
Abstract
Elite parental lines are of great significance to crop breeding. To discover unique genomic loci associated with excellent economic traits in the elite cabbage inbred-line ‘01–20’, we performed comparisons of phenotypes as well as whole-genome insertion-deletion/simple sequence repeat loci between ‘01–20’ and each of its five sister lines. ‘01–20’ has a range of excellent agronomic traits, including early-maturing, and improvements in plant type and leaf colour. Eight unique loci were discovered for ‘01–20’ and ‘01-07-258’, another elite line similar to ‘01–20’ at the whole-genome level. In addition, two excellent double-haploid lines derived from a cross of ‘01–20’ also inherited these loci. Based on the quantitative trait locus association results, five of these loci were found to be associated with important agronomic traits, which could explain why the elite parent ‘01–20’ possesses greener outer leaves, a more compact and upright plant-type, rounder head, shorter core length, and better taste. Additionally, some of these loci have clustering effects for quantitative trait loci associated with different traits; therefore, important genes in these regions were analysed. The obtained results should enable marker-assisted multi-trait selection at the whole-genome level in cabbage breeding and provide insights into significant genome loci and their breeding effects.
Similar content being viewed by others


Introduction
Elite parental lines, which usually possess a range of excellent properties and high combining ability, are important materials for hybrid breeding1. Their use usually results in a batch of excellent hybrid cultivars, which can in turn be used as the basis for new generations of elite parental lines, through combination, optimisation and coordination of genes controlling desirable traits.
With the development of genetics, genomics and molecular biology, elucidating the genetic composition, and its associations with the characteristics, of elite parental lines at the molecular level has become a main approach for improving the accuracy and efficiency of parental selection in plant breeding. At present, studies of elite parents are mainly focused on field crops, such as soybean, barley and maize. Lorenzen et al. reported that 80% of the U.S. soybean germplasm could be traced back to just 12 parental lines2. In addition, Russell et al. reported that 19 elite parental barley lines contained 72% of the genetic variation of modern varieties3. Bernardo et al. also estimated the parental contribution and coefficient of coancestry of 13 inbred maize lines with known pedigrees using restriction fragment length polymorphism and simple sequence repeat (SSR) markers4. Additionally, Kanouni et al. studied high yield and good performance using 35 elite chickpea lines5. Furthermore, Boudiar et al. reported that alleles from the elite Spanish barley cultivar ‘Orria’ contribute to grain yield, reduce plant height and increase 1000-kernel weight, while alleles of chromosome 5H from the landrace ‘SBCC073’ contribute to early vigour, higher grain yield and earlier flowering6.
However, few studies on elite parental lines have been reported in vegetable crops. For cabbage, we reported, and performed a pedigree analysis, on the elite inbred line ‘01–20’7. We bred this line, together with its five sister lines ‘01-07-258’, ‘01-07-251’, ‘01-1-4’, ‘01–88’ and ‘01-16-5’, through a systemic selection from the variety ‘Early Vikings’ introduced into China from Canada. However, the numbers of F1 hybrids resulting from their crosses were 10, 6, 0, 0, 2 and 3, respectively, and the numbers of derived lines (excellent lines selected from their hybrids) were 9, 5, 0, 0, 3 and 0, respectively. To date, some of the hybrids still dominate the market of spring cabbage cultivars in China. The above data show that ‘01–20’ has made a major contribution to cabbage production. Why did ‘01–20’ become an elite parental line, while its sister lines did not? To answer this question, we analysed and compared ‘01–20’ and its five sister lines, as well as two excellent double-haploid (DH) lines derived from ‘01–20’, at the phenotypic and genomic levels, with the objective of identifying the ‘01–20’s distinctive genomic loci associated with excellent agronomic traits.
Materials and Methods
Plant materials and field experiment
‘Early Vikings’, a conventional, early-maturing, spring cultivar introduced from Canada in 1966 by the Institute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences (IVF-CAAS), was used in this study as a population with genetic heterogeneity. Genetic heterogeneity was maintained in this population through open self-pollination every year in a greenhouse with honeybees isolated from the outside using a gauze covering.
‘01–20’, an elite parental line, together with its five sister lines: ‘01-07-258’, ‘01-07-251’, ‘01-1-4’, ‘01–88’ and ‘01-16-5’, were all bred through a method of systemic selection from ‘Early Vikings’. ‘01–20’ is an elite, early-maturing, spring cabbage inbred line having upright and green outer leaves, little wax powder, a green and round head, high general and specific combining abilities, and a tender and crisp taste.
As determined in our previous study, the numbers of inbred lines derived from ‘01–20’ and cultivars generated from it had reached 9 and 10, respectively7. Table 1 shows some of the varieties generated from ‘01–20’ developed by the Cabbage and Broccoli Breeding Group, IVF-CAAS. Of the 10 generated cultivars, there were two particularly excellent varieties: ‘8398’ and ‘Zhonggan No. 21’. ‘8398’, a cultivar suitable for both protected cultivation and open-field cultivation in the spring, won the second prize at the National Science and Technology Progress Awards of China in 1998. ‘Zhonggan No. 21’, another early-maturing spring cabbage cultivar, also won the same prize in 2014 and, after its introduction in 2006, its cumulative harvesting area had reached over 400,000 ha in China by 2015, making it the top spring cabbage variety in China. These two cultivars still dominate and share over 70% of the spring cabbage market in China.
In addition to the six sister lines, ‘D77’ and ‘D83’, two excellent DH lines, selected from over 200 DH lines derived from the cross between ‘01–20’ and ‘96–100’ (‘Zhonggan No. 18’, see Table 1)8, were also used for phenotype measurements and molecular marker assays.
Field trials of the population and the lines mentioned above were carried out at the experimental station of IVF-CAAS in spring 2012 in an open field in Changping District, Beijing. A randomised block design was adopted, with three replications. Each replication/plot consisted of 15 plants. All of the materials were sown on January 27th, transplanted to an open field on March 27th and investigated from mid-May to early June.
Phenotyping
Observations and measurements of the phenotypes were performed for the main agronomic traits of ‘01–20’, and its five sister lines (‘01-07-258’, ‘01-07-251’, ‘01-1-4’, ‘01–88’ and ‘01-16-5’), two DH lines (‘D77’ and ‘D83’) and the parental population. In total, 25 traits from three categories were assessed (Table 2): plant-type-related traits, including plant diameter (Pd), plant height (Ph) and plant type (Pt) measured by two methods (see Table 2); leaf-related traits, including leaf colour (Lc), leaf number, leaf surface (Ls), leaf wax powder, petiole length (Pl) and petiole width (Pw); head-related traits, including head colour, head maturity period (Hm), head weight (Hw), head vertical diameter (Hvd), head transverse diameter (Htd), core length (Cl), ratio of core length to head vertical diameter (Cl/Hvd), ratio of core width to head transverse diameter (Cw/Htd), head shape index (Hsi), head solidity (Hs), dry matter content (Dmc) and crude fibre content (Cfc); and the trait of seed size (Ss).
These traits were assessed following standards described in ‘Descriptors and data standards for cabbage’9 at the rosette or head harvesting stage (Table 2). In addition, Dmc and Cfc were determined following drying and acid, or alkali, digestion methods, respectively, in accordance with the Association of Official Analytical Chemists (AOAC) standards10 (Table 2). For colour-related traits, a CR-400 colour difference meter (Konica Minolta, Shanghai, China) was used to assay the leaf and head colour coordinates a* (redness to greenness), b* (yellowness to blueness) and L (lightness to darkness) (CIE1976_Lab standards) with a standard D65 light source, 0° diffuse illumination and a viewing angle of 2° to CIE 1931 under a dark background.
Average values for each trait of each line were calculated from three randomly selected plants in each plot at the rosette or harvesting stage. Adjusted means for the traits were obtained and used for further analysis. Microsoft Excel 2007 (Microsoft, Seattle, WA, USA) and SPSS 12.0 (SPSS, Chicago, IL, USA) software were used for statistical analyses and multiple comparisons.
Genotyping
Whole-genome scanning for insertion/deletion (indel) and SSR loci was performed for all cabbage materials, using 406 pairs of SSR and indel primers, and the corresponding genetic maps. These markers and maps were developed in our previous study for gene mapping and the detection of quantitative trait loci (QTLs)8,11, in which we discovered robust QTLs and QTL clusters for 24 main agronomic traits using a DH population of 196 lines derived from a cross of ‘01–20’ × ‘96–100’, indicating that the whole-genome indel/SSR loci and the QTLs were highly reliable.
The molecular marker assay protocol was as follows: polymerase chain reaction (PCR) mixture samples with a volume of 20 μl, which contained 2 μl of PCR buffer (10 ×, Mg2+ included), 1.6 μl of dNTP (2.5 mM each), 0.4 μl of Taq DNA polymerase (2.5 U/μl), 5 μl of DNA template (40 ng/μl), 0.6 μl of forward primer (10 μM), 0.6 μl of reverse primer (10 μM) and 9.8 μl of ddH2O, were used. The reaction mixture was incubated in a thermal cycler at 94 °C for 5 min, followed by 35 cycles of 94 °C for 30 s, 55 °C for 30 s and 72 °C for 45 s, and finally 72 °C for 7 min. The PCR products were separated on 8% polyacrylamide gels, which were then subjected to silver staining after running at 160 V for 1.5 h12. The electrophoresis band patterns for each primer pair was investigated: band pattern the same as ‘01–20’ was recorded as ‘a’, band pattern the same as ‘96–100’ as ‘b’, and a third pattern as ‘c’.
Associated traits and candidate gene analysis for the distinctive loci
In a previous study, we mapped 144 QTLs for 24 agronomic traits using a DH population of 196 lines derived from a cross between ‘01–20’ and ‘96–100’13. Based on the map and QTL information, we determined the positions of the distinctive loci that had been identified in the above molecular marker assays on the map constructed for the QTL analysis, to clarify the association between the traits and these loci. QTLs were named using the following criteria: abbreviation of the trait name, followed by chromosome code and QTL code. For example, Ph 1.2 represents the second QTL on chromosome C01 associated with plant height.
The genes located in the regions associated with the distinctive loci were analysed and compared with those in Arabidopsis, using the annotations for the Brassica oleracea reference genome acquired from BRAD (http://brassicadb.org/brad/).
Equipment and settings
Images in Figs 1, 2 and 3, were taken using a SONY DSC-HX30 (Sony Co. Ltd., Tokyo, Japan) camera and were edited using Photoshop CS6 software (Adobe Systems Inc., San Jose, CA, USA). Genetic maps in Fig. 4 were generated using JoinMap 4.0 software14, and integrated using Photoshop and Microsoft Office 2007 software (Microsoft, Seattle, WA, USA).

Field Agronomic performance of ‘Early Vikings’, the parental material of 01–20.

Agronomic Field performance and head traits of 01–20, five of its sister lines, and two DH lines.

This image is from four cropped gels, and full-length gels are presented in Supplementary Figure 2.

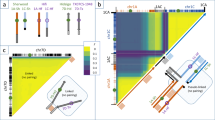
Locations of QTL are indicated by names, bars and arrows to the right of the linkage groups (red: plant type-related traits; blue: leaf-related traits; black: head-related traits). Arrows indicate the relative effect of the 01–20 allele, with downing for positive effects. For abbreviations, see Table 2. Blue blocks on the chromosomes represent QTL cluster regions; orange blocks: the most significant QTL cluster regions; yellow blocks: distinctive loci of 01–20 and 01-07-258.
Results
Clustering analysis of ‘01–20’ and the sister lines
Similarity analyses among the sister lines were conducted using the software NTSYSpc2.1115. The loci types (Supplement Table 1) revealed with 406 markers were employed to calculate the genetic similarity coefficients among 9 accessions, ‘01–20’, ‘01-07-258’, ‘01-07-251’, ‘01-1-4’, ‘01–88’, ‘01-16-5’, ‘96–100’, ‘D77’ and ‘D83’. With a genetic similarity coefficient of 0.27, ‘96–100’ was assigned to one cluster, while the remaining eight accessions were assigned to another cluster. The genetic similarity coefficients between ‘01–20’ and the five sister lines, ‘01-07-258’, ‘01-07-251’, ‘01-1-4’, ‘01-16-5’and ‘01–88’, and the two DH lines, ‘D77’ and ‘D83’, were 0.95, 0.93, 0.67, 0.67, 0.65, 0.73 and 0.68 respectively, while the genetic similarity coefficients between ‘96–100’ and ‘D77’ and ‘D83’ were 0.27 and 0.32 respectively. These results indicated that ‘01-07-258’ and ‘01-07-251’ had genetic backgrounds that were closer to ‘01–20’.
‘01–20’ shows a range of excellent agronomic traits
Phenotype measurements were carried out for all six sister lines (‘01–20’, ‘01-07-258’, ‘01-07-251’, ‘01-1-4’, ‘01–88’ and ‘01-16-5’), a genetically heterogeneous population compromising 28 individuals (Fig. 1), and the two DH lines. Multiple comparison tests showed that there were significant differences among these materials for most traits, including Pt, Lc, Ls, Cl/Hvd, Hw and Hm. Figure 2 shows the differences in Pt, Hs, Lc and Cl among these lines.
Generally, ‘01-07-258’ was the line most similar to ‘01–20’ in most traits, including Pt, leaf- and head-related traits (Fig. 1; Table 3), and combining ability (from our experience). Thus, ‘01-07-258’ was also deemed as an elite line and used frequently in hybrid breeding. The major difference between the two lines was that the Ss of ‘01-07-258’ (1.42 mm in diameter) was smaller than those of ‘01–20’ (1.80 mm) and the other sister lines.
Among the other sister lines, ‘01-07-251’ had the most upright Pt, the largest plant expansion (Pd and Ph), the highest Hw, the lowest Dmc and Cfc, and the largest Ss, but the longest maturing period (58.67 d) and the longest Cl (8.58 cm); ‘01-1-4’ had the most wrinkled leaf surface and the shortest Hm and Cl, but the smallest head (0.55 kg). ‘01–88’ had the most patulous Pt, a higher Hw and a shorter Hm, but the highest Dmc (6.42) and a low tolerance to splitting; and ‘01-16-5’ had a patulous plant type and greener outer leaves, but the highest Cl/Hvd (0.66) and Cfc (0.61), and the lowest tolerance to splitting. In addition, although ‘Early Vikings’ showed a relatively low uniformity, some individuals still showed outstanding performance in terms of Hm, Hw, leaf and head colour, Hsi and Hs (Fig. 2).
Of the two DH lines, ‘D77’ had the most upright Pt, the greyest Lc and Hc, the largest amount of Lw, the fewest leaves and the longest maturing period. ‘D83’ exhibited the largest plant expansion (Pd and Ph), the highest Hw, and the highest levels of both Dmc and Cfc. Both of these lines have excellent combining ability (data not shown), and thus their use in hybrid seed production is promising.
Thus, ‘01–20’ had a range of excellent agronomic traits and no obvious defects, and it not only inherited the early-maturing trait from ‘Early Vikings’, but also exhibited improvements in Pt, Lc, and head- and quality-associated traits. The DH lines also inherited certain excellent traits from ‘01–20’, such as high Hw, low Cl/Hvd and compact Pt.
Eight distinctive indel/SSR loci are present in the elite lines
For assays of indel/SSR loci, 406 pairs of SSR and indel primers were applied for the six sister lines, as well as the two DH lines. In total, 398 primer pairs amplified two alleles and eight amplified three alleles at each locus. Overall, 206 primer pairs were polymorphic, accounting for 50.74% of the total. Figure 3 shows some of the polymorphic markers for the six sister lines.
Whole-genome scanning for indel/SSR loci for the six lines revealed that ‘01–20’ had one unique locus, ‘01-07-258’ had one unique locus, and ‘01–20’ and ‘01-07-258’ had six common loci, which were different from those of the other four sister lines (Table 4). In total, 385 out of the 406 loci (accounting for 95%) were the same for ‘01–20’ and ‘01-07-258’, and the loci on C02, C04 and C07 were all the same between the two lines. This is in accordance them being similar lines, and it also indicates that analysing their unique loci together is appropriate.
The distinctive loci were located on chromosomes C02, C03, C05, C08 and C09 (Fig. 4). Some of them clustered together in the same genomic region. For example, the genetic distance between the loci Indel26 and Indel488 was shown to be 1.3 cM on C02, and the distance between scaffold29640 and Indel64 was 0.7 cM on C03. Figure 4 shows the allele types of all of the distinctive loci for the eight lines.
The seven distinctive loci identified for ‘01–20’ on C02, C03 and C08, were also found in the two excellent DH lines, ‘D77’ and ‘D83”, with the exception of Indel139 on C05 (Fig. 4), further indicating the significant effects of these loci.
Five of the distinctive loci are associated with important agronomic traits
The eight newly discovered loci, described above, may help explain why ‘01–20’ and ‘01-07-258’ became elite parental lines. In a previous study13, we identified the genomic regions associated with 24 important agronomic traits. Thus, the distinctive loci were anchored to the map published by Lv13. We found that five of them, located at the major QTL cluster regions (Fig. 4; indicated in orange: the most significant QTL cluster regions, blue: other major QTL cluster regions) on four chromosomes, were associated with important agronomic traits, including Lc, Hc, Pt, leaf length, Pd and Hs (Table 5). These QTLs were found to explain 6.0–26.1% of the phenotypic variance of these traits.
Several distinctive loci were found to be associated with Lc: Indel26 and Indel353 were associated with Lca*, explaining 8.0–9.5% of its phenotypic variance. An analysis of the allele variance effect showed that this allele in ‘01–20’ contributed to a lower trait value than the other alleles. Thus, it had a negative effect (−a to a: green to red), which explains why ‘01–20’ possesses greener leaves (Table 6). Indel26 was also associated with Lcb*, explaining 12.3% of its phenotypic variance. This allele in ‘01–20’ contributed to a higher trait value than the other alleles. Thus, it had a positive effect (−b to b: blue to yellow), which means that the Lc of ‘01–20’ tends to be yellowish rather than blue (Table 6).
In addition, Indel235 was also associated with Pt, explaining 11.3% of the phenotypic variance. An analysis of the allele variance effect showed that this allele in ‘01–20’ contributed to a higher trait value than the other alleles. That is, it had a positive effect, which means that ‘01–20’ plants tend to be upright rather than patulous (Table 6).
Indel64 was identified to be associated with Pd, Ph, Ll, Lw, Pl, Pw and Hs, explaining 6.4–21.2% of the phenotypic variance of these traits. An analysis of the allele variance effect showed that this allele in ‘01–20’ contributed to lower trait values than the other alleles, except for the case of Hs. Thus, it had a negative effect, which explains why the expansion of ‘01–20’ plants is smaller, and the leaves are short and small. Thus, the plants are compact (Table 6).
In addition to Lc, Indel353, the distinctive loci for ’01-07-258’ on C09 was also associated with the head quality traits, Dmc and Cfc, Hsi and the core trait Cw/Htd. It can explain 10.1% and 13.9% of the phenotypic variance of Dmc and Cfc, respectively. An analysis of the allele variance effect showed that this allele in ‘01-07-258’ (allele type: b, see Table 6) contributed to higher trait values than the other alleles. Thus, it had a positive effect, which means the taste of ‘01-07-258’ is not as tender and crisp as that of ‘01–20’, which has lower Dmc and Cfc levels (Table 6).
In addition, in a previous study, we identified 12 QTL clustering regions associated with different agronomic traits13. Nine of these clusters were possessed by 75% of these lines. These regions may also be key factors in determining whether a line should be maintained for further selection. Thus, the 12 QTL clustering regions on the nine chromosomes may be the selection foundation and these distinctive loci are the core of the foundation.
Thus, the distinctive loci in the elite parental lines are associated with important agronomic traits, which could explain, at the genomic level, why ‘01–20’ has greener leaves, is more compact, has a rounder head and a shorter core length, and tastes better. Loci, like Indel64 on C03, and the clustering of QTLs for different significant agronomic traits may play particularly important roles in the elite parental line ‘01–20’.
Discussion
Application of elite lines: a double-edged sword
Elite lines are of great significance in two ways. First, in plant breeding, they generate a many varieties and contribute greatly to crop production. For example, ‘01–20’, the elite line used in this study has generated as many as 10 varieties, having a spring cabbage market share of over 70%. In rice hybrid production, the elite female parent line ‘Zhenshan’ contributed to a number of widely used rice cultivars in China16. Second, they are good materials for studying the genetic effects of significant genomic loci associated with excellent traits, which in turn provide the basis for breeding programs17. Using two types of PCR-based DNA markers, Mahatma et al. estimated the genetic polymorphisms among nine elite cotton parental lines, which suggested that the genetic constituents of ‘LRA-5166’ are quite different from those of the other eight parental lines18. In addition, Lai et al. detected more than 1,000,000 single nucleotide polymorphisms, 30,000 indels and 101 low-sequence-diversity chromosomal intervals, as well as hundreds of genes showing presence/absence variation in the maize genome by resequencing six elite maize inbred lines19. The current study, for the first time, compared the elite cabbage line ‘01–20’, five sister lines and two derived lines for their phenotypic and genetic constituents, and shed light on the key genomic loci that determined ‘01–20’ as an elite line.
Although elite lines can make major contributions to crop production and quality, excessively applying them may also create problems. During plant breeding, some important agronomic traits, such as disease resistance, high yield and excellent taste, are constantly under directional selection, resulting in significantly reduced genetic diversity20. In a study by Hao et al. 4% of the 340 wheat base collections from the Northwest Spring Wheat Region in China were found to represent more than 70% of their entire variation21. In addition, Ding et al. detected a severe reduction in nucleotide variation at OsAMT1;1, a high-affinity ammonium transporter in rice (Oryza sativa) that controls ammonium uptake capacity, indicating that strong selection on nitrogen uptake-related traits has occurred in rice22. Similar reports have been published on soybean 2, barley 3 and wheat23,24. Additionally, the use of ‘01–20’ as one parent can greatly improve the agronomic traits of hybrids regarding Pt, Lc, Hc, head type, production and taste, among others; however, its lack of disease resistance against Fusarium wilt may be risk crop production as this disease is currently affecting cabbage-producing areas in northern China.
Mining for genes/QTLs associated with important traits using elite parental lines
Elite parental lines are constantly subjected to the directional selection acting on genes that control desirable traits of agronomic importance during their domestication and improvement. Therefore, the genes or loci with a signature of selection from breeders should be identifiable by whole-genome nucleotide polymorphism scanning using DNA markers25,26.
With the development of molecular biology and of the sequencing of numerous genomes, uncovering the genetic basis of important traits of elite parental lines has become the focus of theoretical and applied studies. One way to characterise genes responsible for phenotypic variation is based on the signature of selection in lines with particularly superior characteristics. Vigouroux et al. screened 501 maize genes with a signature of selection and identified 10 as agronomically important candidates because they showed evidence (i.e., the presence of non-neutral SSRs) of exposure to selective pressure. It was further confirmed that one of these, encoding a MADS box transcriptional regulator, experienced a selective sweep during maize domestication27. In another study, Yamasaki et al. identified eight of the sequenced 1,095 maize genes through a selection test in diverse maize landraces and teosintes, and showed that their functions were consistent with agronomic selection for nutritional quality, maturity and productivity. Another way of identifying genes for corresponding traits is association analysis and QTL mapping using elite lines28. For example, Würschum et al. detected several QTLs of important traits in rapeseed, including flowering time, Ph, protein content, oil content, glucosinolate content and grain yield29. In addition, using genome-wide association mapping, Wang et al. identified marker–trait associations in 94 diverse elite wheat lines: marker XwPt-7187 was associated with kernel hardness, XwPt-1250 and XwPt-4628 with test weight, and marker Xgwm512 with Ph30.
In the current study, we compared elite line ‘01–20’ with five of its sister lines by scanning SSR/indel loci across the whole genome. We identified eight loci for which the elite lines were distinctive, which were further found to be associated with important agronomic traits, including Lc, Hc, Pt, leaf length, Pd and Hs. To discover interesting candidate genes at these loci, we performed a preliminary analysis for the genes located in the region for the five distinctive loci associated with important agronomic traits, according to the annotations for the B. oleracea reference genome acquired from BRAD (gene alignment results, Supplement Table 2). We used the Basic Local Alignment Search Tool (BLAST) (https://www.ncbi.nlm.nih.gov/) tool and set the score cutoff value to 400. The annotation of the functions of the genes included transmembrane transporter, transcription factor, ATP binding, kinase and cytochrome. Based on the alignments with Arabidopsis, some of the genes may be associated with related traits. For example, in Region 2 on chromosome C03, which is associated with Pd, Lw, Ll, Pl, Pw and Hs, the homologous gene ubiquitin ligases EOL1 can act with ETO1 and EOL2 collectively to regulate ethylene biosynthesis in Arabidopsis by controlling type-2 ACC synthase levels31. Another homologous gene SPI encodes a WD40/BEACH domain protein and shares a similar actin-regulating ARP2/3 pathway that affects plant growth in various organs32. In Region 4 on chromosome C09, which is associated with Lcb*, Lca*, Hsi, Cw/Htd, Dmc and Cfc, the homologous chloroplast-encoded gene Ycf4 plays an essential role in the Photosystem I complex33. These genes may be associated with related traits. However, further studies are still needed to clarify the connection between the candidate genes and the related traits.
The above results shed light on why ‘01–20’, rather than its sister lines, became an elite parental line. These loci could be useful for the development of whole-genome background markers for cabbage breeding and to promote our understanding of the genetic basis of selected traits.
Additional Information
How to cite this article: Lv, H. et al. Genome-wide indel/SSR scanning reveals significant loci associated with excellent agronomic traits of a cabbage (Brassica oleracea) elite parental line 01-20. Sci. Rep. 7, 41696; doi: 10.1038/srep41696 (2017).
References
Zhuang, Q. S. Chinese Wheat Improvement and Pedigree Analysis. (China Agriculture Press, 2003).
Lorenzen, L. L., Boutin, S., Young, N., Specht, J. E. & Shoemaker, R. C. Soybean pedigree analysis using map-based molecular markers: I. Tracking RFLP markers in cultivars. Crop Science 35, 1326–1336 (1995).
Russell, J. R. et al. A retrospective analysis of spring barley germplasm development from ‘foundation genotypes’ to currently successful cultivars. Molecular breeding 6, 553–568 (2000).
Bernardo, R. et al. Parental contribution and coefficient of coancestry among maize inbreds: pedigree, RFLP, and SSR data. Theoretical and Applied Genetics 100, 552–556 (2000).
Kanouni, H., Farayedi, Y., Mahdieh, M., Shahbazi Dourbash, S. & Behzadi, A. Study on agronomic traits and seed yield production of chickpea elite lines in an international nursery (CIEN-LA-09) in spring and rain-fed planting conditions. International Journal of Infectious Diseases Ijid Official Publication of the International Society for Infectious Diseases 22, 203–204 (2014).
Boudiar, R., Casas, A. M., Cantalapiedra, C. P., Gracia, M. P. & Igartua, E. Identification of quantitative trait loci for agronomic traits contributed by a barley (Hordeum vulgare) Mediterranean landrace. Crop & Pasture Science 67, 37–46 (2016).
Wang, Q. et al. Survey and pedigree analysis of cabbage varieties released in China. Acta Horticulturae Sinica 40, 869–886 (2013).
Lv, H. et al. Development of indel markers linked to fusarium wilt resistance in cabbage. Molecular Breeding 32, 961–967 (2013).
Li, X. & Fang, Z. Descriptors and Data Standards for cabbage (Brassica oleracea L. var. capitata L. and Brassica oleracea L. var. gemmifera Zenk). (China Agriculture Press, 2007)
AOAC. Official Methods of Analysis. (15th ed.) (Association of Official Analytical Chemists, 1995).
Lv, H. et al. Linkage map construction using InDel and SSR markers and QTL analysis of heading traits in cabbage. Mol Breeding 34, 87–98 (2014).
Bassam, B. J., Caetano-Anollés, G. & Gresshoff, P. M. Fast and sensitive silver staining of DNA in polyacrylamide gels. Analytical biochemistry 196, 80–83 (1991).
Lv, H. et al. Whole-genome mapping reveals novel QTL clusters associated with main agronomic traits of cabbage (Brassica oleracea var. capitata L.). Frontiers in Plant Science 7, 989 (2016).
Van Ooijen, J. W., Boer, M. P., Jansen, R. C. & Maliepaard, C. Map QTL 4.0: software for the calculation of QTL positions on genetic maps. Plant Research International 501, 2412–2421 (2002).
Rohlf, F. J. NTSYS-pc 2.1: Numerical Taxonomy and Multivariate Analysis System. New York, America. URL http://www.exetersoftware.com/cat/ntsyspc/ntsyspc.html (1997).
Zhou, P. et al. Simultaneous improvement for four quality traits of Zhenshan 97, an elite parent of hybrid rice, by molecular marker-assisted selection. Theoretical and Applied Genetics 106, 326–331(2003).
Rana, M. K. & Bhat, K. V. RAPD markers for genetic diversity study among Indian cotton cultivars. Curr Sci 88, 1956–1961 (2005).
Mahatma, M. K., Khandelwal, V., Jha, S. K., Kumar, V. & Shah, R. R. Genetic diversity analysis of elite parental lines of cotton using RAPD, ISSR and isozyme markers. Indian Journal of Plant Physiology 14, 105–110 (2009).
Lai, J. et al. Genome-wide patterns of genetic variation among elite maize inbred lines. Nature genetics 42, 1027–1030 (2010).
Palaisa, K., Morgante, M., Tingey, S. & Rafalski, A. Long-range patterns of diversity and linkage disequilibrium surrounding the maize Y1 gene are indicative of an asymmetric selective sweep. The Proceedings of the National Academy of Science of the USA 101, 9885–9890 (2004).
Hao, C. Y. et al. Genetic diversity and core collection evaluations in common wheat germplasm from the Northwestern Spring Wheat Region in China. Molecular Breeding 17, 69–77 (2006).
Ding, Z., Wang, C., Chen, S. & Yu, S. Diversity and selective sweep in the OsAMT1; 1 genomic region of rice. Bmc Evolutionary Biology 11, 61 (2011).
Tian, Q. Z., Zhou, R. H. & Jia, J. Z. Genetic diversity trend of common wheat (Triticum turgidum L.) in China revealved with AFLP markers. Genetic Resours and Crop Evolution 52, 325–331 (2005).
Fu Y. B. et al. Allelic reduction and genetic shift in the Canadian hard red spring wheat germplasm released from 1845 to 2004. Theorerical and Applied Generics 110, 1505–1516 (2005).
Lukens, L. N. & Doebley, J. Molecular evolution of the teosinte branched gene among maize and related grasses. Molecular Biology and Evolution 18, 627–638 (2001).
Andolfatto, P. Adaptive hitchhiking effects on genome variability. Current Opinion in Genetics and Development 11, 635–641 (2001).
Vigouroux, Y. et al. Identifying genes of agronomic importance in maize by screening microsatellites for evidence of selection during domestication. Proceedings of the National Academy of Sciences of the United States of America 99, 9650–9655 (2002).
Yamasaki, M. et al. A large-scale screen for artificial selection in maize identifies candidate agronomic loci for domestication and crop improvement. Plant Cell 17, 2859–2872 (2005).
Würschum, T., Liu, W., Maurer, H. P., Abel, S. & Reif, J. C. Dissecting the genetic architecture of agronomic traits in multiple segregating populations in rapeseed (Brassica napus L.). Theoretical & Applied Genetics 124, 153–61 (2011).
Wang, G. et al. Marker–trait association analysis of kernel hardness and related agronomic traits in a core collection of wheat lines. Molecular Breeding 34, 177–184 (2014).
Matthew, J. et al. The BTB ubiquitin ligases ETO1, EOL1 and EOL2 act collectively to regulate ethylene biosynthesis in Arabidopsis by controlling type-2 ACC synthase levels. The Plant Journal 57, 332–345 (2009).
Saedler, R., Jakoby, M., Marin, B., Galiana-Jaime, E. & Hulskamp, M. The cell morphogenesis gene SPIRRIG in Arabidopsis encodes a WD/BEACH domain protein. The Plant Journal 59, 612–621 (2009).
Krech, K. et al. The plastid genome-encoded Ycf4 protein functions as a nonessential assembly factor for photosystem I in higher plants. Plant Physiol 159, 579–91 (2012).
Acknowledgements
The work reported here was performed at the Key Laboratory of Biology and Genetic Improvement of Horticultural Crops, Ministry of Agriculture, Beijing 100081, China. This work was financially supported by grants from the Key Projects in the National Science & Technology Pillar Program during the Thirteenth Five-Year Plan Period (2016YFD0100300), the Major State Basic Research Development Program (973 Program, 2012CB113906), the National Natural Science Foundation of China (31272180), the Key Projects in the National Science & Technology Pillar Program during the Twelfth Five-Year Plan Period (2012BAD02B01, 2013BAD01B04-4), the Science and Technology Innovation Program of the Chinese Academy of Agricultural Sciences (CAAS-ASTIP-IVFCAAS) and the earmarked fund for the Modern Agro-Industry Technology Research System, China (nycytx-35-gw01).
Author information
Authors and Affiliations
Contributions
H. Lv developed the DH populations and wrote and revised the manuscript. H. Lv and Q. Wang isolated the samples and performed the trait measurements and marker assays. Q. Wang, F. Han and X. Liu analysed the trait and marker data and revised the manuscript. Y. Zhang and Z. Fang conceived the idea and critically reviewed the manuscript. L. Yang, M. Zhuang, Y. Liu and Z. Li coordinated and designed the study. All of the authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Lv, H., Wang, Q., Han, F. et al. Genome-wide indel/SSR scanning reveals significant loci associated with excellent agronomic traits of a cabbage (Brassica oleracea) elite parental line ‘01–20’. Sci Rep 7, 41696 (2017). https://doi.org/10.1038/srep41696
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep41696
This article is cited by
-
A high-quality reference genome for cabbage obtained with SMRT reveals novel genomic features and evolutionary characteristics
Scientific Reports (2020)
-
Development and utilization of an InDel marker linked to the fertility restorer genes of CMS-D8 and CMS-D2 in cotton
Molecular Biology Reports (2020)
-
Identification of a major QTL for seed number per silique in cabbage (Brassica oleracea L. var. capitata) using genotyping by sequencing
Euphytica (2019)
-
Genome-wide characterization of simple sequence repeats in Pyrus bretschneideri and their application in an analysis of genetic diversity in pear
BMC Genomics (2018)
-
Fine mapping and candidate gene identification of the genic male-sterile gene ms3 in cabbage 51S
Theoretical and Applied Genetics (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
