasmalogens in DRMs prepared from FLOT1-knocked down HeLa cells was about 1.5-fold higher than that in mock-treated cells (Fig. 7D), suggesting that plasmalogens are locally enriched in lipid rafts upon reducing the expression of Flot1. We further examined if plasmalogens in the plasma membrane are sensed by sequestering cholesterol in the plasma membrane. To this end, CHO-K1 cells were treated with nystatin, a cholesterol-chelating agent, inducing distortion of the structure and function of cholesterol-rich membrane domain38 and assessed for the degradation of Far1 in the presence of CHX. Degradation of Far1 was significantly suppressed by the treatment with nystatin (Fig. 7E) without reducing total cellular level of plasmalogens and cholesterol (Fig. 7F,G). We interpreted these results to mean that the plasmalogen-induced degradation of Far1 is most likely retarded by interfering with the sensing steps of plasmalogens localised in the plasma membrane.

(A) HeLa cells were transfected with two different dsRNAs (lanes 2 and 3) against FLOT1 and expression of Flot1 was assessed. (B) DRMs (R) and detergent-soluble (S) fractions were prepared as described in Materials and Methods and distribution of Cav1 and Tfr was assessed using respective antibodies. (C,D) Amount of SM (C) and plasmalogens (D) in DRMs prepared as in (B) was determined by LC-ESI-MS/MS and represented by arbitrary units. *p < 0.05; one-way ANOVA with Dunnett’s post hoc test versus mock-treated HeLa cells. n.s., not significant. (E) CHO-K1 cells were cultured for 1 h with nystatin (Nys.), a cholesterol-chelating agent, further incubated for the indicated time in the presence of CHX, and assessed for the expression of Far1 at each time point (upper panel). β–actin was used as a loading control. Relative amounts of Far1 in the presence (triangle) of nystatin at each time point were represented by taking as 100 that at the time point of CHX addition. The relative expression level of Far1 in the absence (square) of nystatin was from Fig. 1A (left panels). **p < 0.01; t test compared with the expression level of Far1 at 9 h in the absence of nystatin. (F) Cellular plasmalogens in CHO-K1 cells cultured in the absence (−Nys.) or presence (+Nys.) of nystatin were analysed by LC-ESI-MS/MS and their relative level was represented by taking as 100 that at (0 h, -Nys.). n.s., not significant by two-way ANOVA. (G) Lipids were extracted from cells cultured as in (F). Cholesterol was detected as described in Fig. 3E. Relative amount of cholesterol is represented by taking as 100 that at (0 h, -Nys.). *p < 0.05 and **p < 0.01; two-way ANOVA with Tukey post hoc test. n.s., not significant. Note that cellular cholesterol was reduced by the treatment of CHX but not with nystatin.
Finally, we assessed if plasmalogens located in the inner leaflet of the plasma membrane are sensed. It is known that aminophospholipids such as phosphatidylserine (PS) and PE are concentrated in the inner leaflet of the plasma membrane, where type-IV P-type ATPases (P4-ATPases) function in the phospholipid transfer from the outer leaflet to the inner leaflet39. Since plasmalogens are also shown located in the inner leaflet of plasma membrane in red blood cells and myelin40,41, we verified the protein level of Far1 by reducing the expression of CDC50A encoding a β-subunit of P4-ATPases that is essential for exiting of most of P4-ATPases from the ER by forming a heterooligomer42,43. Knockdown of CDC50A using two independent dsRNAs elevated the protein level of Far1 (Fig. 8A,B). We also determined distribution of plasmalogens at the outer leaflet with 2,4,6-trinitrobenzene sulfonic acid (TNBS), a membrane impermeable amine-reactive reagent44. TNBS-modified PE was detected with a higher mobility on TLC than PE. Upon the trichloroacetic acid (TCA)-treatment, by which the vinyl ether linkage of plasmalogens is hydrolysed, thereby giving rise to two more slower-migrating bands each corresponding to TNBS-plasmalogens (TNBS-2-acyl-GPE) and unmodified plasmalogens (2-acyl-GPE) (Fig. 8C), where TNBS-plasmalogens and TNBS-PE represented 3.7 ± 0.2% and 6.8 ± 0.5%, respectively, of total plasmalogens and PE (Fig. 8C, lane 4). Knockdown of CDC50A elevated the levels of TNBS-plasmalogens and TNBS-PE (Fig. 8D) and augmented the synthesis of plasmalogen and PE (Fig. 8E) where total amount of PE and plasmalogens was not significantly increased (Fig. 8F). Collectively, these results suggest that plasmalogens located in the inner leaflet of plasma membrane are sensed for detection of the cellular level of plasmalogens.

(A) Expression level of Far1 was assessed in mock-treated HeLa cells (−) or cells transfected with control dsRNA (C) or two different dsRNAs (#22 and #23) against CDC50A (upper panel). β–actin was used as a loading control. Expression level of Far1 was represented by taking as 100 that in mock-treated cell (lower panel). *p < 0.05 and **p < 0.01; one-way ANOVA with Dunnett’s post hoc test versus mock-treated cells. (B) HeLa cells were cultured as described in (A). Expression level of CDC50A relative to RPL363 was determined by real-time PCR using total cell RNA. ***p < 0.001; one-way ANOVA with Dunnett’s post hoc test versus mock-transfected cells (−). (C) HeLa cells were cultured in the presence of 14C-Etn for 18 h and further incubated with (+) or without (−) TNBS. Total cellular lipids were extracted from aliquots of equal amounts of cell proteins before (−) or after (+) treatment with TCA and analysed by TLC. (D) PE and plasmalogens localised at the outer leaflet of plasma membrane were verified as in (C) in mock-transfected HeLa cells (−) or CDC50A-knocked down cells by transfection with dsRNA (#22) (left panel). Relative amounts of TNBS-modified PE (TNBS-PE, open bar) and plasmalogens (TNBS-2-acyl-GPE, solid bar) were represented by taking as 100 those in mock-transfected cells (right panel). *p < 0.05 and **p < 0.01; t test versus mock-transfected cells. (E) Plasmalogen synthesis was assessed in mock-transfected (−) or CDC50A-knocked down (#22) HeLa cells by metabolically labelling with 14C-Etn for 5 h (left panel). Biosynthesis of PE (open bar) and plasmalogens (solid bar) was represented by taking as 100 those in mock-transfected cells (right panel). *p < 0.05 and **p < 0.01; t test versus mock-transfected cells. (F) Relative level of PE (open bar) and plasmalogens (solid bar) in CDC50A-knocked down (#22) HeLa cells was represented by taking as 100 those in mock-transfected cells. n.s., not significant; t test versus mock-transfected cells. Data of unprocessed original blots are available in Supplementary Fig. S2.
Discussion
Plasmalogen synthesis is regulated by a feedback mechanism in a manner dependent on the cellular level of plasmalogens4,5. In the present study, we showed that plasmalogen-induced degradation of Far1 is enhanced by the inhibition of dynamin-, Src kinase-, and Flot1-mediated endocytosis (Figs 3,4 and 5), whereas perturbation of inner leaflet localisation of plasmalogens or sequestering of cholesterol in the plasma membrane suppressed the degradation of Far1 without altering total cellular level of plasmalogens (Figs 7 and 8). Together, these results suggest that plasmalogens localised in the inner leaflet of the plasma membrane is critical for sensing of the cellular level of plasmalogens.
Flot1 is shown to be enriched in lipid rafts of the plasma membrane37,45 and its association with lipid rafts in DHAPAT-knockout mice is reduced46, suggesting that the association of Flot1 with lipid rafts is involved in sensing of plasmalogens. If so, knocking down of FLOT1 would abrogate the sensing system for plasmalogens, stabilize Far1, and elevate plasmalogen synthesis. However, our data showed the converse that knockdown of FLOT1 reduced the expression of Far1 and plasmalogen synthesis, suggesting that another unidentified protein, if any, monitors the cellular level of plasmalogens.
Flot1 is involved in signaling pathway, cell adhesion, and membrane trafficking47. Our findings that the inhibition of dynamin or Src-kinase reduce the expression of Far1 and knocking down of FLOT1 elevates the plasmalogen level in DRMs (Fig. 7D) enriched in cholesterol and SM, both most abundant in the plasma membrane48, suggest that inhibition of the Flot1-mediated endocytic pathway stimulates the degradation of Far1 by increasing the plasmalogen level in the plasma membrane. Based on these findings, we propose that newly synthesized plasmalogens are transported from the ER to the plasma membrane and dynamically endocytosed, rather than being statically localised in plasma membrane. Accordingly, Flot1-mediated endocytosis plays an important role in the plasmalogen homeostasis, while caveolae is less likely involved in the plasmalogen-dependent degradation of Far1 (Fig. 6). Noteworthily, caveolae is not found in neuron49,50 and lymphocytes51. In contrast, Flot1 is expressed in several cell types including neuron52,53 and lymphocytes53,54. Together, our findings suggest that Flot1-mediaed endocytosis plays a crucial role in the plasmalogen homeostasis in most tissues. At the next steps toward elucidation of the flotillin-mediated endocytic pathway in plasmalogen homeostasis, flotillin-2 (Flot2) may need to be verified since Flot2 that is 47% identical to Flot1 in the primary structure55 functions together with Flot1 to generate flotillin microdomains by binding to plasma membrane inner leaflet via SPFH (stomatin, prohibitin, flotillin, Hflk/C) domain and acylation of the cysteine residues in its N-terminal hydrophobic stretch56.
The physiological consequence of the enrichment of plasmalogens in lipid rafts has not been defined. Plasmalogens do not seem to be a structurally essential constituent for the formation of lipid rafts2. The interference with cholesterol-mediated assembly and function of the membrane by nystatin significantly reduces degradation of Far1 (Fig. 7E). Nystatin does not cross the bilayer and enter the cells57, supporting a model for sensing of plasmalogens in the plasma membrane. Nystatin disrupts caveolae morphology and inhibits the function of caveolae without altering clathrin-mediated endocytosis38. However, the elevated level of plasmalogens was sensed, followed by stimulating degradation of Far1 in caveolae-lacking HepG2 cells (Fig. 6). Therefore, plasmalogens that reside within cholesterol-rich domain of the plasma membrane such as non-caveolae rafts seem to be important for the sensing of plasmalogens. Clearly, more precise manipulation of membrane property or lipid constituent in non-caveolae rafts and identification of the molecule sensing plasmalogens are required for full understanding of the sensing step(s) of plasmalogens.
Plasmalogens are located in the inner leaflet of plasma membrane in red blood cells and myelin, which is shown by using spin-labelled plasmalogens in red blood cells40 or by X-ray with mercuric chloride in myelin41, respectively. By a combination of TNBS modification and TCA treatment of the extracted lipids, one-step analysis was made feasible in the detection of TNBS-PE, TNBS-plasmalogens, unmodified PE, and plasmalogens (Fig. 8C). In the analysis using HeLa cells, 5.2 ± 0.2% of total Etn-containing phospholipids corresponding to plasmalogens plus PE were modified with TNBS, although the level of TNBS-modified Etn-phospholipids was slightly lower than that obtained from CHO-K1 in G1 phase44. Therefore, TNBS-modification assay is also useful and a simple procedure for the detection of outer leaflet-localised plasmalogens. In HeLa cells, about 4% of total plasmalogens were modified by TNBS, less than that of PE (Fig. 8C, lane 4), implying that asymmetric distribution of PE and plasmalogens, both types of phospholipids harboring Etn in their head group, is distinctly regulated. To date, fourteen P4-ATPases are identified in humans and the importance of ATP8A1-mediated PE translocation is assessed in cell migration58. We show here that knockdown of CDC50A significantly elevates localisation of plasmalogens at cell-surface, giving rise to the increase in plasmalogen synthesis by reducing plasmalogen-induced degradation of Far1, hence implying that P4-ATPase-mediated asymmetric distribution of plasmalogens is important for sensing the plasmalogens. We should await identification of P4-ATPase(s) responsible for the topogenesis of plasmalogens and delineation of the physiological roles of the P4-ATPase(s) in cellular functions.
In summary, plasmalogen biosynthesis is regulated by modulating the stability of Far1 by sensing the plasmalogens localised in the inner leaflet of plasma membrane, where several mechanisms involving Flot1-mediated endocytosis, P4-ATPase-dependent asymmetric distribution of plasmalogens, and recognition of plasmalogens by yet unidentified proteins, if any, coordinately contribute. Subsequently, a signal emerged from sensing the plasmalogen level is spatially transferred to peroxisomes, where the Far1 stability is modulated. Therefore, plasmalogen synthesis is spatiotemporally regulated via several machineries locating in the plasma membrane and peroxisomes.
Materials and Methods
Cell culture
HeLa4 and HepG2 (a gift from Dr. H. Sumimoto. Kyushu Univ.) were cultured in DMEM supplemented with 10% FBS (Biowest) in 5% CO2 and 95% air4. Chinese hamster ovary (CHO)-K1and adaps ZPEG2512 were cultured in Ham’s F-12 medium supplemented with 10% FBS in 5% CO2 and 95% air2. Cells were cultured in the presence of several types of inhibitors as follows: CHO-K1 cells were cultured in the presence of 80 μM Dynasore (Sigma)23, 25 μM CPZ (Sigma), or 30 μM MiTMAB (TOCRIS) for 5 h. ZPEG251 was treated with 10 μM PP2 (Sigma) for 5 h in the presence of purified plasmalogens4. CHO-K1 cells were treated with 25 μg/ml nystatin (Sigma) for 1 h and further cultured in the presence of 10 μg/ml cycloheximide (Nacalai Tesque) for the indicated period. Knockdown of FLOT1, CAV1, and CDC50A was performed by transfecting dsRNAs against respective genes to HeLa cells and the cells were cultured for 3 days. The target sequences of the dsRNA are as follows: human Flotillin-1–68, 5′-GGGCATCAGTGTGGTTAGCTACACT-3′; human Flotillin-1–69, 5′- CGGGAAGCTAAAGCCAAGCAGGAAA-3′ (Invitrogen); human Caveolin1-9504, 5′-CCCTAAACACCTCAACGATTT-3′ (Sigma); human CDC50A-22, 5′-GAGCTATTGCCAACAGCATTT-3′; and human CDC50A-23, 5′-CGTGTTTATGTATTATGGATT-3′ (Sigma).
Floatation
Cell homogenate (250 μl) containing 200 μg protein was treated with 1% Triton X-100 on ice for 30 min and was adjusted to 40% iodixanol with 500 μl of OptiPrep (Invitrogen) containing 60% of iodixanol. The sample (700 μl) in TLS55 centrifuge tubes (Beckman) was overlaid with 1.2 ml of 30% iodixanol/TNE (150 mM NaCl/2 mM EDTA in 50 mM Tris⋅HCl, pH 7.4) and 0.1 ml of TNE, and centrifuged at 55,000 rpm (259,000 × g) for 2 h at 4 °C. Two 1-ml fractions were collected from the top. Two fractions each containing the detergent-resistant (R) and detergent-soluble (S) materials was collected. Total lipids were extracted from 700 μl of each fraction and analysed for the amount of plasmalogens and SM by liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS)59.
Lipid analysis
Cells were cultured in the presence of 0.1 μCi/ml of 14C-Etn (Moravek). Equal aliquots (100 μg protein) of cell lysates were subjected to lipid extraction by the Bligh and Dyer method60. For the detection of plasmalogens, cell lysates were treated with TCA to hydrolyse vinyl-ether bond of plasmalogens prior to lipid extraction, which generates 2-acyl-GPE2. Lipids were analysed on TLC plates (silica gel 60, Merck) with chloroform/methanol/acetic acid solution (v/v/v: 65/25/10)2 or hexane/diethyl ether/acetic acid solution (v/v/v: 80/20/1.5)16. Total cellular plasmalogens were analysed by LC-ESI-MS/MS59. Distribution of plasmalogens in the outer leaflet of plasma membranes was assessed by TNBS (Wako) modification44. In brief, cells were metabolically labelled with 14C-Etn for 18 h, washed with ice-cold SHT buffer (0.25 M sucrose, 10 mM Hepes-KOH pH 8.5) containing 1 μg/ml taxol (Sigma), incubated on ice for 5 min, and further treated with 10 mM TNBS dissolved in SHT buffer on ice for 30 min in the dark. Excess TNBS was quenched for 15 min with 50 mM Tris-HCl pH 8.0. Lipids were extracted and analysed on TLC as described above. 14C-labelled lipids were detected by autoradiography using a FLA-5000 imaging analyser or TyphoonTM FLA 9500 biomolecular imager and quantified using an image analyser software (Multi Gauge, Fuji Film).
Immunoblot analysis
Cells were harvested in homogenizing buffer2. Equal aliquots of total proteins (3–5 μg) were separated by SDS-PAGE, and subjected to immunoblotting with rabbit polyclonal antibodies to Far14, Pex3p61, Agps2. Rabbit antiserum to human Dhapat was raised against a carboxyl-terminal portion encompassing amino-acid residues between 343 and 680. Guinea pig anti-Pex14pC62, goat anti-lactate dehydrogenase antibody (LDH) (Rockland) and mouse antibodies to β–actin (MBL), Cav1 (Santa Cruz), Flot1 (BD Transduction Laboratories), and Tfr (Zymed Laboratories) were used. After probing with HRP-conjugated secondary antibodies, immunoblots were developed with ECL reagents (GE Healthcare), and visualized by an LAS-4000 Mini luminescent image analyser (Fuji Film). The intensity of bands was quantified by Multi Gauge software version 3.0 software (Fuji Film) within the linear range of detection. Precision Plus Protein™ All Blue Standards (BioRad) was used as standard molecular mass markers.
Real-time PCR
Total RNA was isolated from cells using TRIzol reagent (Invitrogen) and synthesis first-strand cDNA was performed using the PrimeScript RT reagent Kit (Takara Bio). Quantitative real-time RT-PCR was performed in an Mx3000 P QPCR system (Agilent Technologies) using SYBR Premix Ex Taq II (Takara Bio). Primers used were as follows: Chinese hamster FAR1 sense: ClFar1-287Fw. 5′-AAACACCACAAGAGCGAGTG-3′; antisense ClFar1-412Rv. 5′-CGAGTTTAGGTTGGGTGAGTTC-3′, Chinese hamster β–actin sense: ClbAct562Fw. 5′-ACCTGACAGACTACCTCAT-3′; antisense ClbAct630Rv. 5′-ACGCACAATTTCCCTCTC-3′, Human FAR1 sense: HsFar1-149Fw. 5′-AGACACCACAAGAGCGAGTG-3′; antisense HsFar1-253v. 5′-CCAGTTTAGGTTGGGTGAGTTC-3′.
Endocytosis of Tf
CHO-K1 and ZPEG251 cells were cultured in the medium containing FBS plus Dynasore for 4 h and further cultured for 1 h with medium containing Dynasore in the absence of FBS23. The cells were then incubated with 50 μg/ml of Tf tagged with Alexa Fluor 633 (Molecular Probes) for 15 min at 37 °C23. After the fixation, cells were incubated with phalloidin conjugated with Alexa Fluor 568 (Molecular Probes) according to the manufacturer’s instruction.
Immunofluorescence Microscopy
Cells were fixed using 4% paraformaldehyde for 15 min at room temperature, permeabilised with 1% Triton X-100 for 2 min, and blocked with 1% bovine serum albumin (Nacalai Tesque) in PBS (blocking buffer) for 30 min. Cells were then incubated with indicated primary antibodies in blocking buffer for 1 h. Antigen-antibody complexes were visualized using Alexa Fluor 488-goat anti-rabbit or Alexa Fluor 647-labelled goat anti-guinea pig IgG antibody (Invitrogen). Cells were observed using a confocal laser microscope (LSM710; Carl Zeiss). Images were acquired with a ZEN2012 software (Carl Zeiss).
Data presentation
Expression level of Far1 was normalized by the expressed level of β–actin in each sample. Quantitative data were shown as mean ± S.D from at least three independent experiments. Statistical significance was determined by Student’s t-test, or ANOVA with Dunnett’s, Tukey, or Tukey-Kramer post hoc test using R software (http://www.r-project.org).
Additional Information
How to cite this article: Honsho, M. et al. Plasmalogen biosynthesis is spatiotemporally regulated by sensing plasmalogens in the inner leaflet of plasma membranes. Sci. Rep. 7, 43936; doi: 10.1038/srep43936 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Nagan, N. & Zoeller, R. A. Plasmalogens: biosynthesis and functions. Prog. Lipid Res. 40, 199–229 (2001).
Honsho, M., Yagita, Y., Kinoshita, N. & Fujiki, Y. Isolation and characterization of mutant animal cell line defective in alkyl-dihydroxyacetonephosphate synthase: Localization and transport of plasmalogens to post-Golgi compartments. Biochim. Biophys. Acta 1783, 1857–1865 (2008).
Cheng, J. B. & Russell, D. W. Mammalian wax biosynthesis. I. Identification of two fatty acyl-coenzyme a reductases with different substrate specificities and tissue distributions. J. Biol. Chem. 279, 37789–37797 (2004).
Honsho, M., Asaoku, S. & Fujiki, Y. Posttranslational regulation of fatty acyl-CoA reductase 1, Far1, controls ether glycerophospholipid synthesis. J. Biol. Chem. 285, 8537–8542 (2010).
Honsho, M., Asaoku, S., Fukumoto, K. & Fujiki, Y. Topogenesis and homeostasis of fatty acyl-CoA reductase 1. J. Biol. Chem. 288, 34588–34598 (2013).
Braverman, N. E. & Moser, A. B. Functions of plasmalogen lipids in health and disease. Biochim. Biophys. Acta. 1822, 1442–1452 (2012).
Buchert, R. et al. A peroxisomal disorder of severe intellectual disability, epilepsy, and cataracts due to fatty acyl-CoA reductase 1 deficiency. Am. J. Hum. Genet. 95, 602–610 (2014).
Barøy, T. et al. A novel type of rhizomelic chondrodysplasia punctata, RCDP5, is caused by loss of the PEX5 long isoform. Hum. Mol. Genet. 24, 5845–5854 (2015).
Heymans, H. S. A., Schutgens, R. B. H., Tan, R., van den Bosch, H. & Borst, P. Severe plasmalogen deficiency in tissues of infants without peroxisomes (Zellweger syndrome). Nature 306, 69–70 (1983).
Snyder, F. & Wood, R. Alkyl and alk-1-enyl ethers of glycerol in lipids from normal and neoplastic human tissues. Cancer Res. 29, 251–257 (1969).
Smith, R. E. et al. A reliable biomarker derived from plasmalogens to evaluate malignancy and metastatic capacity of human cancers. Lipids 43, 79–89 (2008).
Benjamin, D. I. et al. Ether lipid generating enzyme AGPS alters the balance of structural and signaling lipids to fuel cancer pathogenicity. Proc. Natl. Acad. Sci. USA. 110, 14912–14917 (2013).
Ginsberg, L., Rafique, S., Xuereb, J. H., Rapoport, S. I. & Gershfeld, N. L. Disease and anatomic specificity of ethanolamine plasmalogen deficiency in Alzheimer’s disease brain. Brain Res. 698, 223–226 (1995).
Han, X., Holtzman, D. M. & McKeel, D. W. Jr. Plasmalogen deficiency in early Alzheimer’s disease subjects and in animal models: molecular characterization using electrospray ionization mass spectrometry. J. Neurochem. 77, 1168–1180 (2001).
Wood, P. L., Smith, T., Pelzer, L. & Goodenowe, D. B. Targeted metabolomic analyses of cellular models of pelizaeus-merzbacher disease reveal plasmalogen and myo-inositol solute carrier dysfunction. Lipids Health Dis. 10, 102 (2011).
Honsho, M., Abe, Y. & Fujiki, Y. Dysregulation of plasmalogen homeostasis impairs cholesterol biosynthesis. J. Biol. Chem. 290, 28822–28833 (2015).
Mankidy, R. et al. Membrane plasmalogen composition and cellular cholesterol regulation: a structure activity study. Lipids Health Dis. 9, 62 (2010).
Mandel, H. et al. Plasmalogen phospholipids are involved in HDL-mediated cholesterol efflux: insights from investigations with plasmalogen-deficient cells. Biochem. Biophys. Res. Commun. 250, 369–373 (1998).
Munn, N. J., Arnio, E., Liu, D., Zoeller, R. A. & Liscum, L. Deficiency in ethanolamine plasmalogen leads to altered cholesterol transport. J. Lipid Res. 44, 182–192 (2003).
Thai, T. P. et al. Impaired membrane traffic in defective ether lipid biosynthesis. Hum. Mol. Genet. 10, 127–136 (2001).
Yang, J., Han, X. & Gross, R. W. Identification of hepatic peroxisomal phospholipase A(2) and characterization of arachidonic acid-containing choline glycerophospholipids in hepatic peroxisomes. FEBS Lett. 546, 247–250 (2003).
Kuerschner, L. et al. Polyene-lipids: a new tool to image lipids. Nat. Methods 2, 39–45 (2005).
Macia, E. et al. Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell. 10, 839–850 (2006).
Mayor, S. & Pagano, R. E. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 8, 603–612 (2007).
Liu, D. et al. Role of dihydroxyacetonephosphate acyltransferase in the biosynthesis of plasmalogens and nonether glycerolipids. J. Lipid Res. 46, 727–735 (2005).
Nagan, N. et al. A fibroblast cell line defective in alkyl-dihydroxyacetone phosphate synthase: a novel defect in plasmalogen biosynthesis. Proc. Natl. Acad. Sci. USA. 94, 4475–4480 (1997).
Ivanov, A. I. Pharmacological inhibition of endocytic pathways: is it specific enough to be useful? Methods Mol. Biol. 440, 15–33 (2008).
Itoyama, A. et al. Docosahexaenoic acid mediates peroxisomal elongation, a prerequisite for peroxisome division. J. Cell Sci. 125, 589–602 (2012).
Gao, D. et al. Dynasore protects mitochondria and improves cardiac lusitropy in Langendorff perfused mouse heart. PLoS One 8, e60967 (2013).
Tanaka, A., Kobayashi, S. & Fujiki, Y. Peroxisome division is impaired in a CHO cell mutant with an inactivating point-mutation in dynamin-like protein 1 gene. Exp. Cell Res. 312, 1671–1684 (2006).
Preta, G., Lotti, V., Cronin, J. G. & Sheldon, I. M. Protective role of the dynamin inhibitor Dynasore against the cholesterol-dependent cytolysin of Trueperella pyogenes . FASEB J. 29, 1516–1528 (2015).
Hill, T. A. et al. Long chain amines and long chain ammonium salts as novel inhibitors of dynamin GTPase activity. Bioorg. Med. Chem. Lett. 14, 3275–3278 (2004).
Sharma, D. K. et al. Selective stimulation of caveolar endocytosis by glycosphingolipids and cholesterol. Mol. Biol. Cell 15, 3114–3122 (2004).
Riento, K., Frick, M., Schafer, I. & Nichols, B. J. Endocytosis of flotillin-1 and flotillin-2 is regulated by Fyn kinase. J. Cell Sci. 122, 912–918 (2009).
Fujimoto, T., Kogo, H., Nomura, R. & Une, T. Isoforms of caveolin-1 and caveolar structure. J. Cell Sci. 113, 3509–3517 (2000).
Pike, L. J., Han, X., Chung, K. N. & Gross, R. W. Lipid rafts are enriched in arachidonic acid and plasmenylethanolamine and their composition is independent of caveolin-1 expression: a quantitative electrospray ionization/mass spectrometric analysis. Biochemistry 41, 2075–2088 (2002).
Glebov, O. O., Bright, N. A. & Nichols, B. J. Flotillin-1 defines a clathrin-independent endocytic pathway in mammalian cells. Nat. Cell Biol. 8, 46–54 (2006).
Ros-Baró, A. et al. Lipid rafts are required for GLUT4 internalization in adipose cells. Proc. Natl. Acad. Sci. USA. 98, 12050–12055 (2001).
Lopez-Marques, R. L., Theorin, L., Palmgren, M. G. & Pomorski, T. G. P4-ATPases: lipid flippases in cell membranes. Pflugers Arch. Eur. J. Physiol. 466, 1227–1240 (2014).
Fellmann, P., Herve, P. & Devaux, P. F. Transmembrane distribution and translocation of spin-labeled plasmalogens in human red blood cells. Chem. Phys. Lipids 66, 225–230 (1993).
Kirschner, D. A. & Ganser, A. L. Myelin labeled with mercuric chloride. Asymmetric localization of phosphatidylethanolamine plasmalogen. J. Mol. Biol. 157, 635–658 (1982).
van der Velden, L. M. et al. Heteromeric interactions required for abundance and subcellular localization of human CDC50 proteins and class 1 P4-ATPases. J. Biol. Chem. 285, 40088–40096 (2010).
Bryde, S. et al. CDC50 proteins are critical components of the human class-1 P4-ATPase transport machinery. J. Biol. Chem. 285, 40562–40572 (2010).
Kobayashi, T. & Pagano, R. E. Lipid transport during mitosis. Alternative pathways for delivery of newly synthesized lipids to the cell surface. J. Biol. Chem. 264, 5966–5973 (1989).
Bickel, P. E. et al. Flotillin and epidermal surface antigen define a new family of caveolae-associated integral membrane proteins. J. Biol. Chem. 272, 13793–13802 (1997).
Rodemer, C. et al. Inactivation of ether lipid biosynthesis causes male infertility, defects in eye development and optic nerve hypoplasia in mice. Hum. Mol. Genet. 12, 1881–1895 (2003).
Banning, A., Kurrle, N., Meister, M. & Tikkanen, R. Flotillins in receptor tyrosine kinase signaling and cancer. Cells 3, 129–149 (2014).
Simons, K. & Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1, 31–39 (2000).
Gaudreault, S. B., Blain, J.-F., Gratton, J.-P. & Poirier, J. A role for caveolin-1 in post-injury reactive neuronal plasticity. J. Neurochem. 92, 831–839 (2005).
Masserini, M., Palestini, P. & Pitto, M. Glycolipid-enriched caveolae and caveolae-like domains in the nervous system. J. Neurochem. 73, 1–11 (1999).
Harris, J. et al. Expression of caveolin by bovine lymphocytes and antigen-presenting cells. Immunology 105, 190–195 (2002).
Lang, D. M. et al. Identification of reggie-1 and reggie-2 as plasmamembrane-associated proteins which cocluster with activated GPI-anchored cell adhesion molecules in non-caveolar micropatches in neurons. J. Neurobiol. 37, 502–523 (1998).
Stuermer, C. A. O. et al. Glycosylphosphatidyl inositol-anchored proteins and fyn kinase assemble in noncaveolar plasma membrane microdomains defined by reggie-1 and -2. Mol. Biol. Cell 12, 3031–3045 (2001).
Rajendran, L. et al. Asymmetric localization of flotillins/reggies in preassembled platforms confers inherent polarity to hematopoietic cells. Proc. Natl. Acad. Sci. USA. 100, 8241–8246 (2003).
Edgar, A. J. & Polak, J. M. Flotillin-1: gene structure: cDNA cloning from human lung and the identification of alternative polyadenylation signals. Int. J. Biochem. Cell Biol. 33, 53–64 (2001).
Bodin, S., Planchon, D., Rios Morris, E., Comunale, F. & Gauthier-Rouvière, C. Flotillins in intercellular adhesion - from cellular physiology to human diseases. J. Cell Sci. 127, 5139–5147 (2014).
Horn, R. & Marty, A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. J. Gen. Physiol. 92, 145–159 (1988).
Kato, U. et al. Role for phospholipid flippase complex of ATP8A1 and CDC50A proteins in cell migration. J. Biol. Chem. 288, 4922–4934 (2013).
Abe, Y., Honsho, M., Nakanishi, H., Taguchi, R. & Fujiki, Y. Very-long-chain polyunsaturated fatty acids accumulate in phosphatidylcholine of fibroblasts from patients with Zellweger syndrome and acyl-CoA oxidase1 deficiency. Biochim. Biophys. Acta 1841, 610–619 (2014).
Bligh, E. G. & Dyer, W. J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physio. 37, 911–917 (1959).
Ghaedi, K., Tamura, S., Okumoto, K., Matsuzono, Y. & Fujiki, Y. The peroxin Pex3p initiates membrane assembly in peroxisome biogenesis. Mol. Biol. Cell 11, 2085–2102 (2000).
Mukai, S., Ghaedi, K. & Fujiki, Y. Intracellular localization, function, and dysfunction of the peroxisome-targeting signal type 2 receptor, Pex7p, in mammalian cells. J. Biol. Chem. 277, 9548–9561 (2002).
Wang, B. T. et al. The mammalian target of rapamycin regulates cholesterol biosynthetic gene expression and exhibits a rapamycin-resistant transcriptional profile. Proc. Natl. Acad. Sci. USA 108, 15201–15206 (2011).
Acknowledgements
We thank Y. Nanri and S. Okuno for technical assistance, K. Shimizu for preparing Figures, and the other members of our laboratory for discussions. This work was supported in part by Grants-in-Aid for Scientific Research 23570236 and 26440102 (to M.H.) and 24247038, 25112518, 25116717, 26116007, 15K14511, and 15K21743 (to Y.F.); grants (to Y.F.) from the Takeda Science Foundation, the Naito Foundation, The Japanese Plasmalogen Society, and Japan Foundation for Applied Enzymology.
Author information
Authors and Affiliations
Contributions
M.H. designed the research and performed the experiments. Y.A. performed the analysis by LC-ESI-MS/MS. M.H. and Y.F. interpreted the data and wrote the manuscript. All authors reviewed this manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Honsho, M., Abe, Y. & Fujiki, Y. Plasmalogen biosynthesis is spatiotemporally regulated by sensing plasmalogens in the inner leaflet of plasma membranes. Sci Rep 7, 43936 (2017). https://doi.org/10.1038/srep43936
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep43936
This article is cited by
-
Integrated analysis of gut metabolome, microbiome, and exfoliome data in an equine model of intestinal injury
Microbiome (2024)
-
Lipid composition of the cancer cell membrane
Journal of Bioenergetics and Biomembranes (2020)
-
Plasmalogen lipids: functional mechanism and their involvement in gastrointestinal cancer
Lipids in Health and Disease (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
