
Abstract
A poly-row branched spike (prbs) barley mutant was obtained from soaking a two-rowed barley inflorescence in a solution of maize genomic DNA. Positional cloning and sequencing demonstrated that the prbs mutant resulted from a 28 kb deletion including the inflorescence architecture gene HvRA2. Sequence annotation revealed that the HvRA2 gene is flanked by two LTR (long terminal repeat) retrotransposons (BARE) sharing 89% sequence identity. A recombination between the integrase (IN) gene regions of the two BARE copies resulted in the formation of an intact BARE and loss of HvRA2. No maize DNA was detected in the recombination region although the flanking sequences of HvRA2 gene showed over 73% of sequence identity with repetitive sequences on 10 maize chromosomes. It is still unknown whether the interaction of retrotransposons between barley and maize has resulted in the recombination observed in the present study.
Similar content being viewed by others


Introduction
The architecture of branched inflorescences in grasses depends on the developmental fate of primordia and axis orientation1. The rice (Oryza sativa L.) panicle generates several primary and secondary branches on which spikelets are produced. Sorghum and maize male inflorescences share a structure similar to that of rice. In barley (Hordeum vulgare L.) spikes, however, spikelets are borne directly on the main axis, the rachis, and there are no pedicels. A diagnostic feature of barley is the possession of three one-flowered spikelets at each rachis node2,3. Based on lateral spikelet size and fertility, barley is classified into two-rowed and six-rowed types. Two-rowed barley only has a central fertile spikelet with small and infertile lateral spikelets while the six-rowed barley has three fully-developed fertile spikelets.
The major genes that control row-type variation in barley are Vrs14, Int-c5 and Vrs46. The barley domestication gene Vrs1, located on the long arm of chromosome 2H, encodes a homeodomain-leucine zipper (HD-Zip) transcription factor that suppresses the development of lateral spikelets in two-rowed barley. Mutant vrs1 results in a well-developed six-rowed phenotype4. Int-c, located on chromosome 4H, is an ortholog of the maize (Zea mays. L.) domestication gene, Teosinte branched 1 (TB1), a member of the TCP gene family encoding putative basic helix-loop-helix DNA-binding proteins5. The Int-c gene modifies lateral spikelet fertility in barley, and can influence the phenotypic effect of the Vrs1 locus7.
Vrs4 controls row-type and spikelet determinacy in barley; an induced mutation, vrs4, can convert the two-rowed to a six-rowed phenotype5,8. Vrs4 is an ortholog of the maize inflorescence architecture gene RAMOSA2 (RA2), which encodes a transcriptional regulator that contains the lateral organ boundaries (LOB) domain. Expression analyses by mRNA in situ hybridization and microarray approaches showed that Vrs4 is expressed very early during inflorescence development and controls the row-type pathway through Vrs1 by negatively regulating the lateral spikelet fertility in barley. Moreover, the Vrs4 gene is an important modifier of inflorescence development. Here, we report on a new mutant, poly-row and branched spike (prbs) obtained by soaking a two-rowed barley inflorescence in maize genomic DNA from a single cross hybrid9,10, and characterize its genetics, report its positional cloning, and analyze its origin.
Results
Mutant prbs resulted from deletion of the Vrs4 gene
The poly-row and branched spike (prbs) barley mutant was obtained by soaking a two-rowed barley inflorescence in maize genomic DNA solution9,10. The mutant prbs not only changes two-rowed barley into a poly-rowed form but also adds a spikelet row, forming irregular poly-row and branched spikes (Fig. 1). Genetic analysis indicated that the mutant phenotype was caused by a recessive gene, which has an epistatic effect on Vrs111. The prbs was initially mapped to the centromere of the short arm of chromosome 3H11,12, a location similar to that of vrs4. Furthermore, the immature spikes of the prbs mutant under stereoscope are akin to the scanning electron microscopy images of the Vrs4 immature spikes12. Three molecular markers (DQ327702, Cbic43, and Cbic44), closely linked with the Vrs4 gene, co-segregated with the prbs gene in the prbs/Kunlun12 RIL and prbs/Zangqing 320 F2 populations (Supplementary Fig. 1). No fragment was amplified in the mutant plants using these three molecular markers. Similarly, three other primers covering the Vrs4 gene sequence also failed to amplify a specific DNA fragment from either the prbs mutant or its progeny 11R258-95. Expression analyses revealed that the expression of Vrs4 was not detected and the expression of Vrs1 was significantly down-regulated in immature spikes at lemma primordium stage of the prbs mutant (Fig. 2). These results indicated that the prbs mutant may have resulted from a large deletion around the Vrs4 gene.

(a) normal spikes; (b,c and d) branching spikes; (e) branches of (d,f) mutant prbs; (g) wild parent Pudamai-2.

Constitutively expressed HvActin was used for normalization.
Identification of deletion region in prbs mutant
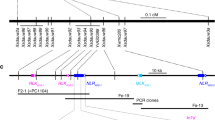
To identify the deletion region in the prbs mutant, a Morex BAC clone was identified that contains the Vrs4 gene. PCR primers were designed at 2 kb intervals from 14 kb upstream to 22 kb downstream of the Vrs4 gene and were tested on the prbs mutant, 11R258-95, Pudamai-2, and Morex. PCR primers located in the region from 3 kb upstream to 10 kb downstream of the Vrs4 gene failed to amplify a specific DNA fragment in the prbs mutant and 11R258-95 but amplified a single band in Pudamai-2 and Morex instead. Sequencing revealed that amplicons represented a single product in Pudamai-2 and Morex. Primers designed from 3 to 13 kb upstream and 10 to 21 kb downstream of the Vrs4 gene amplified a single band in all tested plants, but the amplicons represented multiple products when sequenced. These results did not support these regions arising from a single deletion event in the prbs mutant. However, an additional primer pair, Cbic123, matching a site 14 kb upstream of Vrs4, amplified a single band in all tested plants; the PCR product had 100% sequence identity among the prbs mutant, 11R258-95, Pudamai-2, and Morex. Another primer pair Cbic119, 22 kb downstream of Vrs4, also amplified a single band in all tested plants (sequencing was identical in all tested lines). These results revealed that the deletion sequence was from ~13 to 36 kb in the prbs mutant.
After failure to amplify a single DNA fragment using many PCR primers in the target region, long-range PCR was used to isolate the sequence covering the prbs mutation. Based on the above PCR test results, PCR primers Cbic131 and Cbic132 were designed for this purpose; the forward primer was near the site of primer Cbic123 and the reverse primer near the site of primer Cbic119, as both have been confirmed to amplify a single copy of DNA from the control varieties and mutants. A 15 kb fragment was successfully amplified from both the prbs mutant and 11R258-95 (Fig. 3), whereas the control PCRs using DNA from Pudamai-2 and Morex as a template failed to amplify. The amplification product from prbs was 14,715 bp (accession number KU758926).

PCR-amplification of the 15-kb recombination sequence fragment in the prbs mutant and RIL 11R258-95.
We identified a 48,951 bp sequence from Morex using the 14,715 bp prbs sequence as query in BLASTN searches of the Morex genome database (http://webblast.ipk-gatersleben.de/barley/viroblast.php). Alignment of these two sequences showed that a 27,804 bp sequence in Morex, extending from nt 15,680 to nt 43,769 and containing the entire Vrs4 gene, is deleted in prbs (Fig. 4). Sequence analysis demonstrated that the Vrs4 gene in Morex is flanked by two long terminal repeat (LTR) retrotransposons located, respectively, at nt 10,967 to 19,828 upstream and nt 38,791 to nt 47,684 downstream (Fig. 4). A search of the Triticeae Repetitive Elements (TREP) database revealed high similarity to the retrotransposon RLC BARE1 B consensus-1 (TREPACC = TREP3133) with 90% and 98% of sequence identity, respectively, for the two elements. We refer to the retrotransposon upstream of Vrs4 as BARE up, and the one downstream of Vrs4 as BARE down. The two share 90% identity, are bound by 1.8 kb LTRs, and contain the full-length open reading frame encoding Gag, aspartic proteinase (AP), integrase (IN), reverse transcriptase (RT), and RNaseH (RH) expected for canonical BARE1 elements13 (Fig. 4).

The Vrs4 gene is flanked by two BARE retrotransposons which are bounded by 1.8 kb long terminal repeats (LTRs) and contains coding domains of GAG, integrase (IN), reverse transcriptase (RT), and RNaseH in Morex. The two BAREs sequences are highly conserved and share 89% of sequence identity. The prbs mutant has 28 kb deletion including the entire Vrs4 gene and parts of the two retrotransposons. The non-retrotransposon sequence in the wild parent Pudamai 2 share 100% of sequence identity with Morex.
We further sequenced part of the deletion from the wild parent Pudamai-2, which contains a complete Vrs4 gene. A phylogenetic tree was constructed using the MEGA6 program14 with the minimum evolution method. The results showed that the Vrs4 haplotype in Pudamai-2 was similar to haplotype 8 (Supplementary Fig. 2) which was characterized by an insertion of TA bases in the 5′ UTR of the gene6 and was mainly distributed in Asia. Thus, the prbs mutant resulted from a deletion of the entire functional gene HvRA2, a barley ortholog of the maize inflorescence architecture gene RAMOSA2 (RA2), which thereby transformed a two-row barley into a poly-row branched structure.
Recombination between the integrase genes of two BAREs formed prbs
Sequence analysis of the 15 kb region from prbs identified a single BARE element of the canonical 8.9 kb in length. Alignment revealed that the 5′ part of the prbs (nt 1 to 4,714) BARE was identical to BARE up, whereas the 3′ part (4,972 to 8,918 bp) was the same as BARE down (Fig. 5). The joint between the two halves is between nt 4,715 and nt 4,971 in the prbs BARE (Fig. 5), within the integrase (IN) domain, corresponding to position nt 9,402–9,690 in the cloned prbs fragment (accession number KU758926). Retrotransposon integration generates a direct-repeat target-site duplication (TSD) flanking the individual element, as a consequence of repair of the staggered cut made by the integrase15. BARE up is flanked by imperfect CCAAG TSDs and BARE down by a perfect pair of CTGAA motifs. The BARE in prbs is flanked by CCAAG and CTGAA, supporting the origin of this BARE by recombination. Moreover, the upstream and downstream sequences surrounding the single BARE in prbs correspond, respectively, to that upstream of BARE up and downstream of BARE down. Thus, the HvRA2 gene, flanked by two BARE elements in Pudamai-2, was deleted by recombination between them, thereby generating the prbs mutation.

Red capital letters marked Indel sites and black lower letters marked consensus sequences. Yellow background show BARE up sequences and their homologous sequences in BARE 3. Green background show BARE down sequences and their homologous sequences in BARE 3. Purple red show the recombination region decided by the alignment.
Search the maize genome for sequence similarity
To investigate the possible role of maize DNA in forming the prbs mutation, we used the region spanning the recombination zone in the BARE elements (9,402–9,690 bp) to carry out a BLASTn search the MaizeGDB B73 reference genome sequence. No maize-specific sequence was found in the region. Hence, it appears that no maize DNA has been inserted into the prbs region and that the prbs phenotype results solely from the Vrs4 gene deletion.
As an alternative to insertion, the maize DNA may have played a role through sequence similarity at the recombination point. The region of recombination in prbs corresponds to the most conserved part of the integrase gene, which is the core domain that includes the D-D-35-E active site motif16. The recombination itself took place in the region between the second Asp and the Glu of the active site; a BLASTn search of this region against the MaizeGDB B73 reference genome sequence found more than 120 matches between 73% and 80% identity, containing multiple stretches of ~10 nt perfect identity, dispersed over all maize chromosomes (Table 1). Given their numbers, it is highly likely that these BLAST matches correspond to members of the Copia superfamily in the maize genome, which comprises ~425–485 Mb of the maize genome17, the universal presence of integrase in intact, autonomous LTR retrotransposons. Further research is required whether foreign DNA may induce recombination through sequence similarity, especially when the foreign DNA exist in high concentration or high copy numbers.
Discussion
Barley is classified as two-rowed or six-rowed based on lateral spikelet size and fertility. Two-rowed barley has a central fertile spikelet and two infertile lateral spikelets, and six-rowed barley has three fully-developed fertile spikelets. Vrs4 is an ortholog of the maize (Zea mays. L.) inflorescence architecture gene RAMOSA2 (RA2), which encodes a LOB-domain-containing transcriptional regulator18,19,20. Vrs4 controls row-type variation and modifies inflorescence development in barley (Hordeum vulgare. L)6. Expression analyses of mRNAs by in situ hybridization and microarray analysis revealed that Vrs4 is expressed very early during inflorescence development and controls the row-type pathway in barley through Vrs1, a negative regulator of lateral spikelet fertility.
The prbs mutant was obtained from Pudamai-2, which has the normal Vrs4 gene and a two-rowed phenotype, by soaking the barley inflorescence in maize genomic DNA solution. In the mutant, Vrs4 is deleted through a recombination between two BARE retrotransposons on either side of the Vrs4 gene. LTR retrotransposons are known to recombine; the recombination between two LTRs of a single element, which results in deletion of the internal domain of the retrotransposon and generates a solo LTR, has been studied16. The process of retrotransposon replication generates LTRs that are identical at the moment of integration21; the accumulation of mutations in the LTRs at the neutral rate after that allows for the estimation of the age of the integrated element22. Genome-wide analyses show that the average half-life of a retrotransposon in the Copia superfamily, which includes BARE, is 859,000 years, or a rate of 1.16 × 10−6 events per element per generation, in the grass Brachypodium distachyon, which loses retrotransposons through recombination relatively rapidly23. In barley and other plants, the rate of solo LTR formation varies considerably between retrotransposon families and also between chromosomes and regions20,23,24. Analyses of the frequency of recombination events between internal retrotransposon domains, such as the generated prbs reported here, have not been made and are difficult to identify in the absence of novel phenotypes.
Recombination between the LTRs of two different elements can generate a concatenated structure comprising two internal domains flanking a single, recombinant LTR, which results in the loss of the intervening genomic sequence, including any gene that happens to be there. A quantitative PCR survey of the barley genome for such structures with three LTRs and two internal domains showed that their presence in about 4.3 × 103 copies per haploid genome23. While this indicates the potential for gene loss through recombination of retrotransposons flanking a gene, especially given that the gene islands25 are flanked by retrotransposon “seas” which increases the intervening distance between recombining retrotransposon sequences and appears to be correlated with decreasing recombination frequency20.
The question arises as to whether the maize DNA soaking procedure is connected to the recombination that generated the prbs mutant. Our procedure and the other methods introduce foreign DNA into the megagametophyte before fertilization26,27. Whether or not any foreign DNA is integrated, the presence of extra chromosomal or cytoplasmic DNA triggers a range of defense responses in animals28,29, mediated by DNA recognition by proteins including STING (also called MITA, MPYS, TMEM173, or ERIS)30, specific toll-like receptors (TLRs)31, Z-DNA binding proteins (ZBP-1, DLM-1, or DAI)32, and Mre11 (meiotic recombination 11)33. Mre11 is particularly intriguing because, together with RAD50 and NBS1, is a part of the MRN complex and has been shown to play a vital role in double-strand break (DSB) repair34 in plants, which is an intermediate step in recombination.
The maize genome contains 404,000 Copia superfamily retrotransposons35,36; the integrase domains of these are very similar to the integrase core domain of BARE that underwent recombination in prbs. We speculate whether the homologous maize and barley integrase sequences may have interacted with each other, mediating the recombination. Expression analyses, in situ hybridization and microarrays revealed that Vrs4 is actively expressed during inflorescence development6, corresponding to the stage at which the barley inflorescence was soaked in maize genomic DNA to generate the prbs mutant. Due to its transcriptional activity, this region is likely to have an open chromatin conformation, which could provide an opportunity for maize DNA to interact at the BARE integrase domains and promote the recombination. The high concentration of conserved retrotransposon sequences would make binding and recombination in a retrotransposon sequence more likely than elsewhere. While recombinations between endogenous retroviruses (ERVs)—which are structurally identical to LTR retrotransposons—have caused genic deletions through recombination37, to our knowledge, there has not been an earlier demonstration of this in plants.
Horizontal gene transfer (HGT) is well documented in prokaryotic genome evolution. It is relatively clear that there are several HGT pathways, including transformation, conjugation, and transduction. In eukaryotes, direct DNA exchanges may occur during grafting38, symbiosis39,40, parasitism41, pathogenesis42, and epiphyte or entophyte43. Some vectors, such as pollen43, fungi44, bacteria45, viruses46, plasmids38, insects47 and transposons43, may also be involved in HGT. Transposable elements (TEs) have been recognized as important vectors for the horizontal movement of genes between eukaryotic genomes48,49,50,51. Transposons, with their inherent ability to mobilize, can proliferate and integrate into genomic DNA and generate HGTs with ease52. Transposons have also captured and transduced genomic DNA sequences in both Daphnia pulex53 and Drosophila species52. The transfer of Mu-like transposons between Setaria and rice has been documented48,54. LTR retrotransposons can produce virus-like particles, which may work as more frequent vectors for HGT52,55. Such cases have been demonstrated in LTR-retrotransposon RIRE1 within the genus Oryza56 and the LTR-retrotransposon Route66 in Poaceae50. With the increasing availability of eukaryotic genome sequences, more evidence will be available that plants are also likely to undergo HGT. However, the results have been based on incongruences in molecular phylogenetic trees. On the other hand, there are numerous reports in the literature that have directly introduced foreign DNA by injecting exogenous DNA or directly DNA soaking or pollen tube pathway into rice, barley, wheat, sorghum, maize, cotton, oats, rye, cucumber, pumpkin, kidney bean and soybean to create new genetic variations (Supplementary Table 7). RAPD, AFLP and SSR molecular markers have been used to test DNA transfers between species in several studies57,58,59. However, no study has demonstrated how the exogenous DNA causes genetic variation in other species. Our study provides preliminary evidence that LTR-retrotransposon-mediated gene deletion/insertion may play a role in direct gene transfer between different species.
In addition to act as potential vectors for horizontal gene transfer, transposable elements are also responsive and susceptible to environmental changes. It is well documented that stresses could activate TE to generate new genetic diversity60. This is especially true for LTR-retrotransposons as the LTR is sufficient in itself to activate TE transcription in response to stress. It is possible that soaking the barley spike in the maize DNA solution created stress conditions for the developing spikelets, which activated LTR-TE mediated recombination. In this scenario, the maize genomic DNA may be not essential for the mutation. Further research is required to test this assumption by soaking the developing barley spikes in water or salt solution to provide similar stresses for identification of new mutants.
Materials and Methods
Plant materials
A poly-row branched spike (prbs) barley mutant was obtained by soaking a two-rowed barley inflorescence (cv. Pudamai-2) in maize genomic DNA solution61. The method followed that described earlier for wheat62. Flowering barley spikes were soaked in total maize DNA at 1.6 ug/ul in 0.1 × SSC for 24 hours. After soaking, the head was moved from the solution and air-dried under ambient conditions. Plants were self-pollinated and seeds harvested. The mutant was identified at flowering of the next generation plants.
Genetic mapping was conducted in two populations: one recombinant inbred line (RIL, F2:6) population consisting of 207 plants derived from a cross between the prbs mutant and a six-rowed barley cultivar Kunlun 12, and an F2 population consisting of 285 spike mutant plants derived from a cross between the prbs mutant and a six-rowed barley cultivar Zangqing 320. The prbs mutant, RIL 11R258-95 with a branched spike phenotype, Pudamai-2, and var. Morex were used for DNA sequence analysis.
Genomic DNA extraction and genotype analysis
Genomic DNA was extracted from leaves of individual plants and their parents using a modified CTAB method63. DNA samples were quantified using a Unican UV300 UV/Vis spectrometer (Thermo Electron Corporation, Cambridge, UK), and then adjusted to 25 ng/μl. Because a DQ327702 marker associated with the mutant is closely linked with the Vrs4 gene6, new molecular markers Cbic43 and Cbic44 were designed around the Vrs4 gene using the barley genome sequence14 from the IPK Barley BLAST server (http://webblast.ipk-gatersleben.de/barley/viroblast.php). Primer pairs specific to the Vrs4 gene (AS12, AS34, and AS56)6 were designed for Vrs4 haplotype analysis. Primers were synthesized by Shanghai Sunny Biotechnology (Shanghai, China). PCR reactions were performed in 10 μL volumes containing approximately 25 ng genomic DNA, 0.2 μM of each primer, and 5 μL 2 × Taq Master Mix (Gene Solution, Shanghai, China) using the following program: 94 °C for 3 min, 32 cycles of 94 °C for 30 sec, 55 °C for 45 sec, 72 °C for 1 min, and 72 °C for 5 min. PCR products were separated on 8% polyacrylamide gels.
Cloning of the deletion in mutant prbs
BAC sequences were identified by blasting the Vrs4 against the International Barley Genome Sequencing Consortium database (unpublished data). PCR primer pairs were designed at 2 kb intervals in the region near the Vrs4 locus using the Primer-Blast tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). PCR reactions were described as above. Annealing temperatures were optimized for each primer pair (Supplementary Table 1). PCR products were examined by electrophoresis on 1% agarose gels. Long-range PCRs were performed in 50 μL reactions containing 1 × buffer, 5 μL template DNA, 0.4 μM of each primer, 400 μM each deoxyribonucleotide, and 2.5 U LA Taq DNA polymerase (Takara, Dalian, China) using the following program: 94 °C for 3 min, 32 cycles of 98 °C for 10 sec, 68 °C for 15 min, and 68 °C for 20 min. PCR products were examined by electrophoresis using a 0.8% agarose gel, analyzed by Bio-Red Quantity One gel image analysis system and sequenced by Shanghai RuiDi Biological Technology (Shanghai, China).
Quantitative RT-PCR
RNA was extracted from immature spikes at lemma primordium stage of the prbs mutant and wild parent Pudamai-2 using Spin Column Plant total RNA Purification Kit (Sanggon Biotech (Shanghai) Co.,Ltd). cDNA was prepared from 1 ug RNA using AMV First Strand cDNA Synthesis Kit (Sanggon Biotech (Shanghai) Co., Ltd). qPCR reactions were performed using SYBR Green (SG Fast qPCR Master Mix (HighRox), BBI) and the Applied Biosystems Stepone plus Real-time PCR System. The Real-time PCR assays were performed in triplicate for each cDNA sample. Vrs46, Vrs14 and HvActin6 primer sequences used for quantitative RT-PCR. The HvActin gene was used as reference gene for normalization.
Sequence analysis
Alignments of mutant prbs and barley genomic sequences were constructed using MEGA 6.014 and BLASTN 2.3.0+ 64. The prediction of transposable elements was identified through LTR Finder 1.05 (http://tlife.fudan.edu.cn/ltr_finder/)65 and BLAST66 against the Triticeae Repetitive Element (TREP) database (http://wheat.pw.usda.gov/GG2/blast.shtml). Searches for sequence homology to maize was conducted with MaizeGDB against the sequence database B73 RefGen_v3 (MGSC), using the BLAST program BLASTN63 with an E-value cutoff <1e-50.
Additional Information
How to cite this article: Shang, Y. et al. Gene Deletion in Barley Mediated by LTR-retrotransposon BARE. Sci. Rep. 7, 43766; doi: 10.1038/srep43766 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Doust, A. N. & Kellogg, E. A. Inflorescence diversification in the panicoid “bristle grass” clade (Paniceae, Poaceae): evidence from molecular phylogenies and developmental morphology. Am. J. Bot. 89, 1203–1222 (2002).
Sreenivasulu, N. & Schnurbusch, T. A genetic playground for enhancing grain number in cereals. Trends Plant Sci. 17, 91–101 (2012).
Forster, B. P. et al. The barley phytomer. Ann. Bot. 100, 725–733 (2007).
Komatsuda, T. et al. Six-rowed barley originated from a mutation in a homeodomain-leucine zipper I-class homeobox gene. Proc. Natl. Acad. Sci. USA 104, 1424–1429 (2007).
Ramsay, L. et al. INTERMEDIUM-C, a modifier of lateral spikelet fertility in barley, is an ortholog of the maize domestication gene TEOSINTE BRANCHED 1. Nat. Genet. 43, 169–172 (2011).
Koppolu, R. et al. Six-rowed spike4 (Vrs4) controls spikelet determinacy and row-type in barley. Proc. Natl. Acad. Sci. USA 110, 13198–13203 (2013).
Lundqvist, U. & Lundqvist, A. Induced intermedium mutants in barley: origin, morphology and inheritance. Hereditas. 108, 13–26 (1988).
Lundqvist, U., Franckowiak, J. D. & Konishi, T. New and revised descriptions of barley genes. Barley Genet. Newsl. 26, 22–516 (1997).
Liu, S. et al. Studies on inheritance and spike characters of poly-row-and-branched spike mutant in barley. Fujian Science and Technology of Rice and Wheat. 18, 37–39 (2000).
Ji, H., Chen, Q. & Lin, X. Study on poly-row barley with multi-branches of spikelets emerged by directly of DNAs. Journal of Fujian Agriculture University 24, 9–13 (1995).
Huang, B., Wu, W., Liu, S. & Huang, Z. Genetic Analysis on Poly-row-and-branched Spike Mutant in Barley. Hereditas (Beijing) 26, 903–906 (2004).
Shang, Y. et al. Characterization and mapping of a Prbs gene controlling spike development in Hordeum vulgare L. Genes Genomics 36, 275–282 (2014).
Suoniemi, A., Tanskanen, J., Pentikainen, O., Johnson, M. S. & Schulman, A. H. The core domain of retrotransposon integrase in Hordeum: predicted structure and evolution. Mol. Biol. Evol. 15, 1135–1144 (1998).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Krishnan, L. & Engelman, A. Retroviral integrase proteins and HIV-1 DNA integration. J. Biol. Chem. 287, 40858–40866 (2012).
Vitte, C. & Panaud, O. Formation of solo-LTRs through unequal homologous recombination counterbalances amplifications of LTR retrotransposons in rice Oryza sativa L. Mol. Biol. Evol. 20, 528–540 (2003).
Estep, M. C., DeBarry, J. D. & Bennetzen, J. L. The dynamics of LTR retrotransposon accumulation across 25 million years of panicoid grass evolution. Heredity (Edinb). 110, 194–204 (2013).
Bortiri, E. et al. Ramosa2 Encodes a LATERAL ORGAN BOUNDARY Domain Protein That Determines the Fate of Stem Cells in Branch Meristems of Maize. Plant Cell 18, 574–585 (2006).
Shirasu, K., Schulman, A. H., Lahaye, T. & Schulze-Lefert, P. A contiguous 66-kb barley DNA sequence provides evidence for reversible genome expansion. Genome Res. 10, 908–915 (2000).
Baidouri, M. E. & Panaud, O. Comparative Genomic Paleontology Across Plant Kingdom Reveals The Dynamics Of TE-driven Genome Evolution. Genome Biol Evol. 5, 954–965 (2013).
Schulman, A. H. Retrotransposon replication in plants. Curr Opin Virol. 3, 604–614 (2013).
SanMiguel, P., Gaut, B. S., Tikhonov, A., Nakajima, Y. & Bennetzen, J. L. The paleontology of intergene retrotransposons of maize. Nat. Genet. 20, 43–45 (1998).
Vogel, J. P. et al. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 463, 763–768 (2010).
Mayer, K. F. et al. A physical, genetic and functional sequence assembly of the barley genome. Nature 491, 711–716 (2012).
Liu, R. et al. A GeneTrek analysis of the maize genome. Proc. Natl. Acad. Sci. USA 104, 11844–11849 (2007).
Zhou, G. Y. et al. Introduction of exogenous DNA into cotton embryos. Meth. Enzymol. 101, 433–481 (1983).
Peña, A. D. L., Lörz, H. & Schell, J. Transgenic rye plants obtained by injecting DNA into young floral tillers. Nature. 325, 274–276 (1987).
Barber, G. N. STING-dependent cytosolic DNA sensing pathways. Trends Immunol. 35, 88–93 (2014).
Ma, Z. et al. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc. Natl. Acad. Sci. USA 112, 4306–4315 (2015).
Abe, T. et al. STING Recognition of Cytoplasmic DNA Instigates Cellular Defense. Mol. Cell. 50, 5–15 (2013).
Kawai, T. & Akira, S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity 34, 637–650 (2011).
Takaoka, A. et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448, 501–505 (2007).
Kondo, T. et al. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc. Natl. Acad. Sci. USA 110, 2969–2974 (2013).
Samanic, I., Cvitanic, R., Simunic, J. & Puizina, J. Arabidopsis thaliana MRE11 is essential for activation of the cell cycle arrest, transcriptional regulation and the DNA repair upon the induction of double- stranded DNA breaks. Plant Biol (Stuttg). 18, 681–694 (2016).
Tenaillon, M. I., Hufford, M. B., Gaut, B. S. & Rossibarra, J. Genome size and transposable element content as determined by high-throughput sequencing in maize and Zea luxurians. Genome Biol Evol. 3, 219–229 (2011).
Schnable, P. S. et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 326, 1112–1115 (2009).
Shuvarikov, A. et al. Recurrent HERV-H-mediated 3q13.2-q13.31 deletions cause a syndrome of hypotonia and motor, language, and cognitive delays. Hum. Mutat. 34, 1415–1423 (2013).
Stegemann, S. & Bock, R. Exchange of genetic material between cells in plant tissue grafts. Science 324, 649–651 (2009).
Finan, T. M. Evolving insights: symbiosis islands and horizontal gene transfer. J. Bacteriol. 184, 2855–2856 (2002).
Burger, G. & Lang, B. F. Parallels in genome evolution in mitochondria and bacterial symbionts. IUBMB Life 55, 205–212 (2003).
Yoshida, S., Maruyama, S., Nozaki, H. & Shirasu, K. Horizontal gene transfer by the parasitic plant Striga hermonthica. Science 328, 1128–1128 (2010).
Sharp, A. J. et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat. Genet. 38, 1038–1042 (2006).
Bock, R. The give-and-take of DNA: horizontal gene transfer in plants. Trends Plant Sci. 15, 11–22 (2010).
Richards, T. A. et al. Phylogenomic analysis demonstrates a pattern of rare and ancient horizontal gene transfer between plants and fungi. Plant Cell. 21, 1897–1911 (2009).
Broothaerts, W. et al. Gene transfer to plants by diverse species of bacteria. Nature 433, 629–633 (2005).
Hull, R., Harper, G. & Lockhart, B. Viral sequences integrated into plant genomes. Annu Rev Phytopathol. 5, 362–365 (2000).
Dieterich, C. et al. The Pristionchus pacificus genome provides a unique perspective on nematode lifestyle and parasitism. Nat. Genet. 40, 1193–1198 (2008).
Diao, Y. et al. Next-generation sequencing reveals recent horizontal transfer of a DNA transposon between divergent mosquitoes. PloS One 6, e16743 (2011).
Novick, P., Smith, J., Ray, D. & Boissinot, S. Independent and parallel lateral transfer of DNA transposons in tetrapod genomes. Gene. 449, 85–94 (2010).
Roulin, A. et al. Whole genome surveys of rice, maize and sorghum reveal multiple horizontal transfers of the LTR-retrotransposon Route66 in Poaceae. BMC Evol. Biol. 9, 58 (2009).
Sormacheva, I. et al. Vertical evolution and horizontal transfer of CR1 non-LTR retrotransposons and Tc1/mariner DNA transposons in Lepidoptera species. Mol. Biol. Evol. 9, 3685–3702 (2012).
Loreto, E. L. S., Carareto, C. M. A. & Capy, P. Revisiting horizontal transfer of transposable elements in Drosophila. Heredity. 100, 545–554 (2008).
Schaack, S., Choi, E., Lynch, M. & Pritham, E. J. DNA transposons and the role of recombination in mutation accumulation in Daphnia pulex. Genome Biol. 11, R46 (2010).
Diao, X., Freeling, M. & Lisch, D. Horizontal transfer of a plant transposon. PLoS Biol. 4, 119 (2006).
Silva, J. C., Loreto, E. L. & Clark, J. B. Factors that affect the horizontal transfer of transposable elements. Curr Issues Mol Biol. 6, 57–71 (2004).
Roulin, A., Piegu, B., Wing, R. A. & Panaud, O. Evidence of multiple horizontal transfers of the long terminal repeat retrotransposon RIRE1 within the genus Oryza. Plant J. 53, 950–959 (2008).
Luo, H., Zhong, B., Yang, Z., Li, Y. & He, G. The SSR molecular evidence of rice transformation via pollen tube pathway. Mol. Plant Breed. 2, 501–505 (2004).
Liu, P. & Kang Z. Oat (Avena sat iva L) exogenous DNA introduction into common wheat and PAPD analysis. Agricultural Research in the Arid Areas. 24, 100–103 (2006).
Wang, S. et al. Molecular verification of DNA flow from wild rice (O. minuta) to cultivated rice. Scientia Agricultura Sinica. 39, 2170–2177 (2006).
Casacuberta, E. & Gonzalez, J. The impact of transposable elements in environmental adaptation. Molecular Ecology 22, 1503–1517 (2013).
Huang, B. & Wu, W. Mapping of Mutant Gene prbs Controlling Poly-Row-and-Branched Spike in Barley (Hordeum vulgare L.). Agric. Sci. China 10, 1501–1505 (2011).
Bai, F. et al. Creating a New Wheat Strain of Early Maturity and Another One of Good Dwarf Quality by Introducing Exogeous Maize Nuclear DNA. Acta Agronomica Sinica. 25, 260–264 (1999).
Lu, Y. & Zheng, K. A simple method for isolation of rice DNA. Chinese Journal of Rice Science 6, 47–48 (1992).
Zhang, Z., Schwartz, S., Wagner, L. & Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 7, 203–214 (2000).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, 265–268 (2007).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Acknowledgements
We are grateful to Professor Jing Zhang, Institute of Crop Germplasm Resources, Chinese Academy of Agricultural Science, for providing the mutant prbs, and to Dr Nils Stein Leibniz Institute of Plant Genetics and Crop Plant Research (IPK) for accessing the un-published barley BAC sequence. This work was partially supported by the Natural Science Foundation of Zhejiang Province (LY15C130004), China Agriculture Research System (CARS-05), the National Nature Science Foundation of China (31501309) and the Academy of Finland, Project 266430.
Author information
Authors and Affiliations
Contributions
C.L. and J.Y. conceived, designed and supervised the study; Y.S. and F.Y. are principal investigators; A.H.S., F.Y.W.Z. and Y.J. data analysis; J.Z., J.W., W.H. phenotyping; N.S., X.Z. and Q.J. DNA sequencing; Y.S., F.Y. A.S. and C.L. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Shang, Y., Yang, F., Schulman, A. et al. Gene Deletion in Barley Mediated by LTR-retrotransposon BARE. Sci Rep 7, 43766 (2017). https://doi.org/10.1038/srep43766
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep43766
This article is cited by
-
High-throughput retrotransposon-based genetic diversity of maize germplasm assessment and analysis
Molecular Biology Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
