
Abstract
Use of 10,129 singleton SNPs of known genomic location in tetraploid cotton provided unique opportunities to characterize genome-wide diversity among 440 Gossypium hirsutum and 219 G. barbadense cultivars and landrace accessions of widespread origin. Using the SNPs distributed genome-wide, we examined genetic diversity, haplotype distribution and linkage disequilibrium patterns in the G. hirsutum and G. barbadense genomes to clarify population demographic history. Diversity and identity-by-state analyses have revealed little sharing of alleles between the two cultivated allotetraploid genomes, with a few exceptions that indicated sporadic gene flow. We found a high number of new alleles, representing increased nucleotide diversity, on chromosomes 1 and 2 in cultivated G. hirsutum as compared with low nucleotide diversity on these chromosomes in landrace G. hirsutum. In contrast, G. barbadense chromosomes showed negative Tajima’s D on several chromosomes for both cultivated and landrace types, which indicate that speciation of G. barbadense itself, might have occurred with relatively narrow genetic diversity. The presence of conserved linkage disequilibrium (LD) blocks and haplotypes between G. hirsutum and G. barbadense provides strong evidence for comparable patterns of evolution in their domestication processes. Our study illustrates the potential use of population genetic techniques to identify genomic regions for domestication.
Similar content being viewed by others


Introduction
One of the most remarkable stories in crop domestication is the origin of cultivated cotton1. Gossypium is remarkably diverse, with more than 50 species classified into 8 diploid genome groups and a single, monophyletic tetraploid cluster2,3,4,5,6. For many years, allopolyploid cotton has been used for evolutionary investigations into the genomic mysteries of polyploidy2. Recent phylogeny studies further resolved the monophyletic cluster of tetraploids consisting of six different species; Gossypium mustelinum (AD4) is the basal clade of allotetraploid taxa, all of which are direct descendants of an allopolyploidization event involving ancestral diploid species closely resembling G. arboreum (A2 genome diploid) and G. raimondii (D5 genome diploid) G. tomentosum (AD3), along with two other close species G. ekmanianum (AD6) and G. hirsutum (AD1), form a second clade that is sister to a third clade composed of the Galapagos Islands endemic G. darwinii (AD5) and G. barbadense (AD2)7.
Cultivars of G. hirsutum and G. barbadense produce the overwhelming majority of the world’s cotton fiber and oil. G. hirsutum has a large indigenous range encompassing most of Mesoamerica and the Caribbean, where it shows a diverse array of morphological forms spanning the wild-to-domesticated continuum5,8,9,10. Modern, improved varieties of G. hirsutum (“Upland cotton”) account for about 90% of world cotton commerce, and are day-length neutral annuals derived from subtropical, perennial photoperiodic landraces9,11. Wendel, et al.9 identified two centers of diversity for G. hirsutum, one in southern Mexico–Guatemala and the other in the Caribbean. G. barbadense cultivars are valued for their production of high quality fiber, classified as extra-long staple (ELS), and characterized by being exceptionally long, strong, and fine. These cottons are denoted variously as Pima, Egyptian or Sea Island cotton. G. barbadense, like G. hirsutum, spans a wild-to-domesticated range, and originated as a tropical, photoperiodic (short-day flowering) perennial. Originating in Peru and Ecuador, G. barbadense was domesticated in northwest South America and the Caribbean1,12,13,14.
Groupings and categorizations have been made within the wild to landrace continuum of both G. hirsutum and G. barbadense based upon phenotype and geographic origin. In G. hirsutum, races yucatanense and punctatum are two adjacent populations located in the inferred domestication region, the Yucatan peninsula in Mesoamerica5. Several localized derivatives of race punctatum include richmondi, morilli, palmeri, marie-galante and latifolium15,16, hereafter referred to as G. hirsutum landraces. These landraces have not differentiated at the molecular level. G. barbadense most likely underwent a trans-Andean expansion into northern South America after primary domestication west of the Andes17,18. A secondary stage of dispersal involved expansion into Central America, the Caribbean and the Pacific18. During this dispersal, enough accumulated differentiation occurred within the species to produce the sibling Galapagos Islands endemic species G. darwinii, and the distinctive variety braziliense, formerly recognized as the species G. braziliense13.
The molecular diversity of both G. hirsutum and G. barbadense has been extensively studied with a wide variety of marker tools11,19,20,21,22,23,24,25,26,27,28,29,30,31. However, with the recent release of two independent whole-genome sequences from the same genotype, TM-1, a highly homozygous line of allotetraploid G. hirsutum32,33, researchers have rich opportunities to explore genome-wide constraints on molecular diversity and unravel domestication mechanisms of tetraploid cultivated and landrace cotton.
This report explores at a new level of resolution how breeding histories and selective pressures shaped the gene pools of G. hirsutum and G. barbadense cotton. We used genotyping-by-sequencing (GBS) to generate 10,129 single nucleotide polymorphisms (SNPs) for 440 G. hirsutum and 219 G. barbadense accessions from representative samples of cultivated and wild pools. This report focuses on a comparative study of linkage disequilibrium (LD) among G. hirsutum and G. barbadense chromosomes and constraints in population structure of both allopolyploids. With this genome-wide characterization of SNP variation in domesticated tetraploid cotton, we show that cultivated allotetraploid species experienced both unique and common patterns of genome-wide selection pressure during domestication.
Materials and Methods
Germplasm
A representative sample of 658 cotton accessions (440 of G. hirsutum and 218 of G. barbadense) collected from 85 countries in North America, South America, Europe, Asia, and Africa were obtained from the National Cotton Germplasm Collection (NCGC) maintained by the USDA-ARS in College Station TX34 (Table S1). Improved and unimproved G. barbadense (86 and 132 accessions, respectively) and G. hirsutum (268 and 172 accessions, respectively) were selected for inclusion. Selection of accessions was performed with great attention to achieving a representative sample of the geographic, developmental and historic diversity present in the collection.
Genotyping by sequencing
Genomic DNA isolation from the seedlings involved the DNeasy plant mini kit (QIAGEN, Germany), and GBS was as described35,36. DNA was treated with the restriction enzyme Pst1, barcoded by accession, and sequenced on an Illumina HiSeq 2000 as described35. Raw sequence reads were trimmed with use of Sickle (https://github.com/najoshi/sickle), with minimum PHRED quality threshold of 20 in a sliding window. Then reads were demultiplexed and barcodes were removed. Trimmed reads were mapped to the G. raimondii and G. hirsutum reference sequences by using GSNAP with SNP-tolerant mapping based on >20 million homoeo-SNPs37. Aligned reads were processed by use of SAMtools and classified as the G. hirsutum AT- and DT-genomes by use of PolyCat38,39. Barcoded sequence reads were processed and collapsed into a set of unique sequence tags, with one TagCounts file produced per input FASTQ file40. The separate TagCounts files were then merged to form a “master” TagCounts file, which retained only those tags present at or above an experiment-wide minimum count. This master tag list was then aligned to the TM-1 (G. hirsutum) reference genome33 and a Tags On Physical Map (TOPM) file was generated, containing the genomic position of each tag with a unique, best alignment. The barcode information in the original FASTQ files was then used to tally the number of times each tag in the master tag list was observed in each sample (“taxon”) and these counts were stored in a TagsByTaxa (TBT) file. The information recorded in the TOPM and TBT was then used to discover SNPs at each “TagLocus” (set of tags with the same genomic position) and filter the SNPs based upon the proportion of taxa covered by the TagLocus and minor allele frequency40. For each retained SNP, the allele represented by each tag in the corresponding TagLocus was recorded in the TOPM file, along with its relative position in the locus. The end product of the Discovery Pipeline was a “production-ready” TOPM that was then used by the production Pipeline to call SNPs. To call SNPs and ensure that indels were handled consistently, a de novo multiple sequence alignment of all the tags in each TagLocus was performed using the BioJava 3.0 API41, which implements the CLUSTAL W algorithm42. For each SNP in the resulting “TagLocusAlignment”, the allele represented by each tag is determined and the TBT file was consulted to tally the observed depths of each allele in each taxon.
Population structure analysis
Genetic diversity values were calculated by a neighbor-joining algorithm with TASSEL 5. In a second approach, we used identity by State (IBS) and principle component analysis (PCA) with the SNP & Variation Suite (SVS v8.1.5) (Golden Helix, Inc., Bozeman, MT, USA; www.goldenhelix.com). Observed nucleotide diversity (π) and Tajima’s D was estimated by using TASSEL v5.0 with a sliding-window approach as described43. Estimation of the fixation index (FST) were based on the Wright F statistic44 calculated by use of SVS v8.1.5.
Characterization of LD
We considered only SNPs mapped to the G. hirsutum whole-genome sequence draft, because knowing the chromosome location of SNPs helps prevent spurious LD and characterization of sub-genomic LD. Haplotype blocks were calculated for all markers by using the default settings in SVS v8.1.5 as described in Nimmakayala, et al.45. Adjacent and pairwise measurements of LD were calculated separately for each chromosome. For computing linkage disequilibrium (LD), we used expectation-maximization (EM) algorithm, formalized by46, is a iterative technique for obtaining maximum likelihood estimates of sample haplotype frequencies.
Results and Discussion
GBS of tetraploid cotton and SNP mining
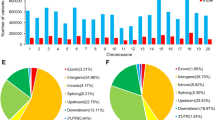
A total of 1,700,626,447 reads were available for analysis after quality trimming; 471,553,537 (33.2%) were classified as from the AT-genome and 460,910,405 (32.4%) from the DT-genome (Fig. S1). The remainder could not be classified by known SNPs between genomes (homoeo-SNPs). The median value of reads per genome (AT and DT) for each accession was 519,563.5; 2,240,812 (46.0%) were aligned to unique positions and 1,020,968 (20.9%) to multiple positions. Overall, 1,612,880 (33.1%) could not be aligned to the G. hirsutum reference genome. When orthologs were below a certain threshold of divergence, sequences aligned to both A and D genomes, which led to multiple-site–aligned sequence tags. In this case, sequence tags from orthologous sequences would collapse into a single locus or two separate loci. The SNPs from these loci showed very high heterozygosity, depending on the sequencing depth. Figure S2 shows the distribution of heterozygosity (in individuals and SNPs) for the SNPs for the G. raimondii and G. hirsutum genomes. The distribution of heterozygosity for the SNPs, when mapped to the G. raimondii genome was higher. In total, 10,129 SNPs were called for uniquely aligned sequence tags and hence are singletons: 314, 240, 327, 205, 641, 247, 315, 448, 326, 441, 567, 454, 367, 309, 364, 203, 317, 643, 296, 326, 451, 422, 434, 548, 485 and 369 were mapped to Chrs. 1–26, respectively. We found SNPs at average intervals of 195 kb across the G. hirsutum genome.
Population structure, nucleotide diversity and demographic history
To investigate genetic differentiation due to population structure among cotton groups as reflected by these genome-wide SNPs, we used PCA and NJ analysis. In a PCA of all analyzed accessions, most cultivated G. hirsutum lines clustered together and 172 photoperiodic landraces of G. hirsutum (TX) clustered into two groups. One group of TX lines was predominantly from South America and the second group was from Central America and the Caribbean (Fig. 1 and Table S3). The cluster of Upland cotton cultivars spread into the cluster of Central America and Caribbean landraces, which indicated that this region might be the center of domestication for Upland cotton. In PCA of 268 Upland cotton cultivars representing worldwide breeding efforts, the distribution of U.S. cultivars spanned the collective distributions of all other national cultivars, indicating a level of diversity equal to the global cultivar diversity. An additional PCA of only 219 G. barbadense lines clearly separated cultivars and landraces (Fig. S3 and Table S4).

See Table S3 for a list of accessions and respective eigen values for respective positions of individual accessions in the figure.
PCA (Fig. 1) and NJ analysis (Fig. 2A, Table S2) revealed two distinct clusters for the G. hirsutum and G. barbadense species, with a clear separation of cultivated and landraces within each species. A focused NJ analysis of only G. hirsutum (Fig. 2B) revealed three distinct clusters within the species, with one cluster comprised of cultivated Upland cottons, an adjacent cluster of landrace accessions from Central America and the Caribbean, and a third cluster of predominantly South American accessions. Overall genetic diversity among the allotetraploids was 0.336, with mean genetic diversity 0.089, 0.22, 0.173 and 0.336 among cultivated G. hirsutum, landraces of G. hirsutum, cultivated G. barbadense, and landraces of G. barbadense, respectively. The diversity among cultivated G. hirsutum accessions is more or less evenly distributed across the world with the highest mean diversity estimates of 0.098, 0.08, 0.071, 0.081, 0.078 and 0.072 occurring among accessions from South America, Central America, the United States, Europe, Asia and Africa, respectively (Fig. 2B). A focused NJ analysis of only G. barbadense (n = 219) lines resolved a monophyletic cultivar cluster containing 86 accessions of cultivated G. barbadense (Fig. 2C) and placed the landraces of G. barbadense into two distinct clusters. The cluster in close proximity to the cultivated forms was a mixture of landraces from the Caribbean and South America. This analysis of population genomic diversity in cultivated allotetraploid genomes of cotton adds resolution to prior knowledge with the inclusion of more accessions and a large genomewide sampling of SNPs. Some studies have developed high-throughput SNPs for tetraploid cultivated cotton; most focused on generating high-resolution genetic maps36,37,38,39,40,41,42,43. Our study involved the use of GBS-generated SNPs mapped to the recently released G. hirsutum genome sequence33 to identify singleton SNPs assigned to chromosome positions.

Phylogenetic trees constructed with neighbor-joining for (A) both Gossypium hirsutum cultivars (red: Upland cultivars; blue: landraces) and Gossypium barbadense cultivars (magenta: cultivated; green: landraces) and (B) cultivars and wild G. hirsutum (green: Upland cultivars; blue and red: two clusters of landraces) and (C) cultivars and landraces of G. barbadense (blue: cultivated; green and red: two clusters of landraces). Distance matrix is in Table S2.
We examined allele sharing across the panel by calculating identity-by-state (IBS) coefficients among all pairs of accessions47. The mean IBS sharing within cultivated and landrace G. hirsutum was 0.95 and 0.85, respectively. The lowest IBS sharing within the cultivated G. hirsutum combinations was noted for Paymaster 54 and Hopi (0.62), Zhong Mian Suo Hao and Hsing-Tai #68–71 (0.63), Express 432 and Hopi (0.63); Rilcot and Hopi (0.63); Rowden #2 and Kekchi (0.63), Blight Master and Anton Stormproof 99 and Lankart Sel.611 (0.63), UKA B1- (72) 047 and UK 64 (0.63), UKA S1 (72) 070 and Deltapine 16 (0.63), UA 7–10 and Hopi (0.63), and 108F with several other G. hirsutum accessions (0.63). The low IBS values obtained between a number of cultivars and Hopi and Kekchi are unsurprising, in that Hopi and Kekchi cottons are probably most appropriately classified as primitive cultivars. Hopi cultivars result from pre-Columbian cultivation by the Hopi Indians of cotton obtained from Mexico, and Kekchi cotton was imported into the U.S. from Mexico in the early 20th century in conjunction with efforts to find boll weevil resistance. These relatively modern introductions to cultivation have contributed to a wider divergence in cultivated cotton. IBS sharing suggests a high degree of genetic similarity between the two accessions/species, either intentionally or unintentionally. The mean IBS sharing between cultivated and landrace G. hirsutum was 0.55. Average IBS sharing between cultivated G. hirsutum and cultivated G. barbadense was 0.41. G. hirsutum cultivated genotypes 108 F and M100 showed high IBS sharing, from 0.92 to 0.95, with Old Pima, Yuma, Bolivia, Pima (Raleigh Stock), Linia1780, Tadla116, Pima32, Pima S-7, 8810, Pima S-3, Early Pima, P76 and Pima S-1, among G. barbadense cultivars. Perhaps some crossing between tetraploid species occurred to produce the 108 F and M100 genotypes because of their high IBS value and historical efforts of trait introgression. Given the high IBS value, an equally probable scenario may be that 108 F and M100 are truly G. barbadense and have been misclassified as G. hirsutum. The mean observed IBS sharing within cultivated G. barbadense was 0.89. G. barbadense cultivars Bahamas 1 and St. Croix consistently showed low IBS sharing (0.53 to 0.58) with the remaining G. barbadense accessions, and may be indicative of low introgression into these two accessions, or their misclassification.
Our diversity and IBS analyses of the two cultivated allotetraploids showed little sharing of alleles between the genomes, with only a few combinations showing high IBS sharing, which indicates sporadic introgression between the two species. Because the two allotetraploids were domesticated independently in geographically isolated locations, each thousands of years ago6,15,48, such divergence is expected.
Because of the strong population structure, we assessed patterns of variation separately for each group when making inferences about the evolutionary dynamics of domestication. Crop domestication is often associated with ‘population bottlenecks’, due to the limited number of founding individuals experiencing domestication events. These bottlenecks may be evident in cotton when comparing diversity between elite cultivars and wild or minimally improved landraces. The within-group polymorphic SNP number was 2,084 (minor allele frequency [MAF] ≥ 0.05) for G. hirsutum, and 2417 (MAF ≥ 0.05) for G. barbadense. To assess levels and patterns of polymorphism in the cultivated (SA) and landrace (TX) G. hirsutum and cultivated (C) and wild (D) G. barbadense gene pools, we estimated nucleotide diversity (π) and Tajima’s D across various chromosomes using the SNPs that have MAF of ≥0.05. The frequency of segregating SNPs as reflected by various chromosomal measures of mean nucleotide diversity and Tajima’s D is presented in Fig. 3. We found a high number of new alleles, representing nucleotide diversity, on chromosomes 1 and 2 in cultivated G. hirsutum as compared with low nucleotide diversity on these chromosomes in landrace G. hirsutum. Subsequent spread of G. hirsutum cultivars across the world and exposure to diverse breeding programs or selection might explain such rapid population size expansion due to overrepresentation of new alleles segregating in cultivars. In addition, we noted biased distribution of Tajima’s D toward negative values on the remaining chromosomes (3 to 26) in cultivated G. hirsutum as compared with its wild ancestor, which indicates a population bottleneck during domestication. In contrast, G. barbadense chromosomes showed negative Tajima’s D on several chromosomes for both cultivated and landrace types, which indicate that speciation of G. barbadense itself might have occurred with relatively narrow genetic diversity. For cultivated G. barbadense, chromosome 4 and 22 appears to have new alleles for nucleotide diversity and Tajima’s D was more positive than for unimproved types.

Frequency spectrum for the chromosomal means of nucleotide diversity (π) and Tajima’s D for cultivated and non-cultivated G. hirsutum and cultivated and non-cultivated G. barbadense.
The presence of high diversity on chromosomes 1 and 2 in cultivated G. hirsutum indicates many relatively new alleles in this region. In contrast, for cultivated G. barbadense, chromosome 4 appears to show increased nucleotide diversity and Tajima’s D. Unusually divergent genomic regions resulting from disproportionate accumulation of novel alleles on particular chromosomes among closely related sub-species such as indica and japonica in rice, G. hirsutum and G. barbadense cottons and Capsicum annuum and C. baccatum peppers could cause reproductive barriers or incompatibility resulting in partial fertility45,49. Similar to current findings, several reports implicated newly recruited polymorphisms as causing highly divergent genomic regions that may control traits associated with reproductive incompatibility or ecological adaptation45,50,51.
To further resolve species differentiation between the two tetraploid taxa, we estimated pairwise fixation index (FST) across all polymorphisms with MAF ≥ 0.05. All FSTs were highly significant (P > 0.001). The FST between wild accessions of G. hirsutum and G. barbadense was 0.66 and 0.53, respectively. The FST between cultivated and wild accessions within G. hirsutum was 0.20 and between cultivated and landraces within G. barbadense was 0.05. Genome-wide means smoothed FST values for various groups of taxa as presented for all chromosomes in Manhattan plots, showing genomic regions with spikes representing the highest FST values and sweeps the lowest (Fig. 4). In recent studies, because of potential FST outliers, adaptive divergence is assessed by comparing neutral genetic variation as outlined by Lewontin-Krakauer method52 with quantitative genetic differentiation (QST) to infer the cause for population differentiation53,54. Whitlock (2008)55 has emphasized that QST values should be compared to the distribution of FST values as Lewontin-Krakauer method by itself does not work well for FST values above 0.1 such as in the current study. We have currently undertaken GWAS in multiple locations and years for fiber components in G. hirsutum and G. barbadense for elucidating evolutionary potential of various genomic regions under divergent selection.

Manhattan plots of chromosome-wise pairwise FST values (mean smoothed) for D_C: (G. barbadense landraces and G. barbadense cultivated); SA_TX (G. hirsutum Upland cultivars and uncultivated G. hirsutum); C_SA (cultivated G. barbadense and cultivated G. hirsutum) and D_TX (landraces of G. barbadense and landraces of G. hirsutum).
Haplotypes, LD decay, and comparative analysis of species-specific versus common LD blocks
Haplotype distribution is important in comparing common and unique patterns of genetic variation of allotetraploid cotton gene pools and has a wide range of applications. Domestication process and breeding history are the two major processes that shape haplotype structure45. We used “Minimize historical recombination”, a block-defining algorithm developed by Gabriel, et al.56. The upper and lower confidence bound were set to 0.98 and 0.70, respectively, and SNPs below MAF of 0.05 were skipped. Maximum block length was set to 160 Kb. The EM (Expectation Maximization) algorithm was used for haplotype estimation with convergence tolerance 0.0001 and frequency threshold of 0.01. Maximum EM iterations were set to 50. We identified 335 SNPs in 143 haplotypes within cultivated G. hirsutum and 689 SNPs in 283 haplotypes (Table S7) in cultivated G. barbadense (Table S8). The location of haplotypes was well conserved in both species, whereas the length of haplotypes was longer for G. barbadense than G. hirsutum, which may indicate narrow genetic diversity in G. barbadense (Fig. 5).

Conserved genomic positions of haplotype formation in both species may indicate conserved patterns of evolution. Correspondence of occurrence of shared haplotype blocks was tested using the hypergeometric probability function (P = 6.29E-26). We estimated LD by using an entire marker set with MAF ≥ 0.05 and identified 1419 and 1293 significant associations in G. hirsutum and G. barbadense respectively (Tables S5 and S6). We also estimated chromosome-wise distribution of LD blocks separately for G. hirsutum and G. barbadense (Figs 6 and S4). Although the magnitude and strength of association differed among the common LD blocks between the cultivated cotton genomes, most of the blocks appeared to be coincident. LD decay in G. hirsutum occurred over intervals averaging 117 Kb. In G. barbadense, 1,293 SNP associations suggested a genome-wide LD decay over intervals averaging 128 Kb (Table S6). Chromosome-wise shared LD blocks between G. hirsutum and G. barbadense and hypergeometric probability significance levels for correspondence between the species are presented in Table 1. The locations of singleton SNPs in the tetraploid genomes provided a unique opportunity to unravel several interesting findings apart from characterizing the global genome-wide diversity of G. hirsutum and G. barbadense. Using genome-wide SNPs, measurements of genetic diversity, haplotype distribution and LD decay patterns in the two genomes shed light on genomic architecture, domestication processes and population demographic history.

Heat map depicts strength of association.
Estimates of LD across the 26 chromosomes of tetraploid cotton provide insights into the haplotype block structure of the various chromosomes, providing researchers with a way to efficiently select markers and infer genotypes based on nearby loci. When a new haplotype-containing accession is used in a breeding program, it can recombine with other diverse haplotypes to create progenies of intermediate relatedness, a genetic basis for novel variation57. Also of interest to breeders, our diversity and population structure analysis provides insights into the domestication process between G. hirsutum and G. barbadense useful for controlling error due to population stratification, which often leads to identifying spurious associations in GWAS experiments. Our analysis of LD revealed considerable similarity within and across the two genomes. In some instances, LD remained elevated over extended genomic regions in both species. In sunflower, Mandel, et al.58 noted that large LD blocks occurred in close proximity to genes and QTL for traits targeted by selection during domestication or improvement. Given that our analysis was based on only 3,708 SNPs, we probably missed several LD blocks. Future resequencing of several diverse cultivated accessions of G. hirsutum and G. barbadense would precisely characterize LD decay and distribution.
Our study illustrates the potential for breeders to use population genetic techniques to identify genomic regions of importance. Performing GWAS for fiber traits with dense SNP panels is feasible and would provide further insights into the complexity of genome organization underlying these two allotetraploid cotton species.
Additional Information
How to cite this article: Reddy, U. K. et al. Genome-wide divergence, haplotype distribution and population demographic histories for Gossypium hirsutum and Gossypium barbadense as revealed by genome-anchored SNPs. Sci. Rep. 7, 41285; doi: 10.1038/srep41285 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Wendel, J. F. & Cronn, R. C. Polyploidy and the evolutionary history of cotton. Advances in agronomy 78, 139–186 (2003).
Adams, K. L. & Wendel, J. F. Exploring the genomic mysteries of polyploidy in cotton. Biological Journal of the Linnean Society 82, 573–581, doi: 10.1111/j.1095-8312.2004.00342.x (2004).
Grover, C., Grupp, K., Wanzek, R. & Wendel, J. Assessing the monophyly of polyploid Gossypium species. Plant systematics and evolution 298, 1177–1183 (2012).
Endrizzi, J., Turcotte, E. & Kohel, R. Genetics, cytology and evolution of Gossypium . Advances in genetics 23, 271–375 (1985).
Brubaker, C. L., Bourland, F. & Wendel, J. F. The origin and domestication of cotton. Cotton: Origin, history, technology, and production. John Wiley & Sons, New York 3–31 (1999).
Hovav, R., Chaudhary, B., Udall, J. A., Flagel, L. & Wendel, J. F. Parallel domestication, convergent evolution and duplicated gene recruitment in allopolyploid cotton. Genetics 179, 1725–1733 (2008).
Grover, C. E. et al. Re-evaluating the phylogeny of allopolyploid Gossypium L. Molecular Phylogenetics and Evolution 92, 45–52, doi: 10.1016/j.ympev.2015.05.023 (2015).
Beasley, J. The origin of American tetraploid Gossypium species. American Naturalist 74, 285–286 (1940).
Wendel, J. F., Brubaker, C. L. & Percival, A. E. Genetic diversity in Gossypium hirsutum and the origin of upland cotton. American Journal of Botany 1291–1310 (1992).
Coppens d’Eeckenbrugge, G. & Lacape, J.-M. Distribution and Differentiation of Wild, Feral, and Cultivated Populations of Perennial Upland Cotton (Gossypium hirsutum L.) in Mesoamerica and the Caribbean. PLoS One 9, e107458, doi: 10.1371/journal.pone.0107458 (2014).
Iqbal, M., Reddy, O., El-Zik, K. & Pepper, A. A genetic bottleneck in the ‘evolution under domestication’ of upland cotton Gossypium hirsutum L. examined using DNA fingerprinting. Theoretical and Applied Genetics 103, 547–554 (2001).
Paterson, A. H. et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 492, 423–427 (2012).
Fryxell, P. A. The natural history of the cotton tribe (Malvaceae, tribe Gossypieae). (Texas A & M University Press., 1978).
Hinze, L. et al. Molecular characterization of the Gossypium Diversity Reference Set of the US National Cotton Germplasm Collection. Theoretical and Applied Genetics 128, 313–327, doi: 10.1007/s00122-014-2431-7 (2015).
Wendel, J. F. In Plant molecular evolution 225–249 (Springer, 2000).
Wendel, J. F., Brubaker, C., Alvarez, I., Cronn, R. & Stewart, J. M. In Genetics and genomics of cotton 3–22 (Springer, 2009).
Wendel, J. F., Brubaker, C. L. & Seelanan, T. In Physiology of cotton 1–18 (Springer, 2010).
Percy, R. G. & Wendel, J. F. Allozyme evidence for the origin and diversification of Gossypium barbadense L. Theoretical and Applied Genetics 79, 529–542, doi: 10.1007/bf00226164 (1990).
Abdalla, A., Reddy, O., El-Zik, K. & Pepper, A. Genetic diversity and relationships of diploid and tetraploid cottons revealed using AFLP. Theoretical and Applied Genetics 102, 222–229 (2001).
Abdurakhmonov, I. Y. et al. Molecular diversity and association mapping of fiber quality traits in exotic G. hirsutum L. germplasm. Genomics 92, 478–487, doi: 10.1016/j.ygeno.2008.07.013 (2008).
Abdellatif, K. F., Khidr, Y. A., El-Mansy, Y. M., El-Lawendey, M. M. & Soliman, Y. A. Molecular diversity of Egyptian cotton (Gossypium barbadense L.) and its relation to varietal development. Journal of Crop Science and Biotechnology 15, 93–99 (2012).
Kantartzi, S. K., Ulloa, M., Sacks, E. & Stewart, J. M. Assessing genetic diversity in Gossypium arboreum L. cultivars using genomic and EST-derived microsatellites. Genetica 136, 141–147 (2009).
Lacape, J.-M., Dessauw, D., Rajab, M., Noyer, J.-L. & Hau, B. Microsatellite diversity in tetraploid Gossypium germplasm: assembling a highly informative genotyping set of cotton SSRs. Molecular Breeding 19, 45–58 (2007).
Liu, B. & Wendel, J. F. Intersimple sequence repeat (ISSR) polymorphisms as a genetic marker system in cotton. Molecular Ecology Notes 1, 205–208 (2001).
Liu, S., Cantrell, R., McCarty, J. & Stewart, J. M. Simple sequence repeat–based assessment of genetic diversity in cotton race stock accessions. Crop Science 40, 1459–1469 (2000).
Lu, H. & Myers, G. Genetic relationships and discrimination of ten influential upland cotton varieties using RAPD markers. Theoretical and Applied Genetics 105, 325–331 (2002).
Multani, D. & Lyon, B. Genetic fingerprinting of Australian cotton cultivars with RAPD markers. Genome 38, 1005–1008 (1995).
Noormohammadi, Z., Hasheminejad-Ahangarani, F. Y., Sheidai, M., Ghasemzadeh-Baraki, S. & Alishah, O. Genetic diversity analysis in Opal cotton hybrids based on SSR, ISSR, and RAPD markers. Genetics and molecular research: GMR 12, 256–269 (2012).
Tyagi, P. et al. Genetic diversity and population structure in the US Upland cotton (Gossypium hirsutum L.). Theoretical and Applied Genetics 127, 283–295 (2014).
Van Deynze, A. et al. Sampling nucleotide diversity in cotton. BMC plant biology 9, 125 (2009).
Wang, X., Feng, C., Lin, Z. & Zhang, X. Genetic diversity of sea-island cotton (Gossypium barbadense) revealed by mapped SSRs. Genet Mol Res 10, 3620–3631 (2011).
Li, F. et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat Biotech 33, 524–530, doi: 10.1038/nbt.3208 http://www.nature.com/nbt/journal/v33/n5/abs/nbt.3208.html#supplementary-information (2015).
Zhang, T. et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat Biotech 33, 531–537, doi: 10.1038/nbt.3207 http://www.nature.com/nbt/journal/v33/n5/abs/nbt.3207.html#supplementary-information (2015).
Frelichowski, J. & Percy, R. In Cotton Agronomy Monograph (American Society of Agronomy, Inc., Crop Science Society of America, Inc., and Soil Science Society of America, Inc., 2015).
Elshire, R. J. et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6, e19379 (2011).
Reddy, U. K. et al. High-Resolution Genetic Map for Understanding the Effect of Genome-Wide Recombination Rate on Nucleotide Diversity in Watermelon. G3: Genes|Genomes|Genetics 4, 2219–30, doi: 10.1534/g3.114.012815 (2014).
Wu, T. D. & Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26, 873–881 (2010).
Page, J. T., Gingle, A. R. & Udall, J. A. PolyCat: a resource for genome categorization of sequencing reads from allopolyploid organisms. G3 3, 517–525 (2013).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Glaubitz, J. C. et al. TASSEL-GBS: A High Capacity Genotyping by Sequencing Analysis Pipeline. PLoS One 9, e90346, doi: 10.1371/journal.pone.0090346 (2014).
Prlic, A. et al. BioJava: an open-source framework for bioinformatics in 2012. Bioinformatics 28, 2693–2695 (2012).
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22, 4673–4680 (1994).
Korneliussen, T. S., Moltke, I., Albrechtsen, A. & Nielsen, R. Calculation of Tajima’s D and other neutrality test statistics from low depth next-generation sequencing data. BMC Bioinformatics 14, 289 (2013).
Weir, B. S. & Cockerham, C. C. Estimating F-statistics for the analysis of population structure. Evolution 1358–1370 (1984).
Nimmakayala, P. et al. Genome-Wide Divergence and Linkage Disequilibrium Analyses for Capsicum baccatum Revealed by Genome-Anchored Single Nucleotide Polymorphisms. Frontiers in Plant Science 7, doi: 10.3389/fpls.2016.01646 (2016).
Dempster, A. P., Laird, N. M. & Rubin, D. B. Maximum likelihood from incomplete data via the EM algorithm. Journal of the royal statistical society. Series B (methodological), 1–38 (1977).
Caicedo, A. L. et al. Genome-Wide Patterns of Nucleotide Polymorphism in Domesticated Rice. PLoS Genet 3, e163, doi: 10.1371/journal.pgen.0030163 (2007).
Wendel, J. F., Schnabel, A. & Seelanan, T. Bidirectional interlocus concerted evolution following allopolyploid speciation in cotton (Gossypium). Proceedings of the National Academy of Sciences 92, 280–284 (1995).
Tang, T. et al. Genomic Variation in Rice: Genesis of Highly Polymorphic Linkage Blocks during Domestication. PLoS Genet 2, e199, doi: 10.1371/journal.pgen.0020199 (2006).
Wu, C.-I. & Ting, C.-T. Genes and speciation. Nature Reviews Genetics 5, 114–122 (2004).
Wu, C. I. The genic view of the process of speciation. Journal of Evolutionary Biology 14, 851–865 (2001).
Lewontin, R. & Krakauer, J. Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics 74, 175–195 (1973).
Leamy, L. J. et al. Large‐scale adaptive divergence in Boechera fecunda, an endangered wild relative of Arabidopsis. Ecology and evolution 4, 3175–3186 (2014).
Storz, J. F. INVITED REVIEW: Using genome scans of DNA polymorphism to infer adaptive population divergence. Molecular ecology 14, 671–688 (2005).
Whitlock, M. C. Evolutionary inference from QST. Molecular ecology 17, 1885–1896 (2008).
Gabriel, S. B. et al. The structure of haplotype blocks in the human genome. Science 296, 2225–2229 (2002).
Platt, A. et al. The Scale of Population Structure in Arabidopsis thaliana . PLoS Genet 6, e1000843, doi: 10.1371/journal.pgen.1000843 (2010).
Mandel, J. R. et al. Association Mapping and the Genomic Consequences of Selection in Sunflower. PLoS Genet 9, e1003378, doi: 10.1371/journal.pgen.1003378 (2013).
Acknowledgements
Funding support is provided by USDA-NIFA Research (2012–02617 and 2013–04053). Authors acknowledge Raman Postdoctoral fellowship extended to CVCM Reddy by Government of India.
Author information
Authors and Affiliations
Contributions
U.K.R., R.G.P., J.A.U. and A.H.P. designed the research and conceived the project. P.N., V.L.A., C.V.C.M.R., J.Z.Y., J.F. and U.K.R. screened and characterized cotton lines and performed the experiments. U.K.R., T.S., P.N., J.T.P., D.Z. and T.S. analyzed the data. U.K.R., P.N., R.G.P., J.Z.Y., J.A.U. and A.H.P. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Reddy, U., Nimmakayala, P., Abburi, V. et al. Genome-wide divergence, haplotype distribution and population demographic histories for Gossypium hirsutum and Gossypium barbadense as revealed by genome-anchored SNPs. Sci Rep 7, 41285 (2017). https://doi.org/10.1038/srep41285
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep41285
This article is cited by
-
Favorable pleiotropic loci for fiber yield and quality in upland cotton (Gossypium hirsutum)
Scientific Reports (2021)
-
Role of SNPs in determining QTLs for major traits in cotton
Journal of Cotton Research (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.

{kind=link}
{kind=link}
{kind=link}