
Abstract
Plant forms display a wide variety of architectures, depending on the number of lateral branches, internode elongation and phyllotaxy. These are in turn determined by the number, the position and the fate of the Axillary Meristems (AMs). Mutants that affect AM determination during the vegetative phase have been isolated in several model plants. Among these genes, the GRAS transcription factor LATERAL SUPPRESSOR (Ls) plays a pivotal role in AM determination during the vegetative phase. Hereby we characterize the phylogenetic orthologue of Ls in Antirrhinum, ERAMOSA (ERA). Our data supported ERA control of AM formation during both the vegetative and the reproductive phase in snapdragon. A phylogenetic analysis combined with an analysis of the synteny of Ls in several species strongly supported the hypothesis that ERA is a phylogenetic orthologue of Ls, although it plays a broader role. During the reproductive phase ERA promotes the establishment of the stem niche at the bract axis but, after the reproductive transition, it is antagonized by the MADS box transcription factor SQUAMOSA (SQUA). Surprisingly double mutant era squa plants display a squa phenotype developing axillary meristems, which can eventually turn into inflorescences or flowers.
Similar content being viewed by others

Introduction
The aerial plant body derives from the Shoot Apical Meristem (SAM), through the iterative production of phytomers. A phytomer unit is comprised of a node, to which a leaf is anchored, the corresponding internode and an Axillary Meristem (AM) at the leaf axil1. AM initiation occurs at the anlagen of the apical meristem, giving rise to new buds, again formed by a set of phytomers that can remain dormant or grow out2. Plants produce new AMs throughout their life cycle, but the nature and properties of AMs change during the different developmental phases. This becomes evident after the floral transition, since the committed floral meristems are indeed products of AMs and are homologous to vegetative axillary buds3.
Two theories have been proposed to explain AM formation. The ‘detached meristem’ theory proposes that the SAM gives rise to AMs during the production of leaf primordia4, therefore the AM founder cells remain undifferentiated. The ‘de novo’ model postulates that AMs are induced from previously differentiated cells in the leaf axis5.
In several plant species, mutants impaired in plant architecture have been described, suggesting a tight genetic control. They are grouped into three main classes: mutants affected in AM initiation, meristem outgrowth, or both6.
An interesting mutant altered in AM initiation is the tomato lateral suppressor (ls)7. Ls function is quite conserved, as indicated by its orthologues in Arabidopsis thaliana (LAS) and in Oryza sativa (MONOCULM1, MOC1)7,8,9. All these mutants fail to produce vegetative AMs. Compared to wild-type plants, tomato ls plants produce fewer flowers (without petals); similarly the rice moc1 rachis forms fewer branches and spikelets9. Conversely, the Arabidopsis las plants do not develop lateral branches during vegetative growth, but inflorescences and flowers appear normal.
The Antirrhinum majus eramosa phenotype is caused by the disruption of the Ls orthologue ERA. era plants are unable to produce AMs during the vegetative phase, whilst during the reproductive phase, era inflorescences develop very few flowers. ERA overexpression in Arabidopsis stimulates branching, strengthening the important role of the Ls clade in controlling agronomic traits. Our phylogenetic reconstruction revealed that leguminous species lack Ls orthologues, suggesting that this family has developed a different means of controlling AM determination.
Results and Discussion
ERA is required for axillary meristem formation in Antirrhinum
Antirrhinum exhibits different types of phyllotaxy during its growth10. During the early vegetative phase, the leaves develop according a decussate phyllotaxy and AMs are formed at the leaf axis11. The reproductive phase is marked by a switch to a spiral phyllotaxy, during which single bracts develop at each node and subtend a new floral AM (Fig. 1a–d)11.

(a–d) Branching of Antirrhinum plants. Wild-type plants possess a main stem and several lateral branches (a) while era plants have a single stem and no lateral branches (b). At the axil of wild-type leaves AMs form buds that give rise to new phytomers (c, arrowheads), while era leaf axils do not subtend new buds (d, arrowheads). (e,f) Histological sectioning of wild-type (e) and era SAM (f) stained with Toludine Blue O. At the axil of wild-type bracts developing buds are present (e) while the axil of era bracts is vacant (f). (g–j) Antirrhinum flower. Wild-type flowers contain five petals with a dorsoventral asymmetry and enclose the flower, hiding the stamens and carpel (g,i). In era flower stamens and carpel are visible and exposed (h,j); supernumerary organs are developed (6 stamens, asterisks). (k–n) SEM analysis of flower ontogenesis. In the first whorl of wild-type flowers, five sepal primordia are formed (k) and, later in development, in the second and third whorls, petal and stamen primordia are developed (m). In era flowers, several primordia at different developmental stages are formed in the first whorl (l). In the inner whorls supernumerary primordia are developed which are not well positioned and not synchronized (n). SAM = Shoot Apical Meristem; br: bracts; fm: floral meristem; s: sepals; p: petals; st: stamens; c: carpel.
The era mutant12 resembles ls/las mutants. era plants do not develop AMs, therefore they have a single stem displaying a very erect growth without secondary branches and inflorescences (Fig. 1a–d). During the vegative phase, era plants develop at each node decussate leaves and after the transition to the reproductive phase bracts are produced according to a spiral phyllotaxis. Histological sections confirm the inability of era plants to produce AMs (Fig. 1e,f). Occasionally, a few abnormal flowers (from 1–2 to 5–6 per plant) are formed while the wild-type inflorescence is an indeterminate raceme (Fig. 1a,b,g–j and Supplementary Fig. S1). Flower ontogeny determined by Scanning Electron Microscopy (SEM) confirmed that era flowers present several defects, such as altered organ position and/or supernumerary organs (Fig. 1k–n). era inflorescences develop very few flowers, with unpredictable spatiotemporal distribution of AMS, and era flowers sometimes develop at the axis of quite well formed bracts.
era and similar mutants in other species
The severity of the era phenotype implicates a pivotal role for this locus in controlling AM formation in either the vegetative or the reproductive phase. ls and las display less severe effects during the reproductive phase, therefore at least in Solanaceae and Brassicaceae, reproductive AMs might be controlled by parallel pathways or by redundant factors.
The morphological defects of the ls mutant are accompanied by changes in hormone levels: ls plants produce more auxin and gibberellic acid, and accumulate less cytokinin13. In addition, the level of abscisic acid (ABA) is affected. indeed tomato ls stems accumulate more ABA than wild type stems14. It has been demonstrated that higher levels of auxin and ABA inhibit bud development14. This inhibition, along with the converse relationship between hormonal content and apical dominance15,16 could explain the ls phenotype.
era decapitation causes plant death and grafting procedures fail, providing indirect evidence for hormonal defects in era plants since hormones like auxin and cytokinin are potential candidates involved in the grafting procedure17 and definitely auxin impairment is strictly related to phyllotaxis and apical dominance and it is clear that auxin depletion is strictly tied to phyllotaxis and apical dominance18,19.
ERA is a GRAS protein and is the orthologue of LAS/Ls
The similarity recorded between era and ls raised the hypothesis that the era phenotype might result from a defective Ls-like gene. We unsuccessfully screened genomic and cDNA libraries using heterologous probes; similar difficulties were encountered isolating Ls7. By using degenerate primers designed with the conserved region of LAS and Ls (Supplementary Table S1) we were able to amplify the first ERA fragment. The complete genomic locus has been retrieved aligning the ERA sequence against the available sequence of the Antirrhinum genome (Xue Yongbiao, Beijing Institute of Genomics, unpublished). The ERA gene is 1239 bp long, intronless and encodes a GRAS protein of 412 aa with all the characteristic domains (Supplementary Fig. S2)20.
PCR analyses using era mutant genomic DNA, amplified with gene specific and generic transposon primers, identified the insertion of a CACTA transposon 561 bp downstream from the ATG. The transposon is a TAM1 -like21 and a deletion in one of the CACTA ends makes the transposon unable to transfer. We backcrossed era on E165 wild-type plants, and then analysed 124 F2 plants. The transposon insertion showed absolute cosegregation with the mutant phenotype (Chi-square test for a 1:2:1 segregation hypothesis = 0.387; P-Value = 0.824).
ERA regulates meristematic tissue formation outside the SAM
The distribution of ERA mRNA was analysed with semi-quantitative RT-PCR and mRNA in situ hybridization. Semi-quantitative RT-PCR showed that ERA is expressed in roots, stem, leaves, inflorescence and flowers at different developmental stages (Supplementary Fig. S3). In situ hybridizations showed ERA transcripts in the SAM, both in the epidermal and subepidermal layers, in the floral dome and sepal primordia (Fig. 2a,b)22. No ERA expression was detected in the era mutant (Fig. 2c). Unlike its orthologues, ERA expression is not restricted to the incipient AMs, as for MOC1, or at the adaxial side of the leaf primordium, as for LAS8,9. This broader expression could explain the severe era phenotype during the reproductive phase.

(a,b) ERA transcript in wild-type plants is detected in the SAM (a), in the floral meristems and in developing flowers (b). (c) In era mutant plants no transcript is detected. (d) In wild-type plants STM is expressed in the SAM and in developing floral meristems. (e) In era mutant plants no STM expression is detected outside the SAM. SAM = Shoot Apical Meristem; br: bracts; fm: floral meristem; s: sepals.
The inability of era mutants to produce new axillary meristems during the reproductive phase might be caused by the absence of meristematic tissue in the axil, or by the inability to produce new flower buds from the meristematic tissue. In order to discriminate between these hypotheses, we analysed the expression of SHOOT MERISTEMLESS (STM) in wild-type and era plants. In wild-type plants STM is expressed in the SAM and in floral meristem, but not in leaf initials, providing an early marker for AMs development (Fig. 2d)23. In era mutants, STM transcripts are detected only in the SAM and no STM expression is detected at the axil of new putative flowers (Fig. 2e). These data clearly indicate that no meristematic tissue is formed during the reproductive phase at the axil of era mutants, highlighting the role of ERA in the production of meristematic tissue outside the SAM. Similarly, the rice orthologue MOC1 is able to control the expression of OSH1 in leaf axils while in Arabidopsis, LAS controls STM accumulation only in old leaf axils8. These data still suggest a fundamental role of ERA in AM formation, and are consistent with the idea that LAS/MOC1/ERA prevent the differentiation of cells in the boundary zone, thus stimulating meristem formation1.
ERA and SQUAMOSA are epistatic after the floral transition
MADS box transcription factors are important regulators of vegetative development. Silencing of the Paulownia kawakamii PkMADS1 stimulates leaf formation with altered phyllotaxy at the expense of meristem maintenance24, whilst potato MADS-box 1 (POTM1) maintains the growth equilibrium between axillary and apical vegetative meristems. POTM1 belongs to the APETALA1/SQUAMOSA (AP1/SQUA) clade25.
Arabidopsis AP1 participates in the establishment of the floral meristem26 by regulating cytokinin homeostasis. In ap1 plants, perturbation in cytokinin content causes the formation of ectopic flowers at the axils of sepals. In Antirrhinum, SQUA encodes a MADS-box transcription factor that shares high sequence similarity with Arabidopsis AP110, SQUA mainly regulates floral meristem identify and proliferation27,28.
We therefore generated era squa double mutants to assay whether these two genes can interact to modulate axillary meristem formation during the reproductive phase. era squa plants resemble era in the vegetative phase, with no AM development at the axis of the decussate leaves. SQUA is epistatic to ERA after the floral transition, since new primordia develop frequently at the axis of bracts (Fig. 3b,d,e). These primordia can give rise to flowers, inflorescences or new seedlings (reversion, Fig. 3d,e). The reversion phenotype can be explained by decreased levels of INCOMPOSITA in the squa mutant28. INCO is homologous to tomato JOINTLESS (J), and it has been reported that j inflorescences revert to a vegetative shoot after the production of few flowers29.

(a) squa single mutant produces a reduced number of abnormal flowers, while the era squa double mutant plants produced more flowers than squa (b). (c) Intriguingly these flowers are similar to the squa mutant flowers and they appear much more regular than the era flowers. (b,d,e) At the axis of the era squa bracts, new primordia develop frequently producing either a flower, an inflorescence or a new seedling (d,e).
Indeed it was clear that squa can recover era defects. Intriguingly, era squa plants produced more flowers than squa (Fig. 3a–c), which completely resembled squa flowers, and appeared much more regularly than those of era (Fig. 1h,j).
ERA forms homodimers and heterodimers
Since ERA and SQUA interact genetically, we asked whether ERA is also able to heterodimerize with SQUA and eventually with other MADS-box transcription factors involved in the establishment of the floral meristems, using yeast two and three hybrid (Y2H and Y3H) and Bimolecular Fluorescence Complementation (BiFC) assays. Y2H tests using ERA as bait demonstrated that this GRAS protein physically interacts with the MADS box transcription factor SQUA (Fig. 4a). Furthermore, the Y2H assays demonstrated that ERA is able to form homodimers (Fig. 4a). In order to reproduce in vivo the interaction among these proteins we exploited the BiFC technique, using leaves of Nicotiana benthamiana for the transient expression of protein chimeras (Fig. 4b–g)30. The results confirmed the data obtained through the Y2H assay, thus ERA is able to interact with SQUA, forming heterodimers, and with itself forming homodimers (Fig. 4d,f,g).

(a) yeast two and three hybrid (Y2H and Y3H) assays demonstrated that ERA physically interacts with SQUA and forms homodimers. The Y3H assay proved that the homodimer INCO-INCO can interact with ERA while all the other combinations tested fail to grow. (b,c) The interaction among INCO and SQUA was used as positive control for testing the interaction: the two proteins are able to interact in planta and to fully reconstruct the YFP signal. The reconstruction of the YFP signal was also observed when ERA interacted with itself (d), suggesting the formation of homodimers, and when ERA interacted with SQUA (f,g), suggesting the formation of heterodimers. No interaction was detected between ERA and INCO as suggested by lack of reconstitution of the YFP signal (e).
SQUA participates in the commitment of the Antirrhinum floral meristem together with the MADS-box INCO. Indeed, genetic and Y2H evidence indicates that the dimer INCO-SQUA promotes flower development28. Interestingly, INCO can homodimerise (although quite weakly) and the INCO-INCO homodimer represses flower development28. We therefore asked whether ERA could interact with INCO, since era squa inflorescences strongly resemble those of inco era plants. To this end, we set up an yeast ternary assay28,31, in which the INCO open reading frame was cloned into pGBKT7 and pTFT1 yeast expression vectors (see material and methods) and these plasmids were used to co-transform yeast strain AH109 with GAL4AD-ERA. The ternary assay scored positively, demonstrating the INCO-INCO dimer can interact with ERA (Fig. 4a) and participates in floral meristem outgrowth. In conclusion, ERA is able to interact with SQUA and with INCO but only when INCO is a homodimer, but the ternary complex ERA-SQUA-INCO was not detected. These data suggest that the interaction between ERA and INCO necessarily relies on INCO homodimer formation, providing a possible explanation for the lack of a ternary complex.
Heterologous overexpression of ERA causes an increase in lateral branching but is not able to complement the las phenotype
To confirm the key role of ERA in AM determination, we overexpressed ERA in A. thaliana (Col-0) under the control of the constitutive promoter 35SCaMV. We analysed the potential shoot branching in the overexpressing lines, compared to wild-type, counting the number of AMs formed from the rosette leaves, without the removal of primary bolts to maintain the correct apical dominance. Half of the analysed lines produced more lateral branches (n = 10). The progeny of two lines with a single T-DNA insertion were used to count the number of lateral branches, confirming the data collected in T1 (Supplementary Fig. S3). Interestingly MOC1 overexpression in rice also stimulates tiller proliferation9.
To understand if ERA is able to complement the Arabidopsis thaliana las phenotype we crossed the wild-type Columbia overexpressing plants with the las mutant. In the T2 generation, we counted the number of lateral branches both in WT, las and las 35S::AmERA (Supplementary Fig. S3). The las plants carrying the 35S::AmERA construct did not show an increase in the number of lateral branches respect to the las mutant. Two possible scenarios can explain this result. First, ERA is not able to complement las, possibly due to the fact that Arabidopsis and Antirrhinum are quite distantly related plant species and the genes could have acquired different functions. Second, and most probably, the regulatory pathways controlling the funtions of ERA and LAS are significanctly different and prevent complementation.
It is also possible that the 35S promoter is unable to express ERA in the founder AM cells. However, we observed that wild-type plants over- (and probably also mis-) expressing ERA produce more lateral branches; therefore we infer that the dimer ERA/LAS is sufficient to activate lateral branch formation, but not ERA alone.
Phylogenetic reconstruction of the Ls subfamily
Previous screens, limited to Arabidopsis and rice, divided the GRAS transcription factor family into eight monophyletic subfamilies32. The Ls subfamily is further split into two main lineages: the first Ls lineage includes Ls, LAS, ERA, MOC1 and its close relative Os-GRAS7, whereas the second lineage includes Os-GRAS19, At-GRAS19 and At-GRAS32 proteins33. At-GRAS19 and At-GRAS32 are allelic to SCL7 (SCARECROW-LIKE7) and SCL4 respectively20. Therefore, we renamed these two lineages the Ls lineage and the SCL4 lineage respectively, from their founding member.
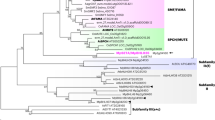
The Ls subfamily originated before the divergence of mosses and vascular plants34. To understand the evolution of the Ls subfamily during land plant diversification, we screened several plant genomes and EST collections (Supplementary Table S2). Protein alignment analyses allowed us to define the Ls subfamily throughout the plant kingdom (Supplementary Fig. S4). Either Ls or SCL4 orthologues are present in the moss Physcomitrella patens, as well as orthologues from other GRAS subfamilies (Supplementary Fig. S4)34. Therefore, the Ls and SCL4 clades diverged before the last common ancestor (LCA) of mosses and Tracheophyta. Our analysis revealed that both the clades are highly conserved within land plants (Fig. 5). Our analysis suggests that SCL4 lineage is sister to Ls, in agreement with previous studies conducted on a few angiosperm species33,35, which is worthy of further investigation.

Both clades appear as monophyletic and conserved in all of the taxa analyzed, without conserved gene duplication events, which seem mostly species-specific. However, Ls orthologues are missing in all of the genomes available from legumes. The scale bar indicates the number of amino acid changes per site.
Blast analysis failed to retrieve GRAS-like proteins from the available green algae genomes (Phytozome). In agreement with Engstrom’s conclusions34, our reconstruction suggests the GRAS family probably arose within the Streptophyta and soon diverged in land plant ancestors.
Within the Ls lineage, most eudicot species possess one Ls/ERA gene, although two Ls-like genes are found in species with recent whole genome duplication events such as Linum usitatissimum, Manihot esculenta and Malus domestica, and there are three Ls-like genes in poplar (Supplementary Fig. S4). The average number of Ls-like factors in monocots is higher: There are two copies in rice, four in Musa acuminata and at least three in Phoenyx dactylifera (Supplementary Fig. S4)36,37.
Surprisingly, no Ls/ERA orthologues were found in Medicago truncatula, Phaseolus vulgaris or Glycine max. We therefore screened the genomes of the other eight legume species currently available. Again, we could not retrieve any Ls-related factors. A new phylogenetic tree has been generated with all the ERAMOSA orthologues currently available (Supplementary Fig. S5), confirming that no ERA homologues can be found in the Leguminosae family. In conclusion, the Ls lineage has been lost before the LCA of Papilionoideae, the largest subfamily of Fabaceae. Therefore the molecular mechanism regulating branching is somehow modified in legumes, and apparently an Ls-like transcription factor is not required. However, it cannot be excluded that other GRAS transcription factors might assume this role.
Micro-synteny of the Ls clade
In the phylogenetic trees the Brassicaceae Ls-like factors are separated from the Ls orthologues of other eudicots, although the eudicot clade includes the homolog from Carica papaya, which belongs to the order Brassicales (Fig. 5). However, the hypothesis that two paralog lineages were differentially lost and retained in Brassicaceae and in all other eudicots seems extremely unlikely. Genome synteny can be used to unambiguously identify orthologous genes in plants38. The chromosome regions downstream of the Ls genes of Arabidopsis and Capsella rubella display conservation in the gene order39; according to our analyses, synteny is also conserved in Eutrema salsugineum, papaya, mimulus, tomato and grape (Fig. 6). To a variable extent, the downstream synteny is conserved in many other core eudicots, or towards the upstream region in the case of Vitis vinifera, but not in the basal eudicot A. coerulea (data not shown). Altogether, the synteny reconstructions confirm that LAS is the true orthologue of Ls and ERA. Thus, the independent clustering of Brassicaceae Ls proteins seems only due to a higher degree of sequence divergence. We extended synteny reconstruction to legumes, and we have been able to identify the “ERA downstream block”, further supporting the absence of the ERA/Ls locus in this family (Fig. 6).

(a) The figure shows the synteny downstream of the Ls homologous locus (represented in black). Genes which are poorly or not syntenic are indicated by white boxes with black margin and, where numerous, gaps have been inserted. In each gap, the number above is relative to the hidden known loci, and the number below is the approximative length of the gap, in kilobase pairs. For detail description, see materials and methods. (b) Schematic representation of the 256 kbp long corresponding region in the woodland strawberry (Fragaria vesca) chromosome LG3. A highly similar, 250 kbp long region is found in the chromosome 6 of peach (not shown). On the genome assembly, the Ls homolog is named mrna01184.1-v1.0-hybrid. Upstream, a gene which is conserved in sequence in core eudicots is encoding for an inosine-5-monophosphate dehydrogenase related protein, indicated in orange (locus gene01181-v1.0-hybrid). Downstream, the syntenic region is formed by the same genes found in (a) and in most other core eudicots, terminating with the gene encoding for the putative Sec14p-like phosphatidylinositol transfer protein (mrna01444.1-v1.0-hybrid). The scheme shows the corresponding genes and regions in the genomes of the legumes Medicago truncatula, Phaseolus vulgaris and the ancient tetraploid Glycine max. Any Ls-like gene or partial sequence is found within 1 Mbp upstream and 1 Mbp downstream from each of these regions, which was further confirmed by NCBI translated nucleotide blast tool (tblastn) using as a query the woodland strawberry Ls homolog protein sequence. In both (a) and (b), the length of the genes and of the intergenic spaces is not to scale. In (b), due to their high number and variability, non-syntenic genes are omitted. The analysis has been done by manual check of the genome browsers available in the Phytozome database.
In conclusion, we identify the phylogenetic snapdragon orthologue of LAS/Ls/MOC1. Altogether our data further support the detached theory, suggesting that axillary meristem founder cells remain undifferentiated4,5,40.
Material and Methods
Plant material and growth conditions
The mutant plant era was originally described by Stubbe12, and the corresponding seeds were obtained from the Gatersleben Centre. Antirrhinum majus wild-type (ecotype E165) and era mutant plants were grown at 22 °C under long-day (16 h light/8 h dark) conditions.
Analytical microscopy: histological analysis, SEM and in situ hybridization
For morphological analysis, wild-type (ecotype E165) and era inflorescences and seedlings were fixed as previously described41. Sections (8 μm) were stained in 0.5% (w/v) toluidine blue O or hybridized with digoxigenin-labelled ERA and STM antisense probes (primers are reported in Supplementary Table S1). DIG-labelled RNA probes were prepared as previously described41. The samples were observed using a Zeiss Axiophot D1 microscope equipped with differential interface contrast (DIC) optics. Images were recorded with an Axiocam MRc5 camera (Zeiss) using the Axiovision program (version 4.1). Scanning electron microscopy (SEM) was performed by producing replicas of flowers and developing inflorescences as described by Green and Linstead42. The samples were observed using a Zeiss DSM 940 scanning electron microscope.
cDNA isolation
In order to isolate the ERA cDNA we screened a cDNA and a genomic DNA Antirrhinum library with probes generated by amplifying the conserved region of LAS and Ls. Subsequently, the ERA cDNA was amplified using degenerate primers to the conserved region of LAS and Ls (Supplementary Table S1). The ERA CDS and protein sequences from this publication have been submitted to the GenBank database (http://www.ncbi.nlm.nih.gov/genbank/) and assigned the identifier KT698108.
Expression Analysis
To analyze ERA expression, semi-quantitative RT-PCR was performed in duplicate on cDNA obtained from two different biological replicates of RNA from different tissues. Total RNA was extracted using the LiCl method43. DNA contamination was removed using the Ambion TURBO DNA-free DNase kit according to the manufacturer’s instructions (Life Technologies). The RNA was reverse transcribed using the ImProm-II reverse transcription system (Promega) and the cDNA was used as template in semi-quantitative RT-PCR reactions. As a control, we simultaneously amplified an ACTIN fragment. Primers used for semi-quantitative RT-PCR are reported in Supplementary Table S1.
Yeast two and three hybrids and BiFC assays
To determine the ability of ERA to interact with other partners, the coding sequences of ERA, INCO and SQUA were cloned into pDONR207 (Life Technologies) and subsequently by Gateway recombination transferred to the GAL4 system (pGADT7 and pGBKT7; Clontech) or pTFT1 Gateway (kindly provided by Marcos Egea Cortines; ref. 44). The two- and three-hybrid assays were performed at 28 °C in the yeast strain AH109 (Clontech) and were tested on selective yeast synthetic dropout medium lacking leucine, tryptophan, adenine and histidine and supplemented with different concentrations of 3-aminotriazole (1, 2 and 5 mM of 3-AT). For BiFC assay, the coding sequences of ERA, INCO and SQUA cloned into pDONR207 (Life Technologies) were transferred by Gateway recombination to the pYFPN43 and pYFPC43 vectors. BiFC was performed as previously described by Belda-Palazon et al.30 using in the co-infiltration procedure the p19 protein of the tomato bushy stunt virus to suppress gene silencing. The abaxial surfaces of infiltrated tobacco leaves were analyzed 5 days after inoculation. Primers used for gene cloning are reported in Supplementary Table S1.
Plasmid Construction and Plant Transformation
To overexpress ERA in Arabidopsis plants, the entire ERA gene was amplified with primers containing attB1 and attB2 recombination sequences (Supplementary Table S1). ERA cDNA was then cloned into the pB2GW7 binary vector, under the regulation of the CaMV 35S constitutive promoter. The vector was introduced into Agrobacterium tumefaciens by electroporation and used to transform Arabidopsis wild-type Col-0 plants using the floral dip method45. Transformants were selected through BASTA selection. The number of AMs of transformed and control plants was counted to verify the role of ERA in AM formation. RNA from overexpressing lines was extracted from seedlings using the NucleoSpin RNA Plant kit (Macherey-Nagel). The RNA was reverse transcribed as above and the cDNA was used as template in semi-quantitative RT-PCR reactions. The semi-quantitative RT-PCR assay was conducted in triplicate. As control, we simultaneously amplified an ACTIN fragment. Primers used for semi-quantitative RT-PCR are reported in Supplementary Table S1.
Phylogenetic analysis
Protein sequences from ERA, known ERA homologues, and the SCL4 clade, were used to conduct tblastn and blastp analyses to screen the plant sequences available at Phytozome (www.phytozome.net) and NCBI Genomes (https://www.ncbi.nlm.nih.gov/genome/). Homologues of Arabidopsis lyrata, Brassica rapa and of grasses, with the exception of O. sativa, were not included in the final phylogenetic analysis. Picea abies46 (www.congenie.org), Amborella trichopoda (www.amborella.org), Musa acuminata47 (http://banana-genome.cirad.fr/) and Phoenyx dactylifera36,37 were included. We also added a cDNA fragment of the Pteridium aquilinum LAS48. In Ricinus communis (www.phytozome.net) synteny reconstruction allowed identification of an ERA homologue (locus 28966.m000535). To confirm the absence of ERA and Ls homologues in the Leguminosae family, we extended blast analysis to the legume genomes currently available at WGS Sequencing Projects (NCBI) and Kazusa DB, which includes two independent genome sequences of Cicer arietinum49,50, Lotus japonicus51, Cajanus cajan52, Lupinus angustifolius53, Vigna angularis54, Arachis duranensis55, Arachis ipaensis55, Glycine max56, Medicago truncatula57, Phaseolus vulgaris58, Trifolium pretense59, andVigna radiate60.
The best blast hits were then aligned using MAFFT (http://mafft.cbrc.jp/alignment/server/). The protein alignments were manually corrected and used to generate phylogenetic trees with the Maximum Likelihood methods with Phylip Package61. The Maximum Likelihood tree has been prepared with the MrEnt software62. We also included in the analysis members of the other GRAS subfamilies reported by Tian et al.33. This allowed us to discard blast hits belonging to different GRAS subfamilies, and to build the Ls subfamily trees presented in this work.
The phylogenetic tree reported in Supplementary Fig. S5 has been constructed using a Maximum Likelihood method through the PhyML software (JTT protein model, bootstrapping of 100). The optimal protein model was previously analyzed using ProTest. Protein datasets were downloaded from Phytozome, GenBank genomes, Sol Genomic Networks, Cucubits Genomic Database and the Kazusa Genomic Data FTP site.
The analysis of synteny between genomes has been conducted manually between the three Brassicaceae species Arabidopsis thaliana (49 kbp of chromosome 1), Capsella rubella (46 kbp of scaffold 1) and Eutrema salsugineum (46 kbp of scaffold 7), the related papaya (Caricaceae, Brassicales; 178 kbp of supercontig 12) and the three asterids: Solanum lycopersicum (Solanaceae, 143 kbp of chromosome 7), Mimulus guttatus (Phrymaceae, 110 kbp of scaffold 13) and Antirrhinum majus (Plantaginaceae, limited to the available sequence of contig 909). The conserved loci considered in the analysis encode the following: a putative ribosomal protein L11 methyltransferase (MT, white boxes with blue margin), a protein phosphatase 2C (PP2C, white boxes with red margin), a F-box and LRR interaction motif protein (F-box,. white boxes with green margin), a WRKY transcription factor (red), a putative leucine-rich receptor serine/threonine-protein kinase, BRASSINOSTEROID INSENSITIVE1-like (BRL, in brown), a CLC chloride channel (yellow), a protein similar to prenylated RAB acceptor 1 (green), a putative HMG-box and ARID/BRIGHT domain DNA-binding protein (grey), a photosystem I subunit G (PSAG, in light blue), a transducin/WD40 domain-containing protein (violet), a putative Sec14p-like phosphatidylinositol transfer protein (blue), a HAPLESS8 protein (HAP, in pink), a peroxisomal targeting signal 1 receptor PEX5 (in lavender color), and a COP9 signalosome subunit 6 (in dark green).
Additional Information
How to cite this article: Mizzotti, C. et al. ERAMOSA controls lateral branching in snapdragon. Sci. Rep. 7, 41319; doi: 10.1038/srep41319 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Ward, S. P. & Leyser, O. Shoot branching. Curr. Opin. Plant Biol. 7, 73–78 (2004).
Beveridge, C. A., Weller, J. L., Singer, S. R. & Hofer, J. M. I. Axillary meristem development. Budding relationships between networks controlling flowering, branching, and photoperiod responsiveness. Plant Physiol. 131, 927–934 (2003).
Long, J. & Barton, M. K. Initiation of axillary and floral meristems in Arabidopsis. Dev. Biol. 218, 341–353 (2000).
Steeves, T. A. & Sussex, I. M. Patterns in Plant Development. (eds Cambridge University Press, 1989).
McConnell, J. R. & Barton, M. K. Leaf polarity and meristem formation in Arabidopsis. Development 125, 2935–2942 (1998).
Wang, Y. & Li, J. Molecular basis of plant architecture. Annu. Rev. Plant Biol. 59, 253–279 (2008).
Schumacher, K., Schmitt, T., Rossberg, M., Schmitz, G. & Theres, K. The Lateral suppressor (Ls) gene of tomato encodes a new member of the VHIID protein family. Proc. Natl. Acad. Sci. USA 96, 290–295 (1999).
Greb, T. et al. Molecular analysis of the LATERAL SUPPRESSOR gene in Arabidopsis reveals a conserved control mechanism for axillary meristem formation. Genes Dev. 17, 1175–1187 (2003).
Li, X. et al. Control of tillering in rice. Nature 422, 618–621 (2003).
Mizzotti, C., Galliani, B. M. & Masiero, S. The backstage of the ABC model: the Antirrhinum contribution. Plant Biosystems 148, 176–186 (2014).
Matkowski, A. & Adler, K. The diversity and ontogenetic changes of phyllotaxis in wild-types and two leaf form mutants of Antirrhinum majus L. Bot. J. Linn. Soc. 131, 235–248 (1999).
Stubbe, H. Genetik und Zytologie von Antirrhinum L. sect Antirrhinum. (eds Veb Gustav Fischer Verlag, 1966).
Tucker, D. J. Endogenous growth regulators in relation to side shoot development in the tomato. New Phytol. 77, 561–568 (1976).
Tucker, D. J. Apical dominance in the tomato: The possible roles of auxin and abscisic acid. Plant Sci. Lett. 12, 273–278 (1978).
Napoli, C. A., Beveridge, C. A. & Snowden, K. C. Reevaluating concepts of apical dominance and the control of axillary bud outgrowth. Curr. Top. Dev. Biol. 44, 127–169 (1999).
Cline, M. Concepts and terminology of apical dominance. Am. J. Bot. 84, 1064 (1997).
Melnyk, C. W. & Meyerowitz, E. M. Plant grafting. Curr. Biol. 25, R183–8 (2015).
Morris, D. A. Transport of exogenous auxin in two-branched dwarf pea seedlings (Pisum sativum L.) : Some implications for polarity and apical dominance. Planta 136, 91–96 (1977).
SNOW, R. O. N. The Nature Of Correlative Inhibition. New Phytol. 36, 283–300 (1937).
Pysh, L. D., Wysocka-Diller, J. W., Camilleri, C., Bouchez, D. & Benfey, P. N. The GRAS gene family in Arabidopsis: sequence characterization and basic expression analysis of the SCARECROW-LIKE genes. Plant J. 18, 111–119 (1999).
Sommer, H. et al. In Plant Transposable Elements (eds Nelson, O., Wilson, C. & Saslaw, C. ) 47, 227–235 (Springer US, 1988).
Vincent, C. A. & Coen, E. S. A temporal and morphological framework for flower development in Antirrhinum majus. Can. J. Bot. 82, 681–690 (2004).
Waites, R., Selvadurai, H. R., Oliver, I. R. & Hudson, A. The PHANTASTICA gene encodes a MYB transcription factor involved in growth and dorsoventrality of lateral organs in Antirrhinum. Cell 93, 779–789 (1998).
Prakash, A. P. & Kumar, P. P. PkMADS1 is a novel MADS box gene regulating adventitious shoot induction and vegetative shoot development in Paulownia kawakamii. Plant J. 29, 141–151 (2002).
Becker, A. & Theißen, G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 29, 464–489 (2003).
Han, Y., Zhang, C., Yang, H. & Jiao, Y. Cytokinin pathway mediates APETALA1 function in the establishment of determinate floral meristems in Arabidopsis. Proc. Natl. Acad. Sci. USA 111, 6840–6845 (2014).
Huijser, P. et al. Bracteomania, an inflorescence anomaly, is caused by the loss of function of the MADS-box gene squamosa in Antirrhinum majus. EMBO J. 11, 1239–1249 (1992).
Masiero, S. et al. INCOMPOSITA: a MADS-box gene controlling prophyll development and floral meristem identity in Antirrhinum. Development 131, 5981–5990 (2004).
Szymkowiak, E. J. & Irish, E. E. JOINTLESS suppresses sympodial identity in inflorescence meristems of tomato. Planta 223, 646–658 (2006).
Belda-Palazón, B. et al. Aminopropyltransferases Involved in Polyamine Biosynthesis Localize Preferentially in the Nucleus of Plant Cells. PLoS One 7, 1–10 (2012).
Egea-Cortines, M., Saedler, H. & Sommer, H. Ternary complex formation between the MADS-box proteins SQUAMOSA, DEFICIENS and GLOBOSA is involved in the control of floral architecture in Antirrhinum majus. EMBO J. 18, 5370–5379 (1999).
Bolle, C. The role of GRAS proteins in plant signal transduction and development. Planta 218, 683–692 (2004).
Tian, C., Wan, P., Sun, S., Li, J. & Chen, M. Genome-wide analysis of the GRAS gene family in rice and Arabidopsis. Plant Mol. Biol. 54, 519–532 (2004).
Engstrom, E. M. Phylogenetic analysis of GRAS proteins from moss, lycophyte and vascular plant lineages reveals that GRAS genes arose and underwent substantial diversification in the ancestral lineage common to bryophytes and vascular plants. Plant Signal. Behav. 6, 850–854 (2011).
Huang, W., Xian, Z., Kang, X., Tang, N. & Li, Z. Genome-wide identification, phylogeny and expression analysis of GRAS gene family in tomato. BMC Plant Biol. 15, 209 (2015).
Al-Mssallem, I. S. et al. Genome sequence of the date palm Phoenix dactylifera L. Nat. Commun. 4, 2274 (2013).
Al-Dous, E. K. et al. De novo genome sequencing and comparative genomics of date palm (Phoenix dactylifera). Nat. Biotechnol. 29, 521–527 (2011).
Causier, B., Castillo, R., Xue, Y., Schwarz-Sommer, Z. & Davies, B. Tracing the Evolution of the Floral Homeotic B- and C-Function Genes through Genome Synteny. Mol. Biol. Evol. 27, 2651–2664 (2010).
Rossberg, M. et al. Comparative sequence analysis reveals extensive microcolinearity in the lateral suppressor regions of the tomato, Arabidopsis, and Capsella genomes. Plant Cell 13, 979–988 (2001).
Teichmann, T. & Muhr, M. Shaping plant architecture. Front. Plant Sci. 6, 233 (2015).
Ambrose, B. A. et al. Molecular and genetic analyses of the silky1 gene reveal conservation in floral organ specification between eudicots and monocots. Mol. Cell 5, 569–579 (2000).
Green, P. B. & Linstead, P. A procedure for SEM of complex shoot structures applied to the inflorescence of snapdragon (Antirrhinum). Protoplasma 158, 33–38 (1990).
Verwoerd, T., Dekker, B. & Hoekema, A. A small-scale procedure for the rapid isolation of plant RNAs. Nucleic Acids Res 17, 2362 (1989).
Egea-Cortines, M., Saedler, H. & Sommer, H. Ternary complex formation between the MADS-box proteins SQUAMOSA, DEFICIENS and GLOBOSA is involved in the control of floral architecture in Antirrhinum majus. EMBO J. 18, 5370–5379 (1999).
Clough, S. J. & Bent, A. F. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743 (1998).
Nystedt, B. et al. The Norway spruce genome sequence and conifer genome evolution. Nature 497, 579–584 (2013).
D’Hont, A. et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488, 213–217 (2012).
Der, J. P., Barker, M. S., Wickett, N. J., dePamphilis, C. W. & Wolf, P. G. De novo characterization of the gametophyte transcriptome in bracken fern, Pteridium aquilinum. BMC Genomics 12, 99 (2011).
Jain, M. et al. A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). Plant J. 74, 715–729 (2013).
Varshney, R. K. et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 31, 240–246 (2013).
Sato, S. et al. Genome structure of the legume, Lotus japonicus. DNA Res. 15, 227–239 (2008).
Varshney, R. K. et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 30, 83–89 (2011).
Yang, H. et al. Draft genome sequence, and a sequence-defined genetic linkage map of the legume crop species Lupinus angustifolius L. PLoS One 8, e64799 (2013).
Kang, Y. J. et al. Draft genome sequence of adzuki bean, Vigna angularis. Sci. Rep. 5, 8069 (2015).
Bertioli, D. J. et al. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat Genet 48, 438–446 (2016).
Schmutz, J. et al. Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183 (2010).
Young, N. D. et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 480, 520–524 (2011).
Schmutz, J. et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 46, 707–713 (2014).
Sato, S. et al. Comprehensive structural analysis of the genome of red clover (Trifolium pratense L.). DNA Res. 12, 301–364 (2005).
Kang, Y. J. et al. Genome sequence of mungbean and insights into evolution within Vigna species. Nat. Commun. 5, 5443 (2014).
Felsenstein, J. PHYLIP (Phylogeny Inference Package). version 3.6. Distributed by the author. Department of Genome Sciences, University of Washington, Seattle. (2005).
Zuccon, A. & Zuccon, D. MrEnt: an editor for publication-quality phylogenetic tree illustrations. Mol. Ecol. Resour. 14, 1090–1094 (2014).
Acknowledgements
We thank Xue Yongbiao (Beijing Institute of Genomics, The Chinese Academy of Sciences) for sharing data in advance of publication. Edward Kiegle (University of Milan) is acknowledged for editing the paper. This work was supported by Cariplo Foundation [grant number 2011-2257 to C.M. and S.M.] and by FP7-PEOPLE-2013-IRSES [FRUIT LOOK to S.M.].
Author information
Authors and Affiliations
Contributions
C.M., L.D., H.S. and S.M. designed research; C.M., B.M.G., A.B. and L.D. performed research; C.M., L.D., A.B. and S.M. analyzed data; C.M., L.D. and S.M. wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Mizzotti, C., Galliani, B., Dreni, L. et al. ERAMOSA controls lateral branching in snapdragon. Sci Rep 7, 41319 (2017). https://doi.org/10.1038/srep41319
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep41319
This article is cited by
-
Molecular mechanisms regulating ornamental traits and scent production in snapdragon (Antirrhinum majus L.)
Horticulture Advances (2023)
-
An efficient protocol for Agrobacterium-mediated genetic transformation of Antirrhinum majus
Plant Cell, Tissue and Organ Culture (PCTOC) (2020)
-
Trans-splicing of plastid rps12 transcripts, mediated by AtPPR4, is essential for embryo patterning in Arabidopsis thaliana
Planta (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
