
Abstract
Expression of the solute carrier (SLC) transporter SLC22A3 gene is associated with overall survival of pancreatic cancer patients. This study tested whether genetic variability in SLC22A3 associates with pancreatic cancer risk and prognosis. Twenty four single nucleotide polymorphisms (SNPs) tagging the SLC22A3 gene sequence and regulatory elements were selected for analysis. Of these, 22 were successfully evaluated in the discovery phase while six significant or suggestive variants entered the validation phase, comprising a total study number of 1,518 cases and 3,908 controls. In the discovery phase, rs2504938, rs9364554, and rs2457571 SNPs were significantly associated with pancreatic cancer risk. Moreover, rs7758229 associated with the presence of distant metastases, while rs512077 and rs2504956 correlated with overall survival of patients. Although replicated, the association for rs9364554 did not pass multiple testing corrections in the validation phase. Contrary to the discovery stage, rs2504938 associated with survival in the validation cohort, which was more pronounced in stage IV patients. In conclusion, common variation in the SLC22A3 gene is unlikely to significantly contribute to pancreatic cancer risk. The rs2504938 SNP in SLC22A3 significantly associates with an unfavorable prognosis of pancreatic cancer patients. Further investigation of this SNP effect on the molecular and clinical phenotype is warranted.
Similar content being viewed by others

Introduction
Pancreatic ductal adenocarcinoma (PDAC, OMIM: 260350) has an extremely poor prognosis1 mostly due to the late diagnosis of disease, when all treatment options are limited. Thus, it is imperative to improve prevention and early detection efforts, such as locating genetic markers of PDAC risk that could inform early detection of the disease.
There are several established epidemiological risk factors for PDAC, e.g., smoking, obesity, personal history of chronic pancreatitis or diabetes, and family history of cancers2. A small fraction of PDACs are caused by high-risk predisposing mutations in DNA repair and damage sensing genes, e.g., BRCA1, BRCA2, PALB2, ATM, CDKN2A, APC, MLH1, MSH2, MSH6, PMS2, PRSS1, and STK113. Several genome-wide association studies (GWAS) have identified several low-penetrance loci associating with PDAC risk4,5,6,7. These authors estimated that the current loci identified in European populations account for approximately only 5% of the inherited pancreatic cancer risk, indicating that a large portion of familial risk alleles remain to be revealed.
The role of the SLC22A subfamily of solute carrier (SLC) transporters in PDAC progression is, at present, not well understood. SLC22A1, SLC22A2, and SLC22A3 mediate the transport of a variety of structurally diverse cations comprising both endogenous and exogenous compounds, e.g., neurotransmitters such as catecholamines and xenobiotics (including drugs), respectively8,9.
Our recent study revealed a highly significant upregulation of SLC22A3 transcripts in PDAC tumors compared with non-neoplastic tissues10. Moreover, a high level of SLC22A3 mRNA in tumors strongly predicted a longer overall survival (P = 0.004) in chemotherapy-treated patients. Association studies also suggest that genetic variability in SLC22A3 is likely to be associated with the risk of different cancer types. A colorectal cancer GWAS reported that rs7758229 in the SLC22A3 gene (Gene ID: 6581) was significantly associated with distal colon cancer risk in Asians11. Interestingly, SNP rs9364554 in intron 5 of SLC22A3 was previously shown to associate with prostate cancer in Caucasian populations12 suggesting a pleiotropic effect of the SNP.
This study tested the hypothesis that genetic variation in the SLC22A3 gene contributes to pancreatic cancer risk and disease survival. The tagging approach of the whole SLC22A3 gene region, including regulatory elements, was used in a two-stage genetic association study of Europeans.
Results and Discussion
Associations of SLC22A3 SNPs with pancreatic cancer risk
In this study, an association analysis of SNPs tagging the SLC22A3 gene with PDAC risk was performed in a two-stage design comprising in total 1,254 cases and 3,391 controls of European descent (Table 1).
We genotyped 208 PDAC cases and 381 controls from the Czech Republic in the discovery phase13,14. Three SNPs (rs2504938, rs9364554, and rs2457571) were significantly associated with PDAC risk in at least one of the genetic models tested (Table 2). These three SNPs were further analyzed in the validation phase comprising 1,046 and 3,010 controls from the PANDoRA (PANcreatic Disease ReseArch), European case-control study of PDAC15. A significant association for rs9364554 (OR = 1.19, 95% CI = 1.02–1.40, p = 0.030) was observed (Table 3), which did not pass the FDR test (q = 0.008) for correction of multiple comparisons. Moreover, combined analysis of both sets rendered all associations as non-significant (Supplementary Table S1). Thus, the results suggest that genetic variability in SLC22A3 probably does not significantly contribute to PDAC risk in the European population.
The trends observed by univariate analyses in the discovery stage did not change in the multivariate analyses adjusted to age, sex, body mass index (BMI), smoking status, and alcohol consumption with the exception of rs4708867 where carriage of the rare G allele was associated with an increased PDAC risk (Supplementary Table S2). Due to the lack of lifestyle data in the validation phase this association could not be replicated.
The present study attempted, for the first time, to find a link between the genetic variability in the SLC22A3 gene and PDAC risk. The recently reported GWAS on PDAC risk did not find any association with SLC22A3 tagging variants at genome wide significance6,7. Thus, despite previous reports on association of rs7758229 SNP with colorectal cancer risk11 and rs9364554 with prostate cancer risk12,16 the present study confirms that common genetic variability in SLC22A3 most probably does not modify PDAC risk. Cancer-specific effects, largely unknown gene-environmental interactions, and inter-population (even between Europeans) heterogeneity may underlie the observed differences.
Associations of SLC22A3 SNPs with pancreatic cancer survival
The second goal of this study was to assess whether genetic variability in SLC22A3 associates with major clinical characteristics of PDAC considering our previously reported association of intratumoral SLC22A3 gene expression with overall survival of PDAC patients10. In the discovery phase, the number of carriers of the T allele or heterozygous genotype in rs7758229 was significantly higher in patients with metastatic disease than in those without distant metastases (OR = 3.63, 95% CI = 1.46–9.06, p = 0.006 for heterozygotes compared with the GG genotype carriers and OR = 2.76, 95% CI = 1.20–6.32, p = 0.016 for T allele carriers compared with the GG genotype carriers, Table 4). Development of metastases is a major sign of cancer spread due to aggressive behavior of the tumor and predicts poor prognosis. Therefore, this SNP was added to the list of SNPs for validation.
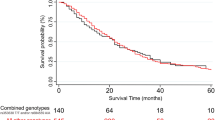
Additionally, patients carrying the AA genotype in rs512077 or T allele in rs2504956 had significantly better overall survival than the other patients (Fig. 1), suggesting that these SNPs might serve as markers of PDAC patient prognosis. None of the other analyzed SNPs in the discovery set were significantly associated with disease outcomes.

The difference in the mean survival between the compared groups of patients was significant (p = 0.034 and p = 0.045 for rs512077 and rs2504956 SNPs, respectively). Hazard ratios, 95% confidence intervals, and p-values calculated by the stage-adjusted Cox regression are presented in Supplementary Table S3.
None of these associations with either metastasis or survival were replicated in the validation phase (all p-values > 0.05). However, patients carrying the TT genotype in rs2504938 had highly significantly worse overall survival than patients with the CC genotype in the validation set (p = 0.002, Fig. 2) which notably retains significance after FDR correction for multiple testing in the validation phase (q = 0.008). However, the combined analysis of discovery and validation sets was not significant (p = 0.073, Supplementary Figure S1). Stage-adjusted analysis of all three SNPs (rs512077, rs2504956, and rs2504938) in both sets separately and combined also showed no significant associations (Supplementary Table S3). When analyzing patients stratified by stage, we observed that patients with stage IV disease carrying the TT genotype in rs2504938 had significantly worse OS than CC genotype carriers (p = 0.012, Fig. 3). In patients with less advanced disease (stages I–III) no such association was found (Fig. 3).

The difference in the mean survival between the compared groups of patients was significant (p = 0.002) in the validation phase. Hazard ratios, 95% confidence intervals, and p-values calculated by the stage-adjusted Cox regression are presented in Supplementary Table S3.

The difference in the mean survival between the compared groups of patients with stage IV disease was significant (p = 0.012).
LD analysis in cases from validation phase suggested that rs2504956 and rs512077 are in high LD (r2 = 0.95), rs2504938 and rs2504956 in strong LD (r2 = 0.81) and rs2504938 and rs512077 in weak LD (r2 = 0.42). This analysis strengthens the observed genetic link of SLC22A3 polymorphisms (rs2504956 in the discovery set and rs2504938 in the validation set) with the OS of PDAC patients. A more refined study of these two loci and surrounding sequences may shed more light into the prognostic importance of SLC22A3 variability in PDAC.
A potential functional effect of the rs2504938 and rs2504956 SNPs was tested by two ways. First, we analyzed the in silico prediction by HaploReg v3 indicating that rs2504938 may alter motifs for DNA binding proteins and transcription factors Hmx_1 (H6 Family Homeobox 1 DNA binding protein, OMIM: 142992) and NF-kappaB_known3 (Nuclear Factor Kappa B, OMIM: 164011). Additionally, rs2504956 may alter motifs E2A_2 and E2A_5 (Transcription Factor 3, OMIM: 147141), Hic1_1 (Hypermethylated in Cancer 1, OMIM: 603825), and ZBTB7A_known1 (Zinc Finger- and BTB Domain-containing Protein 7 A, OMIM: 605878). Second, we analyzed whether rs2504938 allele distribution correlates with gene expression of SLC22A3 in tumor (n = 17) and paired adjacent non-malignant tissues (n = 15) of the subgroup of patients assessed by our previous study10. Although there were modest tissue sample numbers available, the comparison suggests no significant correlation of rs2504938 or rs2504956 with SLC22A3 expression in PDAC tissues (p > 0.05). Thus, any potential influence of the rs2504938 SNP on PDAC survival would not appear to act via the gene expression level. Alternatively, a currently unknown link with other, potentially functional, genetic variation may explain the observed association with survival of PDAC patients.
Moreover, in the light of recent findings demonstrating that neurotransmitters help stimulate prostate tumor growth and metastasis17 and accelerate pancreatic cancer cell growth and invasion18, it would be worthwhile examining whether SLC22A3 might be involved in cancer tumorigenesis through the clearance of these active compounds.
As our previous study showed an association of SLC22A3 gene expression with overall survival only in patients treated with nucleoside analogs10, it would be very interesting to perform a survival analysis stratified by therapy. However, PANDoRA does not yet have sufficiently relevant data for such an analysis at present. Together with this, variations in age in both sets and BMI distribution in the discovery set are limitations of this study. Although, there were no considerable differences between crude and adjusted analyses of both sets, we cannot exclude a potential for some false negative findings. Future meta-analysis of results of this and subsequent independent studies will help to further evaluate these reported associations.
In conclusion, the present study suggests that common genetic variability in the SLC22A3 gene is not significantly associated with risk of PDAC. The rs2504938 SNP was associated with overall survival in the large PANDoRA study when evaluated in univariate manner and especially in stage IV patients, although the biological basis of this correlation remains to be elucidated.
Subjects and Methods
Study populations
We used a two-step strategy with a discovery phase consisting of biological samples from 245 PDAC patients (cases) and 442 controls of Czech Caucasian origin collected in the Czech Republic between 2004 and 2010. Patients were eligible for the study, when they fulfilled at least one of the following criteria: (a) patient had histology- or cytology-confirmed pancreatic adenocarcinoma or (b) at least three of clinical signs of pancreatic cancer (ERCP, EUS with FNAB, mass on CT or MRI, weight loss, anorexia/cachexia, obstructive jaundice). Clinical and pathological data on the cases (date of diagnosis, stage, grade, and histologic diagnosis where available) were collected from their medical records. The controls were included into the study under the condition that the difference in their age was not larger than 5 years from cases recruited in the same period. Basic epidemiological data on all participants (personal history, smoking and drinking history, physical activity, occupational and nutritional information) were collected (for case and control recruitment criteria see refs 13,14). The validation phase consisted of 1,273 cases and 3,466 controls enrolled into the PANcreatic Disease ReseArch (PANDoRA) consortium from three other European countries (Germany, Italy, and Poland). For all cases and controls a DNA sample from blood and/or pancreatic tissue was available, as well as a minimal set of covariates (such as age at diagnosis, sex, disease stage, age of death or at last follow-up for majority of cases). Different region-specific subpopulations of unmatched controls have been selected among the general population, blood donors and among hospitalized subjects with different diagnosis excluding cancer (described in detail in ref. 15).
There are no relevant data concerning chemotherapy treatments and responses so far. Relevant baseline characteristics of the studied populations are shown in Table 1.
Informed consent was obtained from all participating subjects for these studies in accord with the Declaration of Helsinki. All samples were coded to protect patient anonymity. The study was approved by the Ethical Committee of the University of Heidelberg (reference number S-565/2015). All methods were performed in accordance with guidelines and regulations set by the above Ethical Committee.
Selection of polymorphisms
The SLC22A3 gene region together with 10 kb sequences flanking the 5′ and 3′ ends (chr6: chr6:160680000…160806000, NCBI assembly 36) was analyzed by HaploView v4.2 program using a pairwise tagging approach with r2 > 0.819. SNPs with minor allele frequency (MAF) > 0.01 in HapMap CEU sample (International HapMap Project, version 28; http://www.hapmap.org) and at least 75% genotype data were identified. Together 24 SNPs tagging 139 alleles in the analyzed region were selected for analysis in the discovery phase.
The chromosomal locations and minor allele frequencies of the tested SNP variants are listed in Supplementary Table S4.
Genotyping
DNA from the cases and controls in the discovery sample set was isolated from peripheral lymphocytes using a BioSprint 15 DNA Blood kit (Qiagen, Valencia, CA) by KingFisher mL automated system (Thermo Electron Corporation, Vantaa, Finland) according to the manufacturer’s protocol. DNA from participants in the PANDoRA cohort was isolated from whole blood using the Qiagen-mini kit or the AllPrep Isolation kit (both Qiagen) using provider’s protocol. DNA was quantified by Quant-iT PicoGreen DNA Assay Kit (Invitrogen).
In the discovery phase, 24 SNPs were analyzed in DNA from 245 PDAC cases and 442 controls of Czech origin using KASPar technology (LGC Genomics, Hoddesdon, UK). Validation failed for rs12212246 due to unspecific amplification and therefore this SNP was not further analyzed.
During the validation phase, six candidate SNPs (i.e., those that showed an association in the discovery phase with either the risk or clinical outcomes of PDAC in the Czech cohort) were genotyped in the PANDoRA sample set consisting of 1,273 cases and 3,466 controls of European origin. Genotyping was performed at National Institute of Public Health, Prague, Czech Republic by allelic discrimination using TaqMan technology (Life Technologies Corp., Foster City, CA) in a ViiA7 real-time instrument with a 384-well block (Life Technologies). SNP assay reaction conditions are summarized in Supplementary Table S5.
Quality control was performed by determination of duplicate samples for approximately 10% of the samples in both phases. The genotyping concordance between duplicate samples exceeded 99%. All samples with less than 75% successful genotypes for all SNPs were discarded from further analysis. In total, the generated genotypes for 208 cases and 381 controls in the discovery phase and 1,046 cases and 3,010 controls in the validation phase were then analyzed. Together 23 SNPs were successfully genotyped.
Statistical analysis
Hardy Weinberg Equilibrium (HWE) was first examined in control subjects for each SNP. Genotype distribution of the studied SNPs did not deviate from HWE (p > 0.05) with the exception of rs3004079. The rs3004079 variant was therefore excluded from further analyses. Unconditional logistic regression was then used to assess the association of the 22 remaining SNPs with PDAC risk in the discovery phase. Co-dominant, dominant, and recessive genetic models were evaluated. Crude and adjusted for age (continuous), sex, and country of origin odds ratios (OR), 95% confidence intervals (CI), and p-values were calculated for each SNP. Age-, sex-, body mass index-, smoking status-, and alcohol consumption-adjusted analyses were performed by logistic regression in the discovery phase.
Associations of SNPs with prognostic clinical data (tumor size, presence of lymph node and distant metastases) were evaluated by the Cochran’s and Mantel-Hanszel statistics. Overall survival (OS) was defined as time elapsed from diagnosis to patient death, or to the last date at which the patient was known to be alive. Patients lost to follow up were excluded from analyses. Survival functions were plotted by the Kaplan-Meier method and statistical significance was evaluated by the Log-rank test. Stage-adjusted hazard ratios were then calculated by Cox regression. A p-value of less than 0.05 was considered statistically significant. Analyses were conducted by the statistical program SPSS v15.0 (SPSS, Chicago, IL).
The six SNPs genotyped in the validation phase were evaluated using the same statistical methods as for the discovery phase.
In multiple testing adjustments, as Bonferroni’s correction was considered too stringent because of linkage disequilibrium (LD) among the SNPs we tested, the Benjamini-Hochberg false discovery rate (FDR) test20 was used for the evaluation of results in the validation phase.
The functional relevance of the SNP showing significant association (rs2504938) was analyzed in silico by HaploReg v2 and v321. Information about the observed association of this SNP with clinical phenotype of PDAC was submitted to NCBI (The National Center for Biotechnology Information) ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar).
Additional Information
How to cite this article: Mohelnikova-Duchonova, B. et al. SLC22A3 polymorphisms do not modify pancreatic cancer risk, but may influence overall patient survival. Sci. Rep. 7, 43812; doi: 10.1038/srep43812 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Jemal, A. et al. Global cancer statistics. CA Cancer J. Clin. 61, 69–90 (2011).
Maisonneuve, P. & Lowenfels, A. B. Risk factors for pancreatic cancer: a summary review of meta-analytical studies. Int. J. Epidemiol. 44, 186–198 (2015).
Klein, A. P. Genetic susceptibility to pancreatic cancer. Mol. Carcinog. 51, 14–24 (2012).
Amundadottir, L. et al. Genome-wide association study identifies variants in the ABO locus associated with susceptibility to pancreatic cancer. Nat. Genet. 41, 986–990 (2009).
Petersen, G. M. et al. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p.15.33. Nat. Genet. 42, 224–228 (2010).
Wolpin, B. M. et al. Genome-wide association study identifies multiple susceptibility loci for pancreatic cancer. Nat. Genet. 46, 994–1000 (2014).
Childs, E. J. et al. Common variation at 2p13.3, 3q29, 7p13 and 17q25.1 associated with susceptibility to pancreatic cancer. Nat. Genet. 47, 911–916 (2015).
Koepsell, H., Lips, K. & Volk, C. Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm. Res. 24, 1227–1251 (2007).
Cano-Soldado, P. & Pastor-Anglada, M. Transporters that translocate nucleosides and structural similar drugs: structural requirements for substrate recognition. Med. Res. Rev. 32, 428–457 (2010).
Mohelnikova-Duchonova, B. et al. The association between the expression of solute carrier transporters and the prognosis of pancreatic cancer. Cancer Chemother. Pharmacol. 72, 669–682 (2013).
Cui, R. et al. Common variant in 6q26-q27 is associated with distal colon cancer in an Asian population. Gut 60, 799–805 (2011).
Eeles, R. A. et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat. Genet. 40, 316–321 (2008).
Mohelnikova-Duchonova, B. et al. CYP2A13, ADH1B and ADH1C Gene Polymorphisms and Pancreatic Cancer Risk. Pancreas 39, 144–148 (2010).
Mohelnikova-Duchonova, B. et al. Superoxide Dismutase and NAD(P)H Quinone Oxidoreductase Polymorphisms and Pancreatic Cancer Risk. Pancreas 40, 72–78 (2011).
Campa, D. et al. Genetic susceptibility to pancreatic cancer and its functional characterization: The PANcreatic Disease ReseArch (PANDoRA) consortium. Dig. Liver Dis. 45, 95–99 (2013).
Waters, K. M. et al. Generalizability of associations from prostate cancer genome-wide association studies in multiple populations. Cancer Epidemiol. Biomarkers Prev. 18, 1285–1289 (2009).
Magnon, C. et al. Autonomic nerve development contributes to prostate cancer progression. Science 341, 1236361 (2013).
Kim-Fuchs, C. et al. Chronic stress accelerates pancreatic cancer growth and invasion: a critical role for beta-adrenergic signaling in the pancreatic microenvironment. Brain Behav. Immun. 40, 40 (2014).
Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. Haploview, analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005).
Benjamini, Y. & Hochberg, Y. Controlling the False Discovery Rate: A practical and powerful approach to multiple testing. J Roy Statist Soc Ser B 57, 289–300 (1995).
Ward, L. D. & Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acid Res. 40, D930–4 (2011).
Acknowledgements
This study was supported by the projects CSF no.: P301/12/1734, MH CZ no. 16-28375 A, MH CZ - DRO (National Institute of Public Health – NIPH, 75010330), and National Sustainability Program I (NPU I) Nr. LO1503 provided by the Ministry of Education Youth and Sports of the Czech Republic, Prvouk-P27/LF1/1, AIRC 5xmille (grant no. 12182), Italian Cancer Genome Project (FIRB RBAP10AHJB); FIMP, Ministero Salute CUP_J33G13000210001, Pancobank (Prof. M.W. Büchler) and team supported by Heidelberger Stiftung Chirurgie, BMBF grants 01GS08114 and 01ZX1305 C, EU ERA-Net TRANSCAN 01KT1506 as well as BMBH (Prof. P. Schirmacher; BMBF grant 01EY1101), the Italian Ministry of Health grants (RC1503GA42, RC1503GA43) to the Division of Gastroenterology, IRCCS “Casa Sollievo della Sofferenza” Hospital, San Giovanni Rotondo, Italy.
Author information
Authors and Affiliations
Contributions
B.M.D., P.S., D.J.H., and F.C. designed and coordinated the study, performed statistical analyses, and wrote the manuscript. OStrouhal carried out experimental studies and drafted the manuscript. I.H., M.O., Z.K., D.C., C.R., R.P., RT.W., E.M.P., C.S., C.F.Z., S.P., P.F., A.C.M., G.C., G.D.F., R.V., M.G., G.M., R.T.L., OStrobel, T.H., N.G., P.V., L.V., S.L., F.T., D.G., A.P., V.P., A.M., M.P., M.C., F.B., S.E., and N.F. recruited patients, collected clinical data, and drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Mohelnikova-Duchonova, B., Strouhal, O., Hughes, D. et al. SLC22A3 polymorphisms do not modify pancreatic cancer risk, but may influence overall patient survival. Sci Rep 7, 43812 (2017). https://doi.org/10.1038/srep43812
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep43812
This article is cited by
-
Lack of association of CD44-rs353630 and CHI3L2-rs684559 with pancreatic ductal adenocarcinoma survival
Scientific Reports (2021)
-
Glucose metabolism-related gene polymorphisms as the risk predictors of type 2 diabetes
Diabetology & Metabolic Syndrome (2020)
-
Protein expression of ABCC2 and SLC22A3 associates with prognosis of pancreatic adenocarcinoma
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
