
Abstract
Cetacean body structure and physiology exhibit dramatic adaptations to their aquatic environment. Fibroblast growth factors (FGFs) are a family of essential factors that regulate animal development and physiology; however, their role in cetacean evolution is not clearly understood. Here, we sequenced the fin whale genome and analysed FGFs from 8 cetaceans. FGF22, a hair follicle-enriched gene, exhibited pseudogenization, indicating that the function of this gene is no longer necessary in cetaceans that have lost most of their body hair. An evolutionary analysis revealed signatures of positive selection for FGF3 and FGF11, genes related to ear and tooth development and hypoxia, respectively. We found a D203G substitution in cetacean FGF9, which was predicted to affect FGF9 homodimerization, suggesting that this gene plays a role in the acquisition of rigid flippers for efficient manoeuvring. Cetaceans utilize low bone density as a buoyancy control mechanism, but the underlying genes are not known. We found that the expression of FGF23, a gene associated with reduced bone density, is greatly increased in the cetacean liver under hypoxic conditions, thus implicating FGF23 in low bone density in cetaceans. Altogether, our results provide novel insights into the roles of FGFs in cetacean adaptation to the aquatic environment.
Similar content being viewed by others


Introduction
Cetaceans (baleen and toothed whales) were derived from extinct, semi-aquatic, deer-like, even-toed ungulates (artiodactyls) approximately 50 million years ago1 and have successfully re-populated from terrestrial to aquatic environments. After becoming fully aquatic, the Mysticeti (baleen whales) diverged from the Odontoceti (toothed whale) following the development of keratinous sieves that enabled filter-feeding prior to the onset of the Oligocene Epoch, and subsequently lost teeth completely2,3.
The anatomical structures, physiology, and metabolism of cetaceans have changed due to various challenges associated with aquatic life. The body shape has been modified to a streamlined form that could reduce fluid resistance4. Flukes were developed on their tail for propulsion, hindlimbs were degenerated, and forelimbs were modified into diverse forms of flippers with fused elbow joints that were more suitable for steering than paddling5. The hairy fur of their close terrestrial relatives was essentially lost in cetaceans for hydrodynamic reasons4, and the bone mineral density was reduced to allow dynamic buoyancy control in deep water6. The outer ear pinnae were lost in cetaceans, and the outer ears were functionally replaced by the mandible and the mandibular fat pad, which were better adapted for hearing underwater4,7. Cetaceans also exhibit various specializations, such as increased oxygen storage capacity, cardiovascular and metabolic adjustments, and increased levels of antioxidants to adapt to oxygen-limited conditions during diving8. Despite a thorough understanding of cetacean morphological and physiological alterations, the genetic background underlying these adaptations has only begun to be explored9,10,11.
Fibroblast growth factors (FGFs) are a family of highly conserved growth factors with 22 members in mammals (FGF1 to FGF23; FGF15 and FGF19 are the mouse and human orthologs, respectively). FGFs are further divided into the following three groups: the canonical FGFs, hormone-like FGFs (hFGFs), and intracellular FGFs (iFGFs)12. Canonical FGFs function as paracrine factors due to their high affinity for heparin/heparan sulphate proteoglycans (HSPGs) and play essential roles in cell growth and organ development. Members of the hFGF subfamily (FGF19, FGF21, and FGF23) have reduced affinity for HSPGs and function as endocrine factors. The iFGFs (FGF11, FGF12, FGF13, and FGF14) function intracellularly and are independent of FGF receptors12.
FGFs have important roles in the development of internal organs; the formation of the limbs, ears, and teeth during embryonic development; and the regulation of bone density, energy homeostasis, and hair growth in adult life13,14. The diverse functions of FGFs suggest that the evolution of FGF genes in cetaceans may be associated with aquatic adaptations. For example, FGF3 is implicated in ear and tooth development in humans and mice14,15. FGF10 and FGF20 have essential roles in lung and kidney development, respectively14,16. Various FGFs, including FGF5, FGF7, FGF10, FGF18, and FGF22, are expressed at high levels in hair follicles and regulate hair growth13. Mutations causing the fusion of the elbow and knee joints were discovered in human and murine FGF917,18. Furthermore, the emerging roles of FGF19 and FGF21 in the regulation of energy homeostasis and thermogenesis suggest that these factors may be implicated in metabolic modifications in cetaceans12,13. In addition, the increased activity or the overexpression of FGF23 is associated with decreased bone mineral density, as shown in autosomal dominant hypophosphatemic rickets and tumour-induced osteomalacia in humans19. Finally, FGF11, the enigmatic iFGF, was shown to be up-regulated during hypoxia in human endothelial cells and promote capillary-like tube formation when overexpressed20. This finding further triggered interest in the involvement of iFGFs in the acquisition of hypoxia tolerance during cetacean evolution.
Although FGF5, a canonical FGF with a suppressive function in hair growth, was shown to be positively selected in cetaceans21, the general involvement of the FGF family in the acquisition of cetacean-specific traits is largely unknown. Given the various roles of FGFs in mammalian development and physiology, we hypothesized that molecular adaptations of FGF may be associated with the evolution of the cetaceans. Here, we identified coding sequences of all of the 22 mammalian FGF genes from eight representative cetaceans and analysed the molecular adaptation of FGFs in the course of cetacean evolution.
Results
Identification of FGF genes
We sequenced the fin whale (Balaenoptera physalus) genome to a 60x average depth of coverage. We also identified or assembled the coding sequences of all 22 mammalian FGF genes (FGF1 to FGF23) from eight representative cetacean species, including three baleen whales (minke whale (Balaenoptera acutorostrata), bowhead whale (Balaena mysticetus), and fin whale) and five toothed whales (sperm whale (Physeter macrocephalus), killer whale (Orcinus orca), bottlenose dolphin (Tursiops truncatus), finless porpoise (Neophocaena phocaenoides), and baiji (Lipotes vexillifer, also called the Yangtze river dolphin)), using the newly sequenced fin whale genome, recently published genomes from our labs (minke whale, bowhead whale, and finless porpoise)22,23 or public databases (killer whale, baiji, sperm whale, and bottlenose dolphin)24,25.
To confirm the homology of each FGF family, we constructed a maximum likelihood tree with human and mouse FGFs as references. Each of the FGF subfamilies was clustered into a monophyletic group with high bootstrap values (Supplementary Fig. S1). All cetacean FGFs showed the typical features of FGF, including heparin-binding sites, receptor interaction sites, and localization signals, with the exception of FGF22 (Fig. 1). No nonsense or frameshift mutations were detected in cetacean FGFs, except for FGF3 and FGF22. In cetacean FGF3, a gene with essential roles in ear and tooth development, we found a nonsense mutation that removed a conserved and positively charged Arg-rich motif at the C-terminus (Supplementary Fig. S2). Despite the C-terminal deletion, FGF3 seemed to be functional since no deleterious mutations that disrupt the FGF domain were found in cetaceans, and all of the residues involved in receptor binding or heparin binding were strictly conserved.

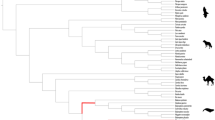
(a) The species tree of mammals was drawn based on previous studies53,54,55,56 to analyse FGF22. Red lines represent lineages with pseudogenized FGF22. (b) Summary of inactivating mutations in FGF22. Nonsense mutations are marked with X in red, and stop codons due to frameshift mutations are marked with X in blue. Indel mutations are expressed with + and −, and exons with frameshift are expressed with red boxes. Animal images were drawn by Oksung Chung and Hana Byun.
Unlike FGF3, independent nonsense or frameshift mutations that destroyed the highly conserved FGF domain were found in FGF22, a gene related with hair follicles, from all cetaceans examined. This finding implies that FGF22 was pseudogenized after the divergence of the toothed and baleen whales (Fig. 1 and Supplementary Figs S3 and S4). The sequences in the vicinity of FGF22 showed a conserved synteny among human, mouse, cow and all of the studied whale species. FGF22 was found between POLRMT and RNF126 without large insertions or deletions in this region (Fig. 1b). FGF22 paralogs were not identified in any other regions. These results exclude the possibility of misassembly due to a lack of callable positions near FGF22 in cetaceans or uncertainty due to the existence of paralogs leading to the misidentification of mutations on FGF22. Furthermore, the lack of FGF22 paralogs indicates that there have been no FGF22 duplication events in the cetacean lineage that could complement the loss of FGF22. Interestingly, we also noticed a loss of FGF22 in African elephant as judged by a complete loss of exon 1 and one nucleotide insertion in exon 2 (Fig. 1 and Supplementary Figs S3 and S4). No deleterious mutations in FGF22 were found in other terrestrial mammals examined here.
Adaptive evolution of FGF genes
The strength of the selective pressure acting on FGF genes was analysed using the nonsynonymous to synonymous substitution rate ratio (dN/dS, also termed ω) based on the species tree shown in Fig. 2a 26. First, we contrasted the strength of selection between whales and other mammals using the branch model27. The average dN/dS ratios of the whale lineages and those of the remaining lineages were calculated under a two ratio model that allows different dN/dS ratios between whale branches and the other branches. The mean dN/dS ratios ranged from 0.0001 (FGF8, FGF12, and FGF13) to 0.6242 (FGF11) in whales and 0.0066 (FGF13) to 0.1855 (FGF21) in the other mammalian species we investigated (Supplementary Table S1). The dN/dS ratio was significantly correlated between whales and other mammals (spearman’s rho = 0.61, p-value = 0.003081). We also performed a likelihood ratio test if the dN/dS ratio was significantly different between whale branches and the other branches for each of FGF genes. None of the FGF genes had a significantly different dN/dS ratio between whales and other mammals, with two exceptions. FGF3 and FGF11 had a significantly higher dN/dS ratio in whales (adjusted p-value = 0.00031 and <2.2 × 10−16, respectively) than in other mammals (Fig. 2b and Supplementary Table S1). This result implies that strong purifying selection has acted on FGFs to a comparable extent between whales and other mammals, except for at FGF3 and FGF11; these genes have experienced relaxed purifying selection or substantial positive selection in the whale lineage.

To test the presence of positive selection in the whale lineage, we used branch site model28. With this model, the phylogenetic tree shown in Fig. 2a was divided into foreground branches, which were composed of ancestral and terminal whale branches and in which the proportion of codons with dN/dS > 1 was estimated, and background branches, which were composed of the other branches and in which no codon was allowed to have dN/dS > 1. We found that no FGF genes showed a significant signature of positive selection in the whale lineage (Likelihood ratio test, Supplementary Table S2). One assumption of the branch site model is that positive selection has not occurred in the non-whale mammals, and there is no reason to believe that this assumption is supported a priori. Thus, we used an alternative approach. To infer signatures of positive selection in whales, we analysed only the whale branches by reconstructing ancestral whale sequences using a maximum likelihood approach and by generating multiple sequence alignments composed of both the inferred ancestral whale sequences and the extant whale sequences. Then, we used the site model29 to test for positive selection (Table 1). For FGF3, the proportion of positively selected codons was greater than zero (corresponding to p2 at M2 in Table 1, 0.644%; raw p-value = 0.0026, and adjusted p-value = 0.055). This result implies that FGF3 may be under positive selection.
To detect possible positive selection that has occurred along whale branches with a resolution of a single amino acid, we used the Mixed Effects Model of Evolution (MEME)30 based on the phylogenetic tree and multiple sequence alignments of extant whales and the inferred ancestral sequences. The MEME estimates the rates of non-synonymous substitution and synonymous substitution for each codon. The difference between these two ratios was tested using a likelihood ratio test and any branch under positive selection was evaluated with an empirical Bayes factor. Of all the FGF genes, only FGF3 and FGF11 showed a significant signal of positive selection (Supplementary Figs S5 and S6). In FGF3, I85L was shown to be under positive selection (p = 0.0235), with the strongest signal observed in minke whale. For FGF11, a significant positive selection was observed at S234T (p = 0.0179), with the strongest signal in baiji. Marginal significance was also observed at 237 of FGF11 (p = 0.0728). This result suggests that positive selection has been acting on FGF3 and FGF11, genes related to ear and tooth development and hypoxia, respectively.
Changes in FGF genes and the emergence of whales
We investigated how phenotypes and physiological aspects of extant whales were generated after diverging from a sister taxa of whales. Among all FGF genes investigated, we identified 13 amino acid replacements in eight FGF genes that occurred after diverging from a sister taxa of whales but prior to the divergence between Mysticeti and Odontoceti (Table 2). The PolyPhen-231 analysis showed that three replacements were predicted to have a drastic function-altering effect, expressed as probably or possibly damaging, and ten replacements were predicted to be neutral. The rate of function-altering substitutions was 162% greater along the ancestral branches of whales than along the other branches (Table 3). However, the neutral substitution rate differed by only 3.8%. We also used two other algorithms, SIFT32 and PROVEAN33, to predict whether the observed mutations had a drastic function-altering effect. The results from these two algorithms showed the same pattern as that from PolyPhen-2 (Table 3). If we assume that the neutral mutations may actually have had slightly deleterious effects, this comparable neutral substitution rate suggests that the strength of purifying selection was largely the same between lineages of ancestral whales and the rest of the mammalian lineage. Thus, higher rates of function-altering substitutions in ancestral whales were likely to be due to stronger positive selection on FGF genes.
The replacement of D203G in FGF9 was predicted to have a damaging effect by all algorithms used (Table 2). Gly203 resides in the short C-terminal helix that is composed of residues 200–203 (Fig. 3a). According to the crystal structure of dimeric human FGF934, the negatively charged side chain of Asp203 forms a salt bridge with the guanidinium group of Arg63 (Fig. 3b,c). The favourable ionic interaction that is mediated by Asp203 cannot occur in the cetacean FGF9 harbouring the D203G substitution. Given that the stability of helices is affected by the side chain-mediated interactions35, the D203G substitution is highly likely to destabilize the short C-terminal helix. Leu200 in the helix and Ile204, the adjacent residue, form a hydrophobic core at the dimeric interface. The helical conformation of residues 200–203 is important to correctly position Leu200 and Ile204 at the dimeric interface. Therefore, the cetacean FGF9 with the D203G substitution might be defective in homodimerization.

(a) Schematic drawing of mammalian FGF9. Cetacean-specific substitutions are marked in red, and residues involved in the homodimerization of FGF9 are marked in green. The C-terminal α helix is underlined, and the salt bridge between Arg63 and Asp203 is shown as a red broken line. Bipartite signal sequences and homodimerization domains are marked with purple and blue boxes. HD, homodimerization domain. (b,c) Ribbon drawing of the dimeric human FGF9 with each monomer in different colours. Side (b) and top (c) views. Residues involved in homodimerization (Leu57, Leu61, Leu200 and Ile204) and stabilization of the C-terminal α helix (Arg63 and Asp203) are shown as sticks. The black dotted line indicates the salt bridge between Arg63 and Asp203. The C-terminal α helix and Ile204 are shown in yellow, and the transparent orange oval represents the hydrophobic core at the dimeric interface. αN, N-terminal α helix; αC, C-terminal α helix. *Function-altering change predicted by PolyPhen-2, SIFT, and PROVEAN.
Induction of FGF11 in the brain and heart of the Cetacea during hypoxia
To understand the role of FGF11 in the evolution of the cetaceans, we examined the expression of FGF11 in various tissues using RNA-seq data from a minke whale and five bowhead whales22,23,36. The expression levels of iFGFs were higher in the brain than in other organs (Supplementary Tables S3 and S4), consistent with results from mouse37. Interestingly, the expression of FGF11 was markedly increased in the brain and heart of the minke whale (Fig. 4a and Supplementary Table S3), but was not detected in the bowhead whales (Supplementary Table S4). The minke whale sample was obtained as bycatch, with drowning being the main cause of mammalian death from bycatch, while the bowhead whales were caught by hunting23,36. Thus, the differential levels of FGF11 between minke and bowhead whales suggest that FGF11 was induced by hypoxia in the minke whale. To examine this possibility, we searched for the presence of Hypoxia-Inducible Factor-1 binding site, the master regulator of hypoxia response, within FGF11 promoters from representative mammals. A consensus hypoxia response element (HRE)38, ACGTG, was identified as being conserved in all the mammals examined, thus implying that FGF11 could be a direct target of Hypoxia-Inducible Factor-1 (Fig. 4b,c).

(a) Increased levels of FGF11 transcripts in the brain and heart of a minke whale revealed by RNA-seq analysis. (b,c) A conserved HRE in FGF11 promoter marked with red line (b) and highlighted in yellow (c). (d) Induction of FGF11 upon hypoxia. SH-SY5Y cells were incubated with normoxia or hypoxia (1% O2) for 18 h, and RNA levels were determined using quantitative RT-PCR (n = 4, mean ± SEM, *p < 0.05).
To determine if FGF11 is induced by hypoxia in nervous tissues, we analysed gene expression profiles in SH-SY5Y human neuroblastoma cells. Under hypoxic conditions, the expression of FGF11 was significantly increased, along with Vascular Endothelial Growth Factor and Glucose Transporter 1, well-known markers for hypoxia (Fig. 4d), suggesting that the increased expression of FGF11 in the minke whale reflects the hypoxic status of the animal. In contrast to FGF11, expression of FGF14 was decreased and expression of FGF13 and FGF17 were unchanged by hypoxia, implying that the induction of FGF11 during hypoxia is not a general feature that is shared with other FGFs. Taken together, our results provide evidence that FGF11 is induced under hypoxic conditions in the brain and the heart of cetaceans, the most sensitive tissues to hypoxic damages.
Induction of FGF23 in the liver during hypoxia and the acquisition of HREs in the promoter of cetacean FGF23
FGF23 is one of the main regulators of bone mineral density and is mainly produced in bone tissue19. Unexpectedly, we found a dramatic increase of FGF23 in the liver of minke whale23 (Fig. 5a and Supplementary Table S3) and two bowhead whales (Fig. 5b and Supplementary Table S4)22,36, with a greater increase in the minke whale. Considering that FGF23 is not expressed in normal liver from other mammals19, we investigated if the expression of FGF23 in cetacean liver was the result of adaption to unique environmental conditions. As the minke whale was obtained as bycatch, whereas the bowhead whales were obtained through hunting, the differential gene expression of FGF23 suggests differences in hypoxic status. In human HepG2 hepatoma cells, we observed that FGF23 was expressed under hypoxia (Fig. 5c), implying that hypoxia can induce the expression of FGF23 in the liver in mammals.

(a,b) RNA-seq analysis showed high levels of FGF23 transcripts in the livers from a minke whale (a) and two bowhead whales (b). (c) Induction of FGF23 upon hypoxia. HepG2 cells were incubated in normoxia or 1% O2 for 18 h, and mRNA levels for FGF23 and VEGF were investigated using quantitative RT-PCR (n = 3, mean ± SEM, *p < 0.05). (d) HREs conserved in at least three species are shown. A conserved HRE across all the mammalian species is shown about −13 kb region in the FGF23 promoter (red line). HREs specific to the Primates and the Cetacea are marked with green and blue lines, respectively. The number of the specific HREs are shown in parentheses. (e) Multiple nucleotide sequence alignment of the promoter region encompassing the conserved HRE is highlighted in yellow.
In the promoter regions, we identified a conserved HRE approximately 13 kb upstream of FGF23 in all the mammals examined (Fig. 5d,e). Interestingly, the number of cetacean specific HREs (twelve) was much higher than that of primates (two) or Artiodactyla (zero) (Fig. 5d). The increased number of HREs in cetaceans suggests the possibility that cetaceans have evolved to produce FGF23 more efficiently under hypoxic conditions during diving.
Although it is well known that cetaceans experienced dramatic bone density reductions during their habitat transition from shallow to deep water resulting in dynamic buoyancy control6, the underlying mechanism is not known. Because the overexpression of FGF23 decreases bone density19, the acquisition of cetacean-specific HREs may have contributed to the development of the extremely low bone density in cetaceans by facilitating FGF23 expression in the liver.
Discussion
Here, we studied the molecular adaptation of FGF genes in the course of cetacean evolution through evolutionary analysis, homology modelling, and expression analysis. We found significant signatures of positive selection in FGF3 and FGF11, a potentially function-altering mutation in FGF9 in ancestral whales, acquisition of cetacean-specific HREs in the promoter region of FGF23, and functional loss of FGF22 in all cetaceans. These evolutionary events appear to be closely correlated with the advent of morphology and physiology of extant cetaceans.
We observed the functional loss of FGF22 in all cetaceans and African elephant (Fig. 1). Each of the FGF subfamilies was derived from an FGF13-like ancestral gene through gene duplications prior to the evolution of vertebrates14, and each subfamily further expanded to multiple members through two rounds of whole genome duplication during the evolution of early vertebrates14,39. The 22 members of the mammalian FGF family were established after the loss of FGF24 in the ancestry of tetrapods39. Although the loss of FGF11, FGF17, and FGF21 were identified in some avian lineages, inactivation of FGF22 has not thus far been reported in other tetrapods39.
FGF22 is preferentially expressed in the inner root sheath of the hair follicle40, but its role in hair development or maintenance is not yet clearly understood41. The independent, damaging mutations in baleen and toothed whales suggest that FGF22 was no longer necessary when the common ancestor of the cetaceans lost its body hair (Fig. 1)9. Interestingly, we also found damaging mutations in African elephant, an animal with very sparse hair (Fig. 1). The African elephant has lost most of its body hair in response to the high demand for efficient heat transfer because it has the largest body volume to surface area ratio among terrestrial mammals and lives in hot environments42. The parallel loss of FGF22 in cetaceans and African elephant indicates the importance of FGF22 in the maintenance or control of the density of hair in mammals. It would be interesting to examine whether FGF22 is maintained in other mammals, e.g., hippopotamus, that have reduced or no skin hairs.
We also observed a significant signature of positive selection on FGF3 in the cetacean lineage (Table 1). FGF3 is known to play important roles in the formation of the ears and teeth in humans and mice15,43. Cetaceans have undergone major changes in the ear and tooth including loss of the external ears, development of mandibular fat body, isolation of the tympanoperiotic complex from the skull, and loss of occlusion of the teeth7,44. Thus, the positive selection on FGF3 is potentially associated with changes in these morphological traits3,4,7,44.
We also found a cetacean-specific loss of positively charged Arg-rich motif in FGF3 (Supplementary Fig. S2). Because all the essential components of the gene, such as signal sequence, receptor binding sites, and heparin-binding sites, are strictly conserved, lack of the Arg-rich motif is likely to modify rather than abrogate the function of FGF3. Given that FGF3 functions as a paracrine factor by binding to negatively charged HSPGs in the extracellular matrix, and Arg binds heparin approximately 2.5-fold more tightly than does Lys45, the Arg-rich motif might be involved in heparan sulphate binding and regulating local concentrations of FGF3 in addition to the canonical heparin binding sites within the FGF domain. Similarly, it was recently shown that modulation of FGF3 dosage is related to the evolution of mammalian dentition46. It would be intriguing to determine if the specific loss of the Arg-rich motif affects the heparin binding affinity of FGF3.
In cetacean FGF9, three amino acids were replaced in the common ancestor of the cetaceans (Table 2). Among these, the D203G substitution was predicted to alter the function of FGF9. Homology modelling further suggested a defective homodimerization of FGF9 because of this substitution (Fig. 3). Interestingly, it is already known that mutations affecting homodimerization of FGF9 are associated with the fusion of the elbow and knee joints in mice and humans17,18. The modification of the cetacean forelimb into an inflexible flipper, resulting in more efficient locomotion, occurred prior to the last common ancestor of extant cetaceans9; however, the genetic mutation underlying this morphological change has not yet been identified. Our results, together with previous reports, suggest that the D203G substitution in FGF9 is responsible for the morphological change in the cetacean joint.
Hypoxia tolerance is one of the main characteristics of the cetaceans8. Recently, evidence of adaptive evolution was found in genes involved in transporting oxygen to the blood and muscle (haemoglobin-α and β, myoglobin) and the regulation of vasoconstriction (endothelin-1, -2, and -3, endothelin receptor type A and B, adrenergic receptor α-1D, and arginine vasopressin)10,11. Here, we also detected a signature of positive selection in FGF11 along the Cetacea (Supplementary Figs S5 and S6), together with a cetacean-specific function-altering amino acid replacements identified by Polyphen-2 (Table 2). Through expression and promoter analyses, we showed an increased expression of FGF11 in the brain and heart in cetaceans and provided compelling evidence of FGF11 induction during hypoxia (Fig. 4). FGF11 belongs to the iFGF subfamily, which is known to be highly expressed in the nervous system37. Other members of the iFGF subfamily were reported to bind voltage-gated sodium channels47 and display altered phenotypes in the nervous system when deleted14. In contrast, gene expression levels of FGF11 in the brain were relatively low, and the function of FGF11 remains largely unknown14. In a previous report, we found the inducible expression of FGF11 in human umbilical vein endothelial cells by hypoxia20. The expression of FGF11 increased capillary-like tube formation in these cells, suggesting a protective role of FGF11 during hypoxia. Increased hypoxia tolerance is one of the main characteristics of the cetaceans, but underlying molecular mechanisms are not yet clearly understood. Our results provide a clue to the function of FGF11 during hypoxia and suggest that molecular adaptation of FGF11 may have played a role in the evolution of the Cetacea by providing hypoxia tolerance in the brain and heart, two pivotal organs to protect during prolonged hypoxic dives.
We also discovered a wide range of gene expression levels of FGF23 in the livers from minke and bowhead whales (Fig. 5). FGF23 is a bone-derived hormone that acts on the kidney to inhibit phosphate reabsorption and vitamin D3 synthesis. FGF23 is induced by increased levels of blood phosphate and vitamin D3, but mechanisms underlying aberrant expression in ectopic tissues during pathological conditions have not yet been clearly understood19. Although Bhattacharyya et al. proposed that hypoxia causes FGF23 induction in osteoblast, HRE in the FGF23 promoter was not identified because only 5 kb of the upstream region was investigated19. By analysing the entire upstream sequence between FGF23 and the nearest gene (~50 kb), we identified a conserved HRE approximately 13 kb upstream of FGF23 and proved inducible expression of FGF23 in a human hepatic cell line. These results explain an important response to hypoxia in the cetacean liver and the general regulatory mechanism of FGF23 expression in other mammals during pathologic conditions, such as tumour-induced osteomalacia and chronic kidney diseases19.
During the first quarter of cetacean evolution, cetacean bone density was transformed dramatically from the typical terrestrial form to osteopetrosis and finally to osteoporosis, depending on the habitat6. Archaeological evidence shows that early archaic cetaceans living in shallow water displayed high bone density that provided static buoyancy control (ballast), while late ancient cetaceans living in deep water exhibited extremely low bone density that allowed for dynamic buoyancy control6. Unlike other morphological changes, such as flippers and flukes that are formed during embryonic stages, maintaining low levels of bone density requires lifetime regulation. Because the overexpression of FGF23 is linked with low bone density19, the induction of FGF23 by hypoxia, which occurs during diving, provides a reasonable explanation for the underlying mechanism of lifetime adjustment and maintenance of low bone density in animals living in deep water.
Surprisingly, we found that cetaceans had a higher number of HREs in the FGF23 promoter than did the other terrestrial mammalian orders studied. This result raises the possibility that cetaceans evolved to produce FGF23 more efficiently in the liver during their transition from shallow water to deep water, concomitant with the increased frequency and intensity of hypoxia during deep and prolonged diving. Although further experiments are required to characterize the contribution of individual HREs to the expression of FGF23, our findings advance the understanding of the molecular basis of cetacean bone evolution during the course of aquatic adaptation.
We also identified cetacean-specific mutations in FGF10 (D43G), FGF20 (R46L), FGF19 (L113S), and FGF21 (E119Q) that were predicted to affect the function of these genes (Table 2). It would be intriguing to determine if these changes are linked with the evolution of the lung (FGF10), or kidney (FGF20), or with the metabolic changes (FGF19 and FGF21) required for aquatic life in cetaceans.
Taken together, our findings provide various novel insights into the roles of FGFs in the aquatic adaptations exhibited by cetaceans. In addition, the whole genome sequences of the fin whale will be an important resource for future studies of cetacean evolution, biology, and genetics.
Methods
Fin whale sequencing and identification of FGF coding sequences
Genomic DNA was extracted from a fin whale that had been accidentally killed and investigated by the Korean maritime police. The whole genome was sequenced to 60x average read depth using Illumina HiSeq2000 with a 400 bp insert library at Theragen BiO Institute (TBI); sequence data have been deposited in GenBank (SRA346104). The genome sequence of the fin whale and the genome sequences of one fin whale and one finless porpoise that had been sequenced previously23 were constructed using the consensus method. The consensus method utilizes the genome sequences of the most closely related species and maps short reads to these sequences resulting in a consensus genome sequence. In this study, short reads from the three cetaceans were mapped to the genome sequences of their closest sister species (Supplementary Table 5). The mapping was conducted using BWA-MEM48 with the default options, and rmdup, implemented in SAMtools 0.1.1949, was used to remove potential PCR duplicates among the short reads. SAMtools 0.1.19 was used for variant calling with the “-d 5” option. The consensus sequences were obtained by replacing nucleotides from the reference genome sequences with the variants. The genome sequence of the bowhead whale was obtained from The Bowhead Whale Genome Resource22 (http://www.bowhead-whale.org/).
We analysed all of the mammalian FGF genes (FGF1 to FGF23) from 24 terrestrial mammals and 8 cetaceans; a list of the species is provided in Supplementary Table 5. Using human FGF coding sequences obtained from the UniGene database (http://www.ncbi.nlm.nih.gov/UniGene) as queries, the FGFs of the terrestrial mammals and 5 cetaceans (minke whale, bottlenose dolphin, killer whale, baiji and sperm whale) were obtained from the GenBank or Ensembl (http://www.ensembl.org) database through BLAST search. The coding sequences of the FGF genes from the fin whale, finless porpoise, and bowhead whale were extracted from their genome sequences.
Minke whale FGF22 genomic regions were amplified using the GC-RICH PCR system (Roche, Basel, Switzerland) with primers FGF22-E1-F, 5′-CACCCACTGTGGCTCTGG-3′ and FGF22-E1-R, 5′-GATCCACACACAGGAAGAAGTG-3′ for exon 1; and FGF22-E2-F, 5′-AGGCGTCTTCTTCGAGTTGCGC-3′ and FGF22-E3-R, 5′-TCAGGAGACCAGGACGGGCAG-3′ for a region spanning exons 2 and 3. The PCR was performed with 1 M GC-RICH resolution solution provided from the supplier. The PCR conditions were as follows: 95 °C for 3 min, followed by 25 cycles of 95 °C for 30 sec, 60 °C for 30 sec and 72 °C for 1 min +5 sec/cycle, and a final extension of 72 °C for 7 min. PCR products were cloned into pGEM-T Easy vector (Promega, USA) and sequenced. A list of the species, sequence identification numbers, and gene identification methods used in this study are summarized in Supplementary Tables S5 and S6.
Phylogenetic tree construction for FGF genes
The coding sequences were aligned using ClustalW in MEGA6, and a phylogenetic tree was constructed using the Maximum Likelihood method based on the General Time Reversible model50, implemented in MEGA651. The initial tree for the heuristic search was obtained automatically by applying the Neighbour-Joining and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach; the topology with the superior log likelihood value was selected. We performed 100 times of bootstrap replications.
Molecular evolutionary and comparative analyses of FGF genes
We used the PRANK program52 for the multiple sequence alignment of the coding sequences of the FGF genes with the empirical codon model option. The CODEML program in PAML 4.526 was used to estimate dN/dS ratios based on the framework of maximum likelihood. The guide tree was drawn based on previous studies53,54,55,56. We used the branch model to calculate the dN/dS ratio for whales and the other mammals. With the two-ratio model27, the dN/dS ratios for whales and the other mammals were estimated separately, and the corresponding likelihoods were calculated. To test if the dN/dS ratios were significantly different between whales and the other mammals, we calculated the mean dN/dS ratio from the entire phylogenetic tree (one ratio model) and the likelihood was calculated. Then, a log likelihood ratio test was performed to test if dN/dS ratios were significantly different between whales and the other mammals (df = 1), with an FDR correction for multiple tests.
To further test for signatures of positive selection, we used a branch-site model28. For each analysis, whale branches composed of ancestral whale branches and terminal whale branches were defined as foreground branches and the remaining branches were defined as background branches. In the selection model, the proportion of codons that had dN/dS ratios greater than 1 in the foreground branch was estimated to be greater than zero. In the neutral model, no codon was allowed to have a dN/dS ratio greater than 1. Then, a likelihood ratio test was performed using the difference in likelihoods between the selection and neutral models to test if the selection model had greater explanatory power than the neutral model to describe the empirical data. The positive selection model was considered unsupported if twice the difference in likelihood did not exceed 3.84, which is the recommended critical value for a significance level of 5% (Yang Z; http://abacus.gene.ucl.ac.uk/software/pamlDOC.pdf).
We also used a site model to identify positive selection29. The ancestral codon sequences of whales were inferred using the CODEML program implemented in the PAML package26. Alignments were made with the extant whale sequences and the inferred ancestral whale sequence and signatures of positive selection were identified from all branches using the site model29. The selection model (M2) in which local dN/dS ratio was allowed to be greater than 1 was compared with the neutral model (M1) in which local dN/dS ratio was not allowed to be greater than 1. In M2, an alignment was categorized into three groups. In group 0, 1, and 2, the dN/dS was lower than 1, equal to 1, and greater than 1, respectively. And the proportions of codons at group 0, 1, and 2 were calculated together with the estimated dN/dS ratios for group 0 and group 2. In M1, there were only group 0 and group 1, thus there was no codon with dN/dS greater than 1. A likelihood ratio test (df = 2) and FDR correction for multiple tests were performed to test for statistical significances.
To detect signatures of positive selection on each codon, we used the MEME algorithms30, implemented in the datamonkey web application57, together with the whale phylogenetic tree shown in Fig. 2a and the multiple sequence alignments for extant whales and their ancestors. The non-synonymous substitution rate (β) and synonymous substitution rate (α) at each codon of an alignment was estimated and the difference between α and β was tested by likelihood ratio test. And Empirical Bayes factors were calculated to estimate the relative likelihood of positive selection at each branch.
Prediction of the functional effects of ancestral cetacean mutations
Function altering amino acid changes were predicted using PolyPhen-231, PROVEAN v1.133, and SIFT32, with the default cutoff values, and human protein sequences were used as queries for the programs. First, amino acid replacements were identified using the multiple sequence alignments of each FGF gene. Second, we classified these replacements into the following two groups: amino acid replacements that were generated in the whale lineages and amino acid replacements arising in the other lineages. Third, we run the web applications of PolyPhen-2 ((http://genetics.bwh.harvard.edu/pph2/) and PROVEAN v1.1 and SIFT (http://provean.jcvi.org/). The predictions ‘possibly damaging’ or ‘probably damaging’ by PolyPhen-2 were regarded as a function-altering replacements. The predictions ‘Deleterious’ and ‘Damaging’ by PROVEAN v1.1 and SIFT, respectively, were also regarded as function-altering replacement.
Promoter analysis
Promoter regions of FGF11 and FGF23 were obtained from NCBI and analysed using the Kalign tool (http://www.ebi.ac.uk/Tools/msa/kalign/). GenBank accession numbers are listed in Supplementary Tables S7 and S8.
Cell culture, hypoxia treatment, and quantitative RT-PCR
HepG2 and SH-SY5Y cells lines were obtained from the American Type Culture Collection and Korean Cell Line Bank, respectively. For hypoxia treatment, cells were incubated in a Modular Incubator Chamber (Billups-Rothenberg Inc., CA, USA) filled with 1% oxygen, 5% CO2, and 94% nitrogen for 18 h. Total RNA was prepared, and cDNA was synthesized using reverse transcriptase. Quantitative PCR was performed using Thunderbird SYBR qPCR Mix reagent (TOYOBO, Japan) and a CFX Connect Real-Time PCR system (BioRad, CA, USA). ACTB was used as an endogenous control. Primer sequences can be found in Supplementary Table S9.
RNA-seq analysis
RNA sequences were obtained from previously published minke23 and a bowhead whale22 data, and from four bowhead whales published by Seim et al.36 and mapped against their own reference genome using the TopHat2 with default options58. The GFF3 annotation files were downloaded from NCBI. The number of reads that were mapped against coding sequences of FGF genes was counted using HTSeq-0.6.159 and normalized by Trimmed Mean of M-values (TMM) using edgeR60.
Additional Information
How to cite this article: Nam, K. et al. Analysis of the FGF gene family provides insights into aquatic adaptation in cetaceans. Sci. Rep. 7, 40233; doi: 10.1038/srep40233 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Thewissen, J. G., Cooper, L. N., Clementz, M. T., Bajpai, S. & Tiwari, B. N. Whales originated from aquatic artiodactyls in the Eocene epoch of India. Nature 450, 1190–1194, doi: 10.1038/nature06343 (2007).
Demere, T. A., McGowen, M. R., Berta, A. & Gatesy, J. Morphological and molecular evidence for a stepwise evolutionary transition from teeth to baleen in mysticete whales. Syst Biol 57, 15–37, doi: 10.1080/10635150701884632 (2008).
Uhen, M. D. Evolution of marine mammals: back to the sea after 300 million years. Anat Rec (Hoboken) 290, 514–522, doi: 10.1002/ar.20545 (2007).
Reidenberg, J. S. Anatomical adaptations of aquatic mammals. Anat Rec (Hoboken) 290, 507–513, doi: 10.1002/ar.20541 (2007).
Cooper, L. N., Dawson, S. D., Reidenberg, J. S. & Berta, A. Neuromuscular anatomy and evolution of the cetacean forelimb. Anat Rec (Hoboken) 290, 1121–1137, doi: 10.1002/ar.20571 (2007).
Gray, N. M., Kainec, K., Madar, S., Tomko, L. & Wolfe, S. Sink or swim? Bone density as a mechanism for buoyancy control in early cetaceans. Anat Rec (Hoboken) 290, 638–653, doi: 10.1002/ar.20533 (2007).
Nummela, S., Thewissen, J. G., Bajpai, S., Hussain, T. & Kumar, K. Sound transmission in archaic and modern whales: anatomical adaptations for underwater hearing. Anat Rec (Hoboken) 290, 716–733, doi: 10.1002/ar.20528 (2007).
Larson, J., Drew, K. L., Folkow, L. P., Milton, S. L. & Park, T. J. No oxygen? No problem! Intrinsic brain tolerance to hypoxia in vertebrates. J. Exp. Biol. 217, 1024–1039, doi: 10.1242/jeb.085381 (2014).
McGowen, M. R., Gatesy, J. & Wildman, D. E. Molecular evolution tracks macroevolutionary transitions in Cetacea. Trends Ecol Evol 29, 336–346, doi: 10.1016/j.tree.2014.04.001 (2014).
Mirceta, S. et al. Evolution of mammalian diving capacity traced by myoglobin net surface charge. Science 340, 1234192, doi: 10.1126/science.1234192 (2013).
Tian, R. et al. Evolutionary Genetics of Hypoxia Tolerance in Cetaceans during Diving. Genome Biol Evol 8, 827–839, doi: 10.1093/gbe/evw037 (2016).
Itoh, N. & Ornitz, D. M. Fibroblast growth factors: from molecular evolution to roles in development, metabolism and disease. J Biochem 149, 121–130, doi: 10.1093/jb/mvq121 (2011).
Imamura, T. Physiological functions and underlying mechanisms of fibroblast growth factor (FGF) family members: recent findings and implications for their pharmacological application. Biol Pharm Bull 37, 1081–1089 (2014).
Itoh, N. & Ornitz, D. M. Functional evolutionary history of the mouse Fgf gene family. Dev Dyn 237, 18–27, doi: 10.1002/dvdy.21388 (2008).
Tekin, M. et al. Homozygous mutations in fibroblast growth factor 3 are associated with a new form of syndromic deafness characterized by inner ear agenesis, microtia, and microdontia. Am J Hum Genet 80, 338–344, doi: 10.1086/510920 (2007).
Barak, H. et al. FGF9 and FGF20 maintain the stemness of nephron progenitors in mice and man. Dev Cell 22, 1191–1207, doi: 10.1016/j.devcel.2012.04.018 (2012).
Harada, M. et al. FGF9 monomer-dimer equilibrium regulates extracellular matrix affinity and tissue diffusion. Nat Genet 41, 289–298, doi: 10.1038/ng.316 (2009).
Wang, Y., Wu, X. L., Wei, D. Q., Li, Y. X. & Wang, J. F. Autoinhibitory mechanism for the mutation-induced impaired FGF9 signaling. J Chem Inf Model 52, 2422–2429, doi: 10.1021/ci3003045 (2012).
Bhattacharyya, N., Chong, W. H., Gafni, R. I. & Collins, M. T. Fibroblast growth factor 23: state of the field and future directions. Trends Endocrinol Metab 23, 610–618, doi: 10.1016/j.tem.2012.07.002 (2012).
Yang, J. et al. Hypoxia-induced fibroblast growth factor 11 stimulates capillary-like endothelial tube formation. Oncol Rep 34, 2745–2751, doi: 10.3892/or.2015.4223 (2015).
Chen, Z., Wang, Z., Xu, S., Zhou, K. & Yang, G. Characterization of hairless (Hr) and FGF5 genes provides insights into the molecular basis of hair loss in cetaceans. BMC Evol Biol 13, 34, doi: 10.1186/1471-2148-13-34 (2013).
Keane, M. et al. Insights into the evolution of longevity from the bowhead whale genome. Cell Rep 10, 112–122, doi: 10.1016/j.celrep.2014.12.008 (2015).
Yim, H. S. et al. Minke whale genome and aquatic adaptation in cetaceans. Nat Genet 46, 88–92, doi: 10.1038/ng.2835 (2014).
Foote, A. D. et al. Convergent evolution of the genomes of marine mammals. Nat Genet 47, 272–275, doi: 10.1038/ng.3198 (2015).
Zhou, X. et al. Baiji genomes reveal low genetic variability and new insights into secondary aquatic adaptations. Nat Commun 4, 2708, doi: 10.1038/ncomms3708 (2013).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24, 1586–1591, doi: 10.1093/molbev/msm088 (2007).
Yang, Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol 15, 568–573 (1998).
Yang, Z. & Nielsen, R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol 19, 908–917 (2002).
Yang, Z., Nielsen, R., Goldman, N. & Pedersen, A. M. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155, 431–449 (2000).
Murrell, B. et al. Detecting individual sites subject to episodic diversifying selection. PLoS Genet 8, e1002764, doi: 10.1371/journal.pgen.1002764 (2012).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat Methods 7, 248–249, doi: 10.1038/nmeth0410-248 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4, 1073–1081, doi: 10.1038/nprot.2009.86 (2009).
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R. & Chan, A. P. Predicting the functional effect of amino acid substitutions and indels. PLoS One 7, e46688, doi: 10.1371/journal.pone.0046688 (2012).
Plotnikov, A. N. et al. Crystal structure of fibroblast growth factor 9 reveals regions implicated in dimerization and autoinhibition. J Biol Chem 276, 4322–4329, doi: 10.1074/jbc.M006502200 (2001).
Shi, Z., Olson, C. A., Bell, A. J. Jr. & Kallenbach, N. R. Stabilization of alpha-helix structure by polar side-chain interactions: complex salt bridges, cation-pi interactions, and C-H em leader O H-bonds. Biopolymers 60, 366–380, doi: 10.1002/1097-0282(2001)60:5<366::AID-BIP10177>3.0.CO;2-5 (2001).
Seim, I. et al. The transcriptome of the bowhead whale Balaena mysticetus reveals adaptations of the longest-lived mammal. Aging (Albany NY) 6, 879–899 (2014).
Smallwood, P. M. et al. Fibroblast growth factor (FGF) homologous factors: new members of the FGF family implicated in nervous system development. Proc. Natl. Acad. Sci. USA 93, 9850–9857 (1996).
Schodel, J. et al. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 117, e207–217, doi: 10.1182/blood-2010-10-314427 (2011).
Oulion, S., Bertrand, S. & Escriva, H. Evolution of the FGF Gene Family. Int J Evol Biol 2012, 298147, doi: 10.1155/2012/298147 (2012).
Nakatake, Y., Hoshikawa, M., Asaki, T., Kassai, Y. & Itoh, N. Identification of a novel fibroblast growth factor, FGF-22, preferentially expressed in the inner root sheath of the hair follicle. Biochim Biophys Acta 1517, 460–463 (2001).
Jarosz, M. et al. Fibroblast growth factor 22 is not essential for skin development and repair but plays a role in tumorigenesis. PLoS One 7, e39436, doi: 10.1371/journal.pone.0039436 (2012).
Myhrvold, C. L., Stone, H. A. & Bou-Zeid, E. What is the use of elephant hair? PLoS One 7, e47018, doi: 10.1371/journal.pone.0047018 (2012).
Krejci, P., Prochazkova, J., Bryja, V., Kozubik, A. & Wilcox, W. R. Molecular pathology of the fibroblast growth factor family. Hum Mutat 30, 1245–1255, doi: 10.1002/humu.21067 (2009).
Armfield, B. A., Zheng, Z., Bajpai, S., Vinyard, C. J. & Thewissen, J. Development and evolution of the unique cetacean dentition. PeerJ 1, e24, doi: 10.7717/peerj.24 (2013).
Munoz, E. M. & Linhardt, R. J. Heparin-binding domains in vascular biology. Arterioscler Thromb Vasc Biol 24, 1549–1557, doi: 10.1161/01.ATV.0000137189.22999.3f (2004).
Charles, C. et al. Modulation of Fgf3 dosage in mouse and men mirrors evolution of mammalian dentition. Proc. Natl. Acad. Sci. USA 106, 22364–22368, doi: 10.1073/pnas.0910086106 (2009).
Goldfarb, M. Fibroblast growth factor homologous factors: evolution, structure, and function. Cytokine Growth Factor Rev 16, 215–220, doi: 10.1016/j.cytogfr.2005.02.002 (2005).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. eprint. arXiv:1303.3997 (2013).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079, doi: 10.1093/bioinformatics/btp352 (2009).
Nei, M. & Kumar, S. Molecular Evolution and Phylogenetics. Oxford University Press, New York (2000).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30, 2725–2729, doi: 10.1093/molbev/mst197 (2013).
Loytynoja, A. & Goldman, N. An algorithm for progressive multiple alignment of sequences with insertions. Proc. Natl. Acad. Sci. USA 102, 10557–10562, doi: 10.1073/pnas.0409137102 (2005).
Fabre, P. H., Hautier, L., Dimitrov, D. & Douzery, E. J. A glimpse on the pattern of rodent diversification: a phylogenetic approach. BMC Evol Biol 12, 88, doi: 10.1186/1471-2148-12-88 (2012).
McGowen, M. R., Spaulding, M. & Gatesy, J. Divergence date estimation and a comprehensive molecular tree of extant cetaceans. Mol Phylogenet Evol 53, 891–906, doi: 10.1016/j.ympev.2009.08.018 (2009).
Perelman, P. et al. A molecular phylogeny of living primates. PLoS Genet 7, e1001342, doi: 10.1371/journal.pgen.1001342 (2011).
Tarver, J. E. et al. The Interrelationships of Placental Mammals and the Limits of Phylogenetic Inference. Genome Biol Evol 8, 330–344, doi: 10.1093/gbe/evv261 (2016).
Pond, S. L. & Frost, S. D. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21, 2531–2533, doi: 10.1093/bioinformatics/bti320 (2005).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36, doi: 10.1186/gb-2013-14-4-r36 (2013).
Anders, S., Pyl, P. T. & Huber, W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169, doi: 10.1093/bioinformatics/btu638 (2015).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140, doi: 10.1093/bioinformatics/btp616 (2010).
Acknowledgements
This work was supported by the Korea Institute of Ocean Science & Technology (KIOST) in-house program to J.-H.L. (PE99413); the National Research Foundation of Korea Grant (NRF-2015R1A2A2A01004168) to S.-S.C.; and the ‘Reference genomes building and application for large scale population genomics’ Research Fund (1.160003.01) of Ulsan National Institute of Science & Technology (UINST) to J.B.
Author information
Authors and Affiliations
Contributions
J.-H.L. and J.-Y.J. designed the research; K.N., J.-Y.J., O.C., and J.J. analysed the data; K.W.L., H.-S.Y., and S.-W.L. performed experiments; S.-S.C. analysed FGF9 structure. J.-Y.J., K.N., S.-S.C., K.W.L., O.C., J.J., S.W.L., Y.S.C., J.B., and J.P.M. wrote the paper. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Nam, K., Lee, K., Chung, O. et al. Analysis of the FGF gene family provides insights into aquatic adaptation in cetaceans. Sci Rep 7, 40233 (2017). https://doi.org/10.1038/srep40233
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep40233
This article is cited by
-
Fibroblast Growth Factor 11 Inhibits Hepatitis B Virus Gene Expression Through FXRα Suppression
Journal of Microbiology (2023)
-
Sex difference on fibroblast growth factors (FGFs) expression in skin and wound of streptozotocin(STZ)-induced type 1 diabetic mice
Molecular Biology Reports (2023)
-
Seascape genomics of common dolphins (Delphinus delphis) reveals adaptive diversity linked to regional and local oceanography
BMC Ecology and Evolution (2022)
-
Expression and purification of intracrine human FGF 11 and study of its FGFR-dependent biological activity
Journal of Microbiology (2022)
-
The Analyses of Cetacean Virus-Responsive Genes Reveal Evolutionary Marks in Mucosal Immunity-Associated Genes
Biochemical Genetics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
