
Abstract
RBM10 is an RNA splicing regulator that is frequently mutated in lung adenocarcinoma (LUAD) and has recently been proposed to be a cancer gene. How RBM10 mutations observed in LUAD affect its normal functions, however, remains largely unknown. Here integrative analysis of RBM10 mutation and RNA expression data revealed that LUAD-associated RBM10 mutations exhibit a mutational spectrum similar to that of tumor suppressor genes. In addition, this analysis showed that RBM10 mutations identified in LUAD patients lacking canonical oncogenes are associated with significantly reduced RBM10 expression. To systematically investigate RBM10 mutations, we developed an experimental pipeline for elucidating their functional effects. Among six representative LUAD-associated RBM10 mutations, one nonsense and one frameshift mutation caused loss-of-function as expected, whereas four missense mutations differentially affected RBM10-mediated splicing. Importantly, changes in proliferation rates of LUAD-derived cells caused by these RBM10 missense mutants correlated with alterations in RNA splicing of RBM10 target genes. Together, our data implies that RBM10 mutations contribute to LUAD pathogenesis, at least in large part, by deregulating splicing. The methods described in this study should be useful for analyzing mutations in additional cancer-associated RNA splicing regulators.
Similar content being viewed by others

Introduction
Large-scale sequencing and computational analysis have enabled systematic identification of genetic mutations in various types of cancer1,2,3,4,5. This information, in particular concerning certain oncogenes, has been successfully used for molecular classification of cancer subtypes, drug development and targeted therapies6. For example, identification of mutations in specific oncogenes has dramatically improved diagnosis and treatment for patients with breast cancer7 and non-small cell lung cancer (NSCLC)8,9. However, the functional and clinical significance of many or even most of the identified mutations in cancer remains unknown1. This lack of knowledge has impeded the development of new diagnostic and therapeutic targets for a large number of cancer patients.
Mutations of uncertain significance (MUS) include those in recently identified candidate cancer genes10,11 and rare mutations in bona fide oncogenes and tumor suppressor genes12,13,14,15. Although algorithms have been developed to predict the functional effects of mutations in cancer, they often produce inconsistent results15,16,17. It is particularly challenging to predict the consequences of missense mutations, and experiments are often required to establish their effects. Robust methods for functional characterization of MUS in cancer-associated genes are therefore needed to develop new clinical applications.
Lung adenocarcinoma (LUAD) is the most common subtype of lung cancer, a heterogeneous set of diseases responsible for the largest number of cancer-related death worldwide8. Although dramatic progress has been made in the application of targeted therapies for patients carrying specific oncogenic mutations, including EGFR, BRAF, ERBB2 mutations and ALK, RET, ROS1 translocations, the majority of LUAD patients do not harbor these currently actionable mutations8,10. Recent sequencing efforts aimed at comprehensively identifying genetic alterations in LUAD have detected frequently occurring mutations in putative cancer genes, including proteins involved in RNA splicing and chromatin modification10,18. Elucidating the functional effects of these genetic mutations should facilitate clinical management of LUAD patients.
RBM10 is an RNA binding protein and splicing regulator located on the X chromosome. Loss-of-function (LOF) mutations in RBM10 have been reported to cause TARP (Talipes equinovarus, atrial septal defect, Robin sequence and persistent left superior vena cava, MIM #311900) syndrome, a congenital, multifaceted developmental disorder19,20,21. In addition, RBM10 mutations have been frequently observed in LUAD, and less frequently in other types of cancers, including colorectal carcinoma22, pancreatic ductal adenocarcinoma (PDA)23 and intraductal papillary mucinous neoplasm (IPMN)24. Notably, RBM10 mutations were found to be significantly enriched in LUAD in men compared with women10,25, suggesting that there may be gender-specific contributions to the development and progression of LUAD.
Our previous study revealed that RBM10 promotes exon skipping in its target genes, including the cancer genes NUMB and CREBBP26. Other studies have demonstrated that RBM10 suppresses LUAD cell proliferation by inhibiting the formation of the long splice variant of NUMB, which in turn represses NOTCH signaling activity27,28. It has also been shown that RBM10 can promote apoptosis by modulating the gene expression of tumor necrosis factor alpha (TNF-α) in breast cancer and leukemia cell lines29. Accordingly, RBM10 has recently been classified as a cancer gene and listed in the Catalog of Somatic Mutations in Cancer (COSMIC) database30. However, it remains largely unclear how RBM10 mutations, especially missense mutations, affect RBM10-mediated regulation of alternative splicing and suppression of LUAD cell proliferation.
In this study, we systematically analyzed the mutational landscape and expression of RBM10 in LUAD. We also designed a robust functional assessment pipeline and used this to elucidate the effects of six representative RBM10 mutations in LUAD. Our findings revealed that RBM10 mutations likely affect its tumor suppressive activity, at least in large part, by interfering with its splicing regulatory functions.
Results
RBM10 mutational spectrum and expression in lung adenocarcinoma (LUAD)
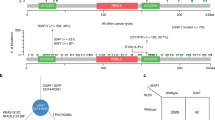
RBM10 mutations were identified in 53 of 1008 LUAD samples (~5.3%) listed in the COSMIC database, which compiles systematic screening data from The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGC) and manual curation data from independent studies. The mutational frequency was higher in TCGA LUAD cohort10,18, with RBM10 mutations identified in 43 out of 546 patients (~7.9%). The majority of RBM10 mutations were protein-truncating variants (PTVs), including nonsense, frameshift and splice site mutations, which often cause loss-of-function (LOF) (Fig. 1a). In addition to PTVs, a large fraction of RBM10 mutations were missense mutations (~30%), for which functional consequences are often uncertain and require experimental confirmation (Fig. 1a). Our survey also showed that mutations occurred throughout the RBM10 coding region rather than concentrating at specific sites (Fig. 1b). This mutational landscape resembles that observed in canonical tumor suppressive genes2.

(a) Distributions of distinct types of RBM10 mutations in LUAD samples in the total COSMIC collection and the TCGA cohort subset. Different mutation types are indicated by different colors. (b) Locations of COSMIC and TCGA LUAD RBM10 mutations within the RBM10 protein sequence. Characterized protein domains are represented by light purple boxes. Arrowheads indicate mutations chosen for experimental assays. RRM: RNA recognition motif; ZF: zinc finger; OCRE, Octamer Repeat; NLS: nuclear localization signals; G-patch: Glycine-patch. Data was downloaded from COSMIC (http://cancer.sanger.ac.uk/cosmic) and TCGA (http://cancergenome.nih.gov/).
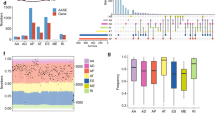
To examine how these mutations affect gene expression, we compared RBM10 mRNA levels in LUAD samples harboring different types of RBM10 mutations with LUAD samples lacking RBM10 mutations and samples from tumor-adjacent normal tissues. For these comparisons, we focused on the TCGA samples, for which RNA sequencing (RNA-seq) data are publically accessible. To rule out possible confounding effects of copy number alterations (CNAs) (Supplementary Fig. S1), we removed samples with RBM10 CNAs from our analysis. As shown in Fig. 2a, LUAD samples carrying three types of PTVs exhibited statistically significant reductions in mRNA expression compared to samples lacking RBM10 mutations or CNAs (P = 2.2e-06, 0.00011, 0.0013 for nonsense, frameshift, and splice site mutations, respectively), or tumor-adjacent normal tissues (P = 0.00035, 0.00035, 0.029 for nonsense, frameshift, and splice site mutations, respectively). By contrast, samples harboring missense mutations did not show statistically significant reductions in RBM10 mRNA levels. These results suggest that PTVs often cause RBM10 LOF, but possible functional consequences of missense mutations cannot be deduced from RNA expression levels alone.

(a) RBM10 mRNA expression in LUAD samples with different types of mutations, without mutation or copy number alteration (no mutation/CNA) and in tumor-adjacent normal tissues (Normal). Boxes represent the median (inside line) and the quartiles (upper and bottom boarder lines). “Whiskers” above and below the boxes represent maximum and minimum values within the 1.5 × IQ (inter-quarter) range. P values were calculated by Mann-Whitney tests followed by Benjamini & Hochberg corrections. Data from TCGA, see Methods for details. (b) Kaplan-Meier analysis of overall survival of LUAD patients, using data from the KM plotter database (http://www.kmplot.com). Patients were stratified into “low” and “high” RBM10 RNA expression groups, based on the auto select best cutoff. P = 0.0017 (log rank test). HR: hazard ratio.
We next examined correlations between RBM10 expression and overall survival of LUAD patients using an online Kaplan-Meier plotter31 (http://www.kmplot.com). This analysis revealed that high levels of RBM10 mRNA expression correlated with longer overall survival (Fig. 2b), suggesting a possible suppressive role of RBM10 in LUAD progression.
Oncogene-negative RBM10 mutations in LUAD patients correlate with significantly reduced RBM10 expression
Mutations in putative cancer genes that occur in the absence of well-defined oncogenes are termed “oncogene-negative” mutations and are considered to be potential “drivers” of carcinogenesis10. To determine whether RBM10 mutations occur in oncogene-negative patient samples, we examined mutation data from a recently published TCGA LUAD cohort study10. This analysis revealed that 4 of the 14 RBM10 mutations reported in the 230 LUAD samples were oncogene-negative10. Interestingly, RBM10 expression levels in each of the 4 oncogene-negative samples were significantly lower than that in LUAD samples lacking RBM10 mutations or CNAs (Fig. 3a, P = 0.0007), suggesting these mutations cause LOF and contribute to LUAD pathogenesis. Inspection revealed that 3 of the 4 RBM10 mutations found in oncogene-negative LUAD patients were unambiguously identified as PTVs, while the remaining one was annotated as a missense mutation (Q416L (c.1247A>T))10 (Supplementary Table S1).

(a) RBM10 mRNA expression in four samples harboring oncogene-negative RBM10 mutations and 201 LUAD samples lacking RBM10 mutations or CNAs (no mutation/CNA). Data obtained from a recently published LUAD cohort study by TCGA10. P = 0.0007 (Mann-Whitney test). (b) Structures of minigene splicing reporters for RBM10 exon 12 wild type and mutant (RBM10-E12M-WT and -1247A>T, respectively). Arrows indicate target specific forward primer (E12M-F) and vector specific reverse primer (BGH-R) used in RT-PCR. Asterisk indicates the position of mutation. E: Exon, In: intron. (c) RT-PCR analysis of exon 12 inclusion levels for RBM10-E12M-WT and -1247A>T in HEK293 cells. Left panel: a representative gel image. GAPDH: internal control. The full-length gel image is presented in Supplementary Fig. S9a. Right panel: quantification of exon 12 inclusion levels using PSI (percent-spliced-in) values. Data present means ± SEM. n = 4 biological replicates. ***P ≤ 0.001 (Student’s t-test).
The oncogene-negative RBM10 missense mutation disrupts RBM10 splicing
To determine how the c.1247A>T mutation reduces expression of RBM10, we examined its DNA sequence context and found that the mutation is located in the second-to-last nucleotide of RBM10 exon 12 (NM_005676.4). The A to T transversion reduced the 5′ splice site MAXENT score32 from 7.04 to 2.63, predicting decreased exon inclusion. To test this prediction, we generated a minigene splicing reporter, RBM10-E12M-WT, containing the entire genomic sequence from upstream exon 11 to downstream exon 13, and used site-directed mutagenesis to introduce the point mutation, producing RBM10-E12M-1247A>T (Fig. 3b). We independently transfected HEK293 cells with each of the two minigene reporters and examined exon 12 splicing levels using reverse transcription (RT)-PCR. Strikingly, WT construct showed approximately 71% inclusion of exon 12, estimated from PSI (percent-spliced-in) values of RT-PCR products, the point mutation completely abolished inclusion of exon 12 derived from the mutant construct (Fig. 3c).
RBM10 exon 12 skipping produces a premature termination codon (PTC) positioned more than 55 nt upstream of the last exon-exon junction, and thus is predicted to trigger nonsense-mediated mRNA decay (NMD)33 and reduce RBM10 mRNA expression level. These findings demonstrate that the main effect of the c.1247A>T missense mutation is not to produce a protein with an altered amino acid sequence, but rather significantly reduce RBM10 expression. In fact, RBM10 Q416L (c.1247A>T) overexpressed in HEK293 cells from a plasmid containing its coding sequence localized to the nucleus (Supplementary Fig. S5b) and produced splicing changes in four RBM10 target genes similar to those observed following overexpression of RBM10 WT (Supplementary Fig. S4). These observations suggest that the RBM10 Q416L mutant protein itself has no effect on the splicing of RBM10-target genes. As this example demonstrates, caution is required when annotating coding mutations located close to splice sites.
Pipeline for functional assessment of RBM10 mutations
To systematically assess the effects of RBM10 mutations, in particular missense mutations, we designed a functional assessment pipeline (Fig. 4a). Briefly, we constructed expression plasmids containing wild type (WT) or mutant (MUT) RBM10 proteins with enhanced green fluorescent protein (EGFP) reporters fused to their C-termini. These plasmids were transiently transfected into HEK293 cells to assess the effects of the mutations on RBM10 protein expression, subcellular localization and splicing regulatory functions. In addition, the effects of RBM10 mutations on LUAD cell proliferation and RNA splicing of RBM10 target genes were assessed in LUAD-derived A549 cells.

(a) Functional assessment pipeline. (b) Detection of endogenous RBM10 and exogenous RBM10-EGFP WT and MUTs by western blot analysis using an antibody against RBM10. α-Tubulin: loading control. The full-length blot image is presented in Supplementary Fig. S9b. Results shown are a representative of three biological replicates. (c) Fluorescent images showing subcellular localizations of RBM10-EGFP WT and MUTs. Magnification: 40x. Scale bar: 10 μm. Nuclei were visualized by DAPI staining. (d) Structure of NUMB exon 9 minigene splicing reporter. Arrowheads represent target specific forward primer (NUMB-F) and vector specific reverse primer (BGH-R) used in RT-PCR. (e) Effects of RBM10-EGFP WT and MUTs on splicing of NUMB exon 9. Splice variants were detected by RT-PCR and the inclusion levels of exon 9 were quantified as PSI (percent-spliced-in) values. PSI means ± SEM from 3 independent experiments are plotted below the gel image. GAPDH: internal control. The full-length gel image is presented in Supplementary Fig. S9c. All conditions were compared to EGFP control. ns: not significant, **P ≤ 0.01, ***P ≤ 0.001 (One-way ANOVA followed by Dunnett’s test for multiple comparisons to a single control).
We applied the pipeline to the analysis of six RBM10 mutations located in different conserved protein domains of RBM10 (Fig. 1b) and predicted to be pathogenic by the FATHMM algorithm used in the COSMIC database34,35. The six mutations comprised: a nonsense mutation (E177* (c.529G>T)), a frameshift mutation (F227fs*39 (c.678delC)) and four missense mutations (G153C (c.457G>T), I316F (c.946A>T), S781L (c.2342C>T), G896V (c.2687G>T)).
Following transfection of HEK293 cells with RBM10-EGFP WT, the fusion protein was readily detected by western blot analysis (Fig. 4b) and predominantly localized to the nucleus (Fig. 4c). Overexpression (OE) of RBM10-EGFP WT in HEK293 cells also increased exon skipping in five known RBM10 target genes (Supplementary Fig. S2), in agreement with previous studies26,36,37. These observations confirm that fusing EGFP to the C-terminus of RBM10 does not interfere with its function, and thus demonstrate that RBM10 fusion proteins can be used in functional assays.
RBM10 missense mutations have distinct effects on alternative splicing
Following independent transfection of RBM10-EGFP WT or MUT expression plasmids into HEK293 cells, no fusion protein or fluorescence was detected for the nonsense and frameshift MUTs (Fig. 4b and Supplementary Fig. S5a), consistent with the predicted disruption of protein expression. By contrast, each of the four RBM10 missense fusion proteins had RNA and protein levels comparable to those of the WT fusion protein (Supplementary Fig. S3 and Fig. 4b) and localized primarily to the nucleus like the RBM10 WT protein (Fig. 4c).
Because RBM10 suppresses LUAD cell proliferation, at least partially by promoting exon 9 skipping of NUMB27,28, we performed NUMB minigene splicing reporter assays to explore the effects of the mutations on splicing of RBM10 target genes (Fig. 4d). Compared with the EGFP control, cells transfected with RBM10-EGFP nonsense or frameshift MUT expression plasmids did not show significant changes in splicing of the NUMB minigene (Fig. 4e), consistent with the observed lack of expression of these fusion proteins (Fig. 4b). Intriguingly, OE of each of the four missense MUTs displayed distinct outcomes for NUMB exon 9 splicing. Similar to RBM10 WT, OE of the G153C and G896V mutants produced significantly lower levels of exon 9 inclusion compared to the EGFP control (Fig. 4e, P ≤ 0.01), indicating that these mutations do not affect RBM10-regulated splicing. By contrast, OE of RBM10-S781L produced no significant difference compared to the EGFP control (Fig. 4e), indicating a LOF with regard to splicing. Of note, OE of RBM10-I316F produced significant higher levels of exon inclusion compared to the EGFP control, suggesting a change-of-function (Fig. 4e, P ≤ 0.001).
To further characterize the effects of OE of RBM10 MUTs on splicing, we examined splicing changes of four RBM10 target genes. OE of the above MUTs yielded results consistent with the trend obtained from NUMB minigene assays, except for RBM10-I316F, which produced no observable changes in splicing of the target genes compare to the EGFP control (Supplementary Fig. S4). This discrepancy may result from the low efficiency and heterogeneity of transient transfection assays, which produce mixtures of transfected and non-transfected cells. Together, these observations demonstrate that the two truncation RBM10 mutations produced LOF, while the four missense mutations had differential effects on splicing, despite showing protein expression and subcellular localization patterns resembling RBM10 WT.
RBM10 missense mutations exhibit correlated effects on LUAD cell proliferation and RNA splicing
To further investigate how the RBM10 missense mutations affect LUAD cell proliferation, we used LUAD-derived A549 cells to construct stable tet-on cell lines inducibly expressing RBM10-EGFP WT or one of the three missense MUTs that showed distinct effects on splicing. Upon overexpression in A549 cells, each of the three mutant fusion proteins displayed nuclear localizations similar to RBM10-EGFP WT (Supplementary Fig. S6), consistent with observations in HEK293 cells (Fig. 4c).
Cell counting kit-8 (CCK-8) assays revealed that OE of RBM10-EGFP WT significantly decreased the rate of cell proliferation (Fig. 5a, P ≤ 0.001), while RBM10 depletion had the opposite effect (Supplementary Fig. S7). These results are consistent with previous findings that RBM10 negatively regulates LUAD cell proliferation27,28. Among the three missense MUTs, expression of RBM10-EGFP G896V significantly decreased the rate of LUAD cell proliferation resembling WT (Fig. 5b, P ≤ 0.001), suggesting no alteration of function. Expression of RBM10-EGFP S781L caused no significant changes (Fig. 5c), indicating LOF. By contrast, expression of RBM10-EGFP 316 F significantly increased the rate of LUAD cell proliferation (Fig. 5d, P ≤ 0.001), indicating change-of-function.

(a–d) Growth curves of tet-on A549 cells inducibly overexpressing RBM10-EGFP wild type (a) or three missense mutants (b–d) using CCK-8 assays. Error bars indicate ± SEM, n = 3 biological replicates. ***P ≤ 0.001 (two-way ANOVA followed by Bonferroni correction for multiple testing). Insertions in the graphs show levels of endogenous RBM10 and RBM10-EGFP proteins measured by western blot analysis using an antibody against RBM10. α-Tubulin: loading control. The full-length blot image is presented in Supplementary Fig. S9d. Fusion protein overexpression was achieved by 1 μg/ml doxycycline treatment (see Methods).
Notably, the effects of these RBM10 missense mutations on LUAD cell proliferation correlated with that on the splicing of NUMB exon 9 minigene reporter (Fig. 4e). Additionally, we investigated the effects of overexpressing RBM10-EGFP WT or MUTs on the splicing of four RBM10 target genes in the tet-on A549 cells. We found that OE of RBM10-EGFP G896V enhanced skipping of each of four RBM10-regulated exons similar to RBM10-EGFP WT, OE of RBM10-EGFP S781L did not induce prominent changes in splicing of these exons, and OE of RBM10-EGFP I316F dramatically increased levels of exon inclusion in two of the four target genes: NUMB and SAT1 (Supplementary Fig. S8). These results are in agreement with the trend obtained using the NUMB exon 9 minigene splicing reporter (Fig. 4e). The other two target genes, CREBBP and POLDIP3, did not show dramatic increases in exon inclusion levels upon RBM10-EGFP I316F OE, very likely because their basal inclusion levels were close to 100%, as observed in controls (Supplementary Fig. S8). Collectively, these observations indicate that the suppressive functions of RBM10 in LUAD are, at least in large part, mediated by splicing regulation, and the effects of LUAD-associated RBM10 mutations can be predicted from their effects on the splicing of RBM10 target genes.
Discussion
Large-scale sequencing studies in cancer have generated data concerning genetic mutations, gene expression, and epigenetic alterations for various types of cancer. These data are valuable resources for both basic and clinical cancer investigations6,38. Predicting the consequences of functionally uncertain genetic mutations is often aided by integrative analysis, in particular by integrating information about mutations with gene expression data10,39. As described in our study, the mutational architecture and corresponding patterns of RNA expression revealed by integrative data analysis support a role of RBM10 as a tumor suppressor in LUAD.
In our study, LUAD samples with RBM10 PTVs showed dramatically lower RBM10 expression levels compared to both LUAD samples lacking RBM10 mutations or CNAs and tumor-matched normal tissues, the former of which displayed higher RBM10 expression levels relative to the latter (Fig. 2a). These observations suggest that RBM10 PTVs contributes to LUAD primarily via mutation induced decreases in RNA expression, which often indicates LOF. It should be noted that PTVs can also produce truncated proteins with toxic functions rather than protein LOF40, particularly for PTVs that do not show prominent reductions in RNA expression. For this reason, experiments are often required to determine the effects of PTVs. In addition, our observation that oncogene-negative RBM10 coding variants reduced its mRNA expression (Fig. 3a), thereby potentially producing LOF, provided a compelling reason to investigate the underlying molecular mechanism, as shown in the case of RBM10 c.1247A>T mutation (Fig. 3b,c).
Using our functional assessment pipeline, we successfully demonstrated distinct functional outcomes for four RBM10 missense mutations and confirmed LOF as the functional outcome for one nonsense mutation and one frameshift mutation (Figs 4 and 5). The fact that the true functional consequences of the four missense mutations could not be predicted by the FATHMM algorithm34,35 or by measurements of RNA and protein expression demonstrates the usefulness of our pipeline.
Importantly, the observation that the effects of these missense mutations on LUAD cell proliferation (Fig. 5) correlated with their effects on splicing (Fig. 4e and Supplementary Figs S4 and S8) suggests that functional consequences of RBM10 mutations can be predicted based on splicing of RBM10 target genes. Of note, overexpression of one mutant (I316F) showed an enhanced inclusion of NUMB exon 9 and increased rate of A549 cell proliferation compared with overexpression of wild type RBM10 (Figs 4e and 5 and Supplementary Fig. S8), suggesting a possible contribution to LUAD via change-of-function. Given the high concordance between alterations of splicing in endogenous target genes and the NUMB minigene reporter (Fig. 4e and Supplementary Figs S4 and S8), we propose that examining the effects of RBM10 mutations on the splicing of NUMB exon9 minigene in HEK293 cells by itself may be sufficient for predicting functional consequences of the mutations. Use of our pipeline for the analysis of additional RBM10 mutations in LUAD and other cancers may provide supporting evidence to this proposal.
EGFP fusion proteins are commonly used for evaluating the expression, subcellular localization and other functional aspects of genetic variants because they allow direct and dynamic observations. It should be noted, however, that fusion with EGFP can potentially interfere with protein function. In our study, we confirmed that RBM10-EGFP retained normal functions of RBM10 by assessing subcellular localization and splicing of RBM10 target genes (Fig. 4c and Supplementary Fig. S2). These results support the argument that this fusion protein is appropriate for investigating the properties of RBM10 mutations.
Genetic mutations within or close to core splicing cis-elements (e.g. 5′ and 3′ splice sites) of cancer genes have been found to promote tumorigenesis by disrupting splicing41,42,43,44,45. For mutations within core cis-elements, it is straightforward to predict changes in splicing10,39. However, it is more difficult to predict the effects of mutations located close to core cis-elements46. Again, quantifying exon splicing patterns and gene expression levels using RNA-seq is of great help in elucidating splicing effects10,39. In addition, minigene splicing reporter assays are also useful for determining the effects of mutations on splicing, as shown by our analysis of the oncogene-negative c.1247A>T mutation of RBM10 (Fig. 3b,c) and examples from other studies47,48.
Our results, together with those in previous investigations10,18,27,28, suggest RBM10 and its target genes as attractive candidates for intervention in LUAD. For example, given that RBM10 depletion can activate NOTCH signaling by promoting the formation of the long isoform of NUMB, it is possible that NOTCH inhibitors or specific modulators of NUMB splicing may be helpful for LUAD patients with RBM10 LOF mutations. Because RBM10 regulates the splicing of hundreds of genes26, further studies are required to fully elucidate its functional and regulatory roles in LUAD. These analyses can be expected to provide additional opportunities for clinical translation.
In conclusion, our study revealed that RBM10 mutations contribute to LUAD via genetic mutation and subsequent splicing deregulation. The methods we employed should be useful for the functional characterization of additional RBM10 mutations and mutations in other cancer-associated splicing regulators.
Methods
LUAD data in Public databases
LUAD mutation and copy number alteration (CNA) data were downloaded from the Catalogue of Somatic Mutation in Cancer (COSMIC) website http://cancer.sanger.ac.uk/cosmic)30. LUAD mutation and expression data were obtained from The Cancer Genome Atlas (TCGA) website (http://cancergenome.nih.gov/). Data concerning RBM10 mutations, CNA and its RNA expression levels from these databases were extracted and combined. RNA expression levels in TCGA were estimated from RNA-Seq data using the “RNA-Seq by Expectation Maximization” (RSEM) algorithm and presented as “normalized counts” following upper quartile normalization49,50. RBM10 mRNA expression levels in each group of LUAD samples with different types of RBM10 mutations or CNAs were compared to expression levels in samples lacking RBM10 mutations or CNAs or normal tissues adjacent to tumors, respectively. Survival analysis of LUAD patients was performed using data deposited in the Kaplan Meier plotter online database (http://www.kmplot.com)31. All the parameters were set as “default” in the online tool for lung cancer analysis, except for the following specifications. We used Affy ID: “215089_s_at” as the JetSet probe set (recommended by the online tool as the best probe set), selected “Auto select best cutoff”, and chose Histology: “lung adenocarcinoma.”
Cell culture
Human embryonic kidney (HEK) 293 and HEK293-FT cells were cultured in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). The human lung adenocarcinoma cell line A549 was cultured in F-12K medium supplemented with 10% FBS. All cultures were maintained under standard culture conditions (37 °C, 5% CO2).
Constructs
RBM10-EGFP was generated by amplifying RBM10 from pFRT-TO-RBM1026, followed by double digestion of the PCR Product with NheI and EcoRI (NEB) and ligation into NheI/EcoRI linearized pEGFP-N3 vector (Clontech) using T4 DNA ligase (NEB). The tet-on lentiviral plasmids carrying RBM10-EGFP were constructed by amplifying the target sequence from RBM10-EGFP, cutting the resulting PCR product with NotI/MluI and ligating this to NotI/MluI linearized pLVX-Tight-Puro Vector (Clontech).
Two minigene splicing reporters were constructed. For the RBM10-E12M minigene vector, genomic segments from the RBM10 exon 11 to exon 13 (chrX: 47039611–47040800, GRCh37) were amplified from HEK293 genomic DNA and inserted into the pcDNA3.1(-) expression vector (Invitrogen) using the Clone Express II reagent (Vazyme) following the manufacturer’s protocol. For the NUMB-exon9 minigene, DNA fragments containing NUMB exon 8, intron 8, exon 9, intron 9 and the first 100 bp of exon 10 (chr14: 73743902–73749213, GRCh37) were amplified and inserted into pcDNA3.1(-) using the Gibson Assembly Cloning Kit (NEB) following instructions in the manufacturer’s manual.
Mutagenesis
Mutations were introduced into the RBM10-EGFP plasmid and RBM10-E12M minigene using Q5 Site-Directed Mutagenesis Kit (NEB) or Mut Express II Fast Mutagenesis Kit V2 (Vazyme) following the manufacturer’s instructions.
Plasmids and siRNAs transfection
For plasmids, cells were seeded in 6-well plates at approximately 50% confluency and transfected with 1 μg total plasmid DNA per well using Lipofectamine 3000 reagent (Thermo Scientific). For siRNAs, cells were transfected with Lipofectamine RNAiMax reagent (Thermo Scientific) using the reverse transfection method according to the manufacturer’s instructions. Cells were harvested 24 hours for minigene splicing reporters and 48 hours for RBM10 expression plasmids after transfection. The harvested cells were used for RNA and/or protein extraction.
Total RNA preparation, reverse transcription and PCR (RT-PCR)
Total RNA was extracted from cell lysates in TRIzol (Invitrogen) and treated with DNase I using the TURBO DNA-free kit (Ambion). 1 μg DNase I-treated total RNA from each sample was reverse transcribed using HiScript II Reverse Transcriptase (Vazyme) and oligo dT20 primers. RBM10 mRNA expression level and splicing changes of target genes were assessed by semi-quantitative RT-PCR as described previously26. The RT products from minigene splicing reporters were amplified by gene-specific forward primer and vector specific reverse primer. GAPDH was used as an internal control. All PCR primers were listed in Supplementary Table S2. PCR products were mixed with loading buffer containing GelRed fluorescent DNA stains (GENEray), resolved in 1.5 or 2% agarose gels and visualized using a UV image system. The signal intensity of each band was quantified using Image J software (https://imagej.nih.gov/ij/). The percent-spliced-in (PSI) value was defined as: PSI = (intensity of the upper band)/[intensity of the upper band + (LL/LS) × (intensity of the lower band) × 100%, where LL and LS represent the lengths of the long and short PCR products that include or exclude the alternative exon, respectively.
Western blotting
Western blot analysis was performed as previously described26. Briefly, cells were lysed in RIPA buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 0.1% SDS and 0.1% sodium deoxycholate) followed by brief sonication. Lysates were cleared by centrifugation and the supernatant fractions with added loading buffer were boiled at 95 °C for 5 min. Denatured protein lysates were resolved by SDS-PAGE and transferred to a PVDF membrane (Millipore). The membranes were blocked with 5% milk in TBST (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Tween 20) and sequentially probed with primary antibodies and horseradish peroxidase (HRP)-conjugated secondary antibodies. Bands were detected with an enhanced chemiluminescence (ECL) system (Millipore) and analyzed using the ChemiDocTM XRS+ image system (BioRad). The following antibodies were used: rabbit anti-RBM10 (cat. No. HPA034972, 1:10000, Sigma), rabbit anti-α-tubulin (cat. No. 11224-1-AP, 1:5000, proteintech), Goat anti-rabbit IgG-HRP (cat. No. 458, 1:5000, MBL).
Fluorescence imaging
Cells grown on coverslips were fixed in 4% PFA and stained with 1 μg/ml DAPI (Invitrogen). Fluorescent images were acquired using a Leica DM IL LED fluorescent microscope with a 40x objective and DFC450 C camera (Leica) and processed using Image J software (https://imagej.nih.gov/ij/).
Generation of tet-on RBM10-EGFP A549 cell lines
Inducible overexpression of RBM10-EGFP wild type (WT) or mutants (MUTs) in A549 cells was achieved using the Lenti-X Tet-On Advanced Inducible Expression System (Clontech) according to the manufacturer’s manual. Briefly, lentiviral plasmids (pLVX-tight-puro) carrying RBM10-EGFP WT or MUTs or TetR (pLVX-Tet-On) were transfected into HEK293-FT cells together with three packaging plasmids (pLP1, pLP2, and pLP/VSVG, Invitrogen). Medium containing the lentiviral particles was harvested 48 or 72 hrs after transfection.
Lentiviral particles carrying TetR was added into the A549 cell cultures and selected by 200 μg/ml G418 (Invitrogen). The G418 selected cells were transfected with lentiviruses carrying RBM10 WT or MUTs followed by selection in medium containing 2 μg/ml puromycin (Sigma). Stably transfected A549 cells were maintained under standard conditions and overexpression of RBM10-EGFP proteins were induced by 1 μg/ml doxycycline treatment (Sigma) for 2 days unless otherwise stated.
Cell proliferation assays
Tet-on RBM10-EGFP A549 cells were seeded in 96-well plates (1000 or 1500 cells/well) and treated with 1 μg/ml doxcycline to induce the overexpression of WT or MUTs. Cells not exposed to doxcycline were used as controls. Numbers of viable cell were measured each day for 5 days using Cell Counting Kit-8 (CCK-8) following the manufacturer’s manual (Dojindo). For each measurement, cells were incubated with CCK-8 reagents for 1 hour at 37 °C and the optical absorption at 450 nm was measured using a spectrophotometer (MULTISKAN MK3, Thermo Scientific). For each time point and condition, we included 4 wells of cells seeded from the same set of cultured cells as technical replicates. CCK-8 assays were carried out three times using three independently prepared sets of cells as biological replicates. The number of viable cells is proportional to the absorption values.
Statistical analysis
Statistical analyses were carried out using tests when the data satisfied the assumptions of the tests. For comparisons of RBM10 mRNA levels in LUAD samples, P values were calculated by Mann-Whitney tests followed by Benjamini & Hochberg corrections for multiple comparisons using the open source statistical programming environment ‘R’. For experimental data, statistical tests were carried out using GraphPad Prism 6 (GraphPad software). For comparisons of sample pairs, two-tailed Student’s t tests were used. For experiments with one variable and multiple conditions, one-way ANOVA followed by Dunnett’s multiple comparisons test was used. For experiments with two independent variables, two-way ANOVA followed by Bonferroni correction for multiple testing was used.
Primer and oligonucleotide
All primer and oligonucleotide sequences are listed in Supplementary Table S2.
Additional Information
How to cite this article: Zhao, J. et al. Functional analysis reveals that RBM10 mutations contribute to lung adenocarcinoma pathogenesis by deregulating splicing. Sci. Rep. 7, 40488; doi: 10.1038/srep40488 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Pon, J. R. & Marra, M. A. Driver and Passenger Mutations in Cancer. Annual Review of Pathology: Mechanisms of Disease 10, 25–50, doi: 10.1146/annurev-pathol-012414-040312 (2015).
Vogelstein, B. et al. Cancer Genome Landscapes. Science 339, 1546–1558, doi: 10.1126/science.1235122 (2013).
Martincorena, I. & Campbell, P. J. Somatic mutation in cancer and normal cells. Science 349, 1483–1489, doi: 10.1126/science.aab4082 (2015).
Gonzalez-Perez, A. et al. Computational approaches to identify functional genetic variants in cancer genomes. Nature methods 10, 723–729, doi: 10.1038/nmeth.2562 (2013).
Lawrence, M. S. et al. Mutational heterogeneity in cancer and the search for new cancer genes. Nature 499, 214–218, doi: 10.1038/nature12213 (2013).
Roychowdhury, S. & Chinnaiyan, A. M. Translating Genomics for Precision Cancer Medicine. Annual Review of Genomics and Human Genetics 15, 395–415, doi: 10.1146/annurev-genom-090413-025552 (2014).
Polyak, K. & Metzger Filho, O. SnapShot: Breast Cancer. Cancer Cell 22, 562–562.e1, doi: 10.1016/j.ccr.2012.06.021 (2012).
Chen, Z., Fillmore, C. M., Hammerman, P. S., Kim, C. F. & Wong, K.-K. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer 14, 535–546, doi: 10.1038/nrc3775 (2014).
Heist, R. S. & Engelman, J. A. SnapShot: Non-Small Cell Lung Cancer. Cancer Cell 21, 448–448.e2, doi: 10.1016/j.ccr.2012.03.007 (2012).
The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550, doi: 10.1038/nature13385 (2014).
Guan, B., Gao, M., Wu, C.-H., Wang, T.-L. & Shih, I.-M. Functional Analysis of In-frame Indel ARID1A Mutations Reveals New Regulatory Mechanisms of Its Tumor Suppressor Functions. Neoplasia 14, 986–993 (2012).
Greulich, H. et al. Functional analysis of receptor tyrosine kinase mutations in lung cancer identifies oncogenic extracellular domain mutations of ERBB2. Proceedings of the National Academy of Sciences 109, 14476–14481, doi: 10.1073/pnas.1203201109 (2012).
Parikh, C. et al. Disruption of PH–kinase domain interactions leads to oncogenic activation of AKT in human cancers. Proceedings of the National Academy of Sciences 109, 19368–19373, doi: 10.1073/pnas.1204384109 (2012).
Yi, K. H., Axtmayer, J., Gustin, J. P., Rajpurohit, A. & Lauring, J. Functional analysis of non-hotspot AKT1 mutants found in human breast cancers identifies novel driver mutations: implications for personalized medicine. Oncotarget 4, 29–34 (2013).
Kim, E. et al. Systematic Functional Interrogation of Rare Cancer Variants Identifies Oncogenic Alleles. Cancer Discovery 6, 714–726, doi: 10.1158/2159-8290.cd-16-0160 (2016).
Martelotto, L. G. et al. Benchmarking mutation effect prediction algorithms using functionally validated cancer-related missense mutations. Genome Biology 15, 1–20, doi: 10.1186/s13059-014-0484-1 (2014).
Dogruluk, T. et al. Identification of Variant-Specific Functions of PIK3CA by Rapid Phenotyping of Rare Mutations. Cancer Research 75, 5341–5354, doi: 10.1158/0008-5472.can-15-1654 (2015).
Imielinski, M. et al. Mapping the Hallmarks of Lung Adenocarcinoma with Massively Parallel Sequencing. Cell 150, 1107–1120 (2012).
Johnston, J. J. et al. Expansion of the TARP Syndrome Phenotype Associated with De Novo Mutations and Mosaicism. American journal of medical genetics. Part A 0, 120–128, doi: 10.1002/ajmg.a.36212 (2014).
Gripp, K. W. et al. Long Term Survival in TARP Syndrome and Confirmation of RBM10 as the Disease Causing Gene. American journal of medical genetics. Part A 155, 2516–2520, doi: 10.1002/ajmg.a.34190 (2011).
Johnston, J. J. et al. Massively Parallel Sequencing of Exons on the X Chromosome Identifies RBM10 as the Gene that Causes a Syndromic Form of Cleft Palate. The American Journal of Human Genetics 86, 743–748, doi: 10.1016/j.ajhg.2010.04.007 (2010).
Giannakis, M. et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Reports 15, 857–865, doi: 10.1016/j.celrep.2016.03.075 (2016).
Witkiewicz, A. K. et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 6, doi: 10.1038/ncomms7744 (2015).
Furukawa, T. et al. Whole-exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Sci. Rep. 1, 161, doi: 10.1038/srep00161 (2011).
Yuan, Y. et al. Comprehensive Characterization of Molecular Differences in Cancer between Male and Female Patients. Cancer Cell 29, 711–722, doi: 10.1016/j.ccell.2016.04.001 (2016).
Wang, Y. et al. Integrative analysis revealed the molecular mechanism underlying RBM10-mediated splicing regulation. EMBO Molecular Medicine 5, 1431–1442, doi: 10.1002/emmm.201302663 (2013).
Bechara, Elias G., Sebestyén, E., Bernardis, I., Eyras, E. & Valcárcel, J. RBM5, 6, and 10 Differentially Regulate NUMB Alternative Splicing to Control Cancer Cell Proliferation. Molecular Cell 52, 720–733, doi: 10.1016/j.molcel.2013.11.010 (2013).
Hernández, J. et al. Tumor suppressor properties of the splicing regulatory factor RBM10 . RNA Biology 13, 466–472, doi: 10.1080/15476286.2016.1144004 (2016).
Wang, K. et al. RBM10 Modulates Apoptosis and Influences TNF-α Gene Expression. Journal of Cell Death 5, 1–19, doi: 10.4137/JCD.S9073 (2012).
Forbes, S. A. et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Research 39, D945–D950, doi: 10.1093/nar/gkq929 (2011).
Szász, A. M. et al. Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget 7, 49322–49333, doi: 10.18632/oncotarget.10337 (2016).
Yeo, G. & Burge, C. B. Maximum Entropy Modeling of Short Sequence Motifs with Applications to RNA Splicing Signals. Journal of Computational Biology 11, 377–394, doi: 10.1089/1066527041410418 (2004).
Lykke-Andersen, S. & Jensen, T. H. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol 16, 665–677, doi: 10.1038/nrm4063 (2015).
Shihab, H. A., Gough, J., Cooper, D. N., Day, I. N. M. & Gaunt, T. R. Predicting the functional consequences of cancer-associated amino acid substitutions. Bioinformatics 29, 1504–1510, doi: 10.1093/bioinformatics/btt182 (2013).
Shihab, H. A. et al. Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions using Hidden Markov Models. Human Mutation 34, 57–65, doi: 10.1002/humu.22225 (2013).
Xiao, S. J. et al. S1‐1/RBM10: Multiplicity and cooperativity of nuclear localisation domains. Biology of the Cell 105, 162–174, doi: 10.1111/boc.201200068 (2013).
Inoue, A. et al. S1‐1 nuclear domains: characterization and dynamics as a function of transcriptional activity. Biology of the Cell 100, 523–539, doi: 10.1042/BC20070142 (2008).
Forbes, S. A. et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Research 43, D805–D811, doi: 10.1093/nar/gku1075 (2015).
Seo, J. S. et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome research 22, 2109–2119, doi: 10.1101/gr.145144.112 (2012).
Scekic-Zahirovic, J. et al. Toxic gain of function from mutant FUS protein is crucial to trigger cell autonomous motor neuron loss. The EMBO Journal 35, 1077–1097, doi: 10.15252/embj.201592559 (2016).
Sveen, A., Kilpinen, S., Ruusulehto, A., Lothe, R. A. & Skotheim, R. I. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 35, 2413–2427, doi: 10.1038/onc.2015.318 (2015).
Chen, J. & Weiss, W. A. Alternative splicing in cancer: implications for biology and therapy. Oncogene 34, 1–14, doi: 10.1038/onc.2013.570 (2015).
Oltean, S. & Bates, D. O. Hallmarks of alternative splicing in cancer. Oncogene 33, 5311–5318, doi: 10.1038/onc.2013.533 (2014).
Zhang, J. & Manley, J. L. Misregulation of Pre-mRNA Alternative Splicing in Cancer. Cancer Discovery 3, 1228–1237, doi: 10.1158/2159-8290.cd-13-0253 (2013).
Wojtuszkiewicz, A. et al. Pre-mRNA splicing in cancer: the relevance in oncogenesis, treatment and drug resistance. Expert opinion on drug metabolism & toxicology 11, 673–689, doi: 10.1517/17425255.2015.993316 (2015).
Supek, F., Miñana, B., Valcárcel, J., Gabaldón, T. & Lehner, B. Synonymous Mutations Frequently Act as Driver Mutations in Human Cancers. Cell 156, 1324–1335, doi: 10.1016/j.cell.2014.01.051 (2014).
Liu, C. et al. The UPF1 RNA surveillance gene is commonly mutated in pancreatic adenosquamous carcinoma. Nat Med 20, 596–598, doi: 10.1038/nm.3548 (2014).
Sebestyén, E. et al. Large-scale analysis of genome and transcriptome alterations in multiple tumors unveils novel cancer-relevant splicing networks. Genome research 26, 732–744, doi: 10.1101/gr.199935.115 (2016).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 1–16, doi: 10.1186/1471-2105-12-323 (2011).
Bullard, J. H., Purdom, E., Hansen, K. D. & Dudoit, S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics 11, 1–13, doi: 10.1186/1471-2105-11-94 (2010).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31371299 to Y.W.). We thank Dr. Ning Sun in School of Basic Medical Sciences, Fudan University for providing the lentiviral packaging plasmids.
Author information
Authors and Affiliations
Contributions
Y.W., W.L. and Z.S. planned the experiments. J.Z. (Jiawei Zhao) and Y.S. performed the experiments, interpreted the data and prepared the figures under the supervision of Y.W., W.L. and J.Z. (Ji Zuo). Y.H. and F.S. performed the genomic data analysis under the supervision of Z.S. and D.S. Z.H. and Y.B. assisted with the experiments. Y.W. wrote the manuscript, with contributions from Y.S. and D.S. D.S. edited the final draft. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhao, J., Sun, Y., Huang, Y. et al. Functional analysis reveals that RBM10 mutations contribute to lung adenocarcinoma pathogenesis by deregulating splicing. Sci Rep 7, 40488 (2017). https://doi.org/10.1038/srep40488
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep40488
This article is cited by
-
RBM10 C761Y mutation induced oncogenic ASPM isoforms and regulated β-catenin signaling in cholangiocarcinoma
Journal of Experimental & Clinical Cancer Research (2024)
-
Alternative splicing and related RNA binding proteins in human health and disease
Signal Transduction and Targeted Therapy (2024)
-
Functional insight into a neurodevelopmental disorder caused by missense variants in an RNA-binding protein, RBM10
Journal of Human Genetics (2023)
-
Frequently mutated genes in predicting the relapse of stage I lung adenocarcinoma
Clinical and Translational Oncology (2023)
-
RNA-binding proteins in human genetic disease
Nature Reviews Genetics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
