
Abstract
The bacterium Bacillus thuringiensis produces Crystal (Cry) proteins that are toxic to a diverse range of insects. Transgenic crops that produce Bt Cry proteins are grown worldwide because of their improved resistance to insect pests. Although Bt “pyramid” cotton that produces both Cry1A and Cry2A is predicted to be more resistant to several lepidopteran pests, including Spodoptera exigua, than plants that produce Cry1Ac alone, the mechanisms responsible for the toxicity of Cry2Aa in S. exigua are not well understood. We identified several proteins that bind Cry2Aa (polycalin, V-ATPase subunits A and B, actin, 4-hydroxybutyrate CoA-transferase [4-HB-CoAT]), and a receptor for activated protein kinase C (Rack), in S. exigua. Recombinant, expressed versions of these proteins were able to bind the Cry2Aa toxin in vitro assays. RNA interference gene knockdown of the Se-V-ATPase subunit B significantly decreased the susceptibility of S. exigua larvae to Cry2Aa, whereas knockdown of the other putative binding proteins did not. Moreover, an in vitro homologous competition assay demonstrated that the Se-V-ATPase subunit B binds specifically to the Cry2Aa toxin, suggesting that this protein acts as a functional receptor of Cry2Aa in S. exigua. This the first Cry2Aa toxin receptor identified in S. exigua brush-border membrane vesicles.
Similar content being viewed by others
Introduction
The Crystal (Cry) toxins produced by Bacillus thuringiensis (Bt) are a diverse group of proteins that are used to control a broad range of insect pests1. Not only are Bt compounds used worldwide as pesticides, but Cry genes have been used to create transgenic crops with enhanced resistance to pest insects. Of the Cry2A subfamily, both Cry2Aa and Cry2Ab have been successfully incorporated into plants to produce transgenic insect-resistant crops2,3.
In China, transgenic Bt cotton expressing the Cry2Ab toxin has not been commercialized. In contrast, transgenic Cry1Ac cotton, which was first cultivated in 1997, is now grown on more than 3 million hectares in 20154. Adoption of this Bt cotton variety has resulted in the decline of several important pest populations at the landscape level in China, as well as reductions in the application of broad-spectrum insecticides5. Nonetheless, the continued large-scale planting of Bt cotton has led to new problems, including the evolution of resistance among target pests6,7 and rapid increases in non-target hemipteran8 and lepidopteran pests9,10,11. Developing plants that express more than one Cry toxin could, however, both delay insect resistance to Bt crops and increase the target pest spectrum12,13. For example, transgenic plants that express both Cry1Ac and Cry2Ab toxin would be expected to be much more resistant to lepidopteran pests, especially the beet armyworm Spodoptera exigua3,14.
S. exigua (Hübner; Lepidoptera: Noctuidae) is a polyphagous insect that has not been a significant crop pest in China for some time11. However, because of the recent reduction in pesticide usage in cotton fields, and because it is insensitive to Cry1Ac, the beet armyworm has once again become a major economic pest of cotton in China3,15,16,17. Although some studies suggest that S. exigua is less sensitive to Cry2Aa/b than to Cry1B, Cry1C or other toxins18,19, Bt crops producing both Cry1Ac and Cry2Aa/b (Cry2Ab in the case of cotton) are predicted to be more resistant to S. exigua, and several other lepidopteran pests, than those currently cultivated in China which produce only Cry1Ac3,15,20,21,22. However, except for cadherin23, little is known about the receptor proteins that mediate the toxicity of the Cry2A subfamily of proteins in the Lepidoptera.
In this paper, we present the first analysis of Cry2Aa receptor proteins in S. exigua brush-border membrane vesicles (BBMVs). Because the Cry2Aa protein has 87% sequence homology with Cry2Ab, and similar toxicity to both the Lepidoptera and Diptera, we chose Cry2Aa to represent the Cry2A subfamily24,25. In addition, and possibly more important, the purified toxin (purity > 98%) is only commercially available for Cry2Aa at present. The goal of this study was to identify Cry2Aa binding proteins in S. exigua BBMVs using two-dimension gel electrophoresis (2DE) and LC-MS (liquid chromatography-mass spectrometry)/MS techniques. The utility of using such a combination of protein binding assays and RNA interference to analyze the receptor function of binding proteins is also evaluated and discussed.
Results
Binding of Cry2Aa to S. exigua BBMVs
Proteins of S. exigua BBMVs were separated by 2DE and silver stained (Fig. 1a). Proteins ranging in size from 10 kDa to 130 kDa were isolated using pH 3–10 IPG strips and 8% SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) gels. Activated Cry2Aa toxin and a polyclonal antibody were used to identify specific proteins binding to Cry2Aa. An antibody-specificity test was conducted before the binding assays to confirm that the Cry2Aa antibody recognizes Cry2Aa but not Cry1Ac (Supplementary Fig. S1).

(a) S. exigua BBMV proteins (100 μg) separated by 2DE, marker positions are indicated on the left of the gel. The pH 3–10 IPG strip used for isoelectric focusing is shown at the bottom. (b) S. exigua Cry2Aa-binding proteins are the spots numbered 1 to 7; spot positions correspond to those in Fig. 1a.
Cry2Aa bound to seven proteins of approximately 100, 110, 65, 50, 30, 35 and 15 kDa (protein spots numbered 1 through 7 in Fig. 1b). To the best of our knowledge, this is the first evidence that Cry2Aa binds to S. exigua BBMV proteins. Protein spots were excised from the silver-stained gel based on PVDF (polyvinylidene fluoride) membrane signals and analyzed by LC-ESI (electrospray ionization)-MS/MS. After searching protein databases, the protein spots in the silver-stained gel (Table 1) were identified as polycalin, V-type ATPase subunit A, V-type ATPase subunit B, actin, 4-hydroxybutyrate CoA-transferase (4-HB-CoAT), and a receptor for activated protein kinase C (Rack). Among these, 4-HB-CoAT and Rack were not previously known to bind to Cry toxin.
Cloning and sequence analysis of S. exigua genes encoding Cry2Aa-binding proteins
We cloned the full-length of Se-polycalin cDNA (GenBank accession no. KU234093) from the midguts of S. exigua larvae. The 3,339-bp open reading frame (ORF) encodes a protein of 1,113 residues with a predicted mass of 122 kDa. The deduced protein sequence includes a signal peptide, glycosylphosphatidylinositol (GPI)-anchoring site, N-glycosylation sites and O-glycosylation sites (Supplementary Fig. S2). Phylogenetic analysis shows that Se-polycalin clusters with lepidopteran polycalin (Supplementary Fig. S3). Alignment using DNAMAN software indicates that Se-polycalin has highest homology with that of Mamestra configurata and Helicoverpa armigera (47.0% and 46.2%, respectively).
S. exigua V-ATPase subunits A and B were also cloned, and their sequences submitted to GenBank (KX685519 and KX685520, respectively). Their respective cDNAs contained ORFs of 1,848 and 1,482 bp, encoding 616- and 494-amino acid proteins with estimated molecular weights of 68 kDa and 55 kDa. The conserved domains walker A motif/ATP-binding site, walker B motif, N-glycosylation sites, and O-glycosylation sites, are shown in Supplementary Figs S4 and S6. Phylogenetic analysis placed both proteins in the lepidopteran clade. High identity of V-ATPase subunits A and B among diverse insect species was detected; for example, S. exigua V-ATPase subunit A has 95.3% identity with that of Ostrinia furnacalis and Bombyx mori and 96.3% identity with that of Manduca sexta, but lower identity with that of Ceratitis capitata (11.1%) (Supplementary Fig. S5). S. exigua V-ATPase subunit B also has high identity with that of Manduca sexta (99.2%) and H. armigera (99.2%) (Supplementary Fig. S7).
The sequence of 4-hydroxybutyrate CoA-transferase was also cloned and submitted to GenBank (KX685521). The 1,431-bp cDNA obtained encodes one 52-kDa protein of 477 residues with N-glycosylation and O-glycosylation sites (Supplementary Fig. S8). It had highest identity with that of Papilio xuthus (87%; Supplementary Fig. S9).
Specific expression profiles in different developmental stages and tissues
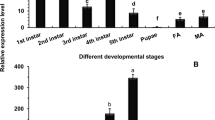
We tested the expression patterns of all genes encoding binding proteins in different developmental stages of S. exigua (Fig. 2). The Se-polycalin gene was highly expressed in 3rd instar larvae but was hardly expressed in pupae and adults (Fig. 2a). However, the expression of Se-V-ATPase subunit A, Se-actin and Se-4-HB-CoAT was not significantly different among different developmental stages (Fig. 2b,d,e). Expression of Se-V-ATPase subunit B progressively decreased from the 1st to 5th instar, and its expression in adults and 5th instar larvae was moderate (Fig. 2c). Expression of the Rack gene was stable in different larval instars and low in adults (Fig. 2f).

cDNA templates were derived from 1st, 2nd, 3rd, 4th and 5th instar larvae, pupae and adults. Three independent samples were examined for relative transcript levels using the 2−∆∆CT method. a = Se-polycalin, b = Se-V-ATPase subunit A, c = Se-V-ATPase subunit B, d = Se-actin, e = Se-4-HB-CoAT, f = Se-Rack. Expression levels were normalized to those of the reference genes Se-RpL10 and Se-GAPDH. Bars with different letters indicate P values < 0.05 (ANOVA).
Differential expression of the binding protein genes in different tissues was also measured (Fig. 3). Se-polycalin and Se-4-HB-CoAT were most highly expressed in the midgut (Fig. 3a,e) and Se-V-ATPase subunit A and Se-V-ATPase subunit B were most highly expressed in the Malpighian tubules (Fig. 3b,c). Se-actin and Se-Rack were most highly expressed in the remaining tissues (Fig. 3d,f).

cDNA templates were derived from the foregut (FG), midgut (MG), hindgut (HG), fat body (FB), Malpighian tubules (MT), and the remainder (R), of 4th instar larvae. Three independent samples were examined for relative transcript levels using the 2−∆∆CT method. a = Se-polycalin, b = Se-V-ATPase subunit A, c = Se-V-ATPase subunit B, d = Se-actin, e = Se-4-HB-CoAT, f = Se-Rack. Expression levels were normalized to those of the reference genes Se-RpL10 and Se-GAPDH. Bars with different letters indicate P values < 0.05 (ANOVA).
RNA interference knockdown of binding proteins
Compared to dsEGFP or H2O, larval ingestion of dsRNAs specific for the Se-V-ATPase subunit A, Se-V-ATPase subunit B, Se-actin, Se-4-HB-CoAT, Se-Rack, and Se-polycalin, significantly reduced transcript levels of these genes by 46.6%, 36.7%, 39.1%, 45.8%, 45.9% and 37.4%, respectively (Fig. 4a). Corrected mortalities following ingestion of Cry2Aa toxin for each of the above dsRNA treatment groups were 86.4%, 47.9%, 78.9%, 81.1%, 67.7% and 87.1%, respectively. The mortality of larvae fed dsRNA specific for Se-V-ATPase subunit B was significantly lower than that of the water or dsEGFP control groups (Fig. 4b).

(a) Relative expression of target gene transcript levels in S. exigua neonate larvae that had been allowed to feed for 48 h on an artificial diet to which either water, EGFP dsRNA, or Se-target genes dsRNA, had been added. Relative estimates of target gene transcript levels were obtained by qRT-PCR and normalized to those of RpL10 and GAPDH. *P < 0.05 (ANOVA). (b) Corrected mortality of different treatment groups after feeding on a diet containing 2.6 μg/cm2 Cry2Aa toxin. The corrected mortality is based on 5 replicates. Bars with different letters indicate P values < 0.05 (ANOVA).
Production of recombinant peptides and binding assays
Expressed peptides were purified and separated by 8% SDS-PAGE gels (Fig. 5a). The results of an ELISA (enzyme-linked immunosorbent assay) indicate that the Se-V-ATPase subunit A, Se-V-ATPase subunit B, Se-actin, Se-4-HB-CoAT, Se-Rack and three partial fragments of Se-polycalin, all bound to Cry2Aa toxin (Fig. 5b).

(a) Positions of purified, recombinant peptides after staining with Coomassie Blue on SDS–PAGE gel. Peptides had been bacterially expressed and purified in a nickel-nitrilotriacetic acid (Ni-NTA) affinity column. Lanes 1 to 8 are: the Se-V-ATPase subunit A, Se-V-ATPase subunit B, Se-actin, Se-4-HB-CoAT, Se-Rack, and three truncated recombinant Se-polycalin peptides (6 = peptide1, 7 = peptide2, 8 = peptide3). (b) Degree of binding, as indicated by optical density (OD), of Cry2Aa to different Se-peptide fragments. Se-V-ATPase subunit A (■), Se-V-ATPase subunit B (♦), Se-actin (▲), Se-4-HB-CoAT (★), Se-Rack (○), Se-polycalin peptide1 (◇), Se-polycalin peptide2 (▼) and Se-polycalin peptide3 (●).
Dot blot analysis of the Cry2Aa receptor in S. exigua
Based on the previous bioassays, we conducted homologous, competitive binding assays to test the specificity of binding between Cry2Aa and the recombinant Se-V-ATPase subunit B peptide. Binding between Cry2Aa and the Se-V-ATPase subunit B peptide was markedly reduced at higher concentrations of un-labelled Cry2Aa (Fig. 6).

Weight ratios of 1:0, 1:50 and 1:500 of unlabeled Cry2Aa toxin were used to compete with the Se-V-ATPase subunit B in binding to biotinylated-Cry2Aa.
Discussion
Our results indicate that S. exigua V-ATPase subunit B is associated with Cry2Aa toxicity. Identifying this novel putative Cry2Aa receptor is potentially crucial to understanding how Cry2Aa is toxic to lepidopteran species. Since V-ATPase was first reported in Saccharomyces cerevisiae26, a substantial amount of evidence has demonstrated that V-ATPases, which are located in the goblet cell apical membrane and rely on ATP hydrolysis to actively pump their substrates across membranes, are involved in energy production and conversion27,28. V-ATPase in the insect midgut mediates pH to create an alkaline environment and participates in ion-transport processes29. V-ATPase up-regulation has been found to be related to Cry1Ac resistance in Plodia interpunctella and P. xylostella30,31. Although V-ATPase has been identified as a Cry toxin-binding protein in H. virescens (Cry1Ac), H. armigera (Cry1Ac) and O. furnacalis (Cry1Ab)32,33,34, little is known about its function with regard to Cry toxins in other insects. Interestingly, RNA silencing of the A. aegypti ATP synthase subunit beta increased larval mortality to Cry toxins, which suggests that this protein is involved in Cry toxin resistance35.
The model proposed by Jurat-Fuentes et al.36 postulates that the Cry toxin binds to cadherin and is then inserted into the cell membrane, facilitating interaction of the toxin with other molecules such as V-ATPase. In fact, Cry1Ac binds to V-ATPase and disturbs H+/K+ transport, thereby destabilizing pH37. Moreover, Cry1Ac has been found to inhibit (Na+, K+)-ATPase in mammals38. Two main classes of active transporters comprise the active transmembrane transport system. Sodium solute symporter (SSS) is driven by proton or sodium transmembrane gradients39, and has been shown to act as a Cry toxin receptor40. Secondary active transporters include ATP-binding cassette (ABC) proteins and V-ATPase, both of which are ATP-dependent electrogenic proton pumps that actively move substrates across cell membranes. ABC proteins are also involved in Cry toxicity41,42,43 and our results suggest that V-ATPase interacts with Cry2Aa toxin. However, further research is required to determine whether the V-ATPase subunit B interacts with Cry toxin in a manner similar to that of the ABC transporter.
Our results show that Se-Polycalin was most highly expressed in the midgut of the 3rd to 5th larval instars of S. exigua, which may protect the larval midgut from viruses or oxidative damage44. Se-V-ATPase subunit B was most highly expressed in 1st instar larvae and adults, and Se-V-ATPase subunit A, Se-V-ATPase subunit B, and Se-4-HB-CoAT, were most highly expressed in the gut and Malpighian tubules, which suggests that they may be involved in energy metabolism27,28,45. Se-Actin and Se-Rack showed high transcript levels in other larval tissues, perhaps because their proteins comprise part of the cytoskeleton46. We also performed in vitro binding assays which showed that the polycalin peptides 2 and 3 had the highest binding affinities with Cry2Aa, followed by V-ATPase subunit B. These different binding affinities may have occurred because different binding proteins have different binding sites for the Cry toxin47.
Although cadherin is a crucial receptor in lepidopterans, including S. exigua23, we did not identify cadherin as a receptor in this study. This could be because LC-MS technology is relatively insensitive to proteins that are not abundant; we found that cadherin was not abundant in a previous study. Another possible explanation is that 2DE technology does not effectively resolve proteins of high molecular weight; Se-cadherin has a molecular weight of approximately 200 kDa48.
Although we identified seven putative Cry2Aa-binding midgut proteins, the results of our RNAi knockdown experiment suggest that only the Se-V-ATPase subunit B is a functional receptor for Cry2Aa in S. exigua.
Materials and Methods
Insect rearing and BBMV preparation
S. exigua larvae were collected from the campus greenhouse of Huazhong Agricultural University in June 2012 and reared on an artificial diet according to the protocol described by Qiu et al.23. S. exigua BBMVs were prepared from fourth-instar larvae according to the method described by Wolfersberger et al.49, with minor modifications. Protein concentrations were determined using the method described by Bradford (1976)50 with bovine serum albumin (BSA) as the standard.
2DE and ligand blotting
Proteins were extracted and precipitated with a ReadyPrepTM 2-D Clean-up Kit (BIO-RAD). Precipitated proteins were dissolved in solubilization buffer (7 M urea, 2 M thiourea, 4% CHAPS, 65 mM dithiothreitol (DTT), 0.2% (w/v) Bio-Lyte, 0.001% bromophenol blue). For 2D electrophoresis, ReadystripTM IPG strips (11 cm long, pH 3–10, nonlinear; BIO-RAD) were rehydrated overnight with solubilized protein. Following rehydration, the strips were subjected to isoelectric focusing using a PROTEAN IEF CELL following the manufacturer’s recommendations (BIO-RAD). The focused strips were equilibrated for 12 min in equilibration buffer (6 M urea, 2% SDS, 20% glycerol, 0.375 M Tris-HCl [pH 8.8]) containing 1% DTT followed by a second equilibration for 12 min in equilibration buffer plus 4% iodoacetamide. For 2D separation, the strips were overlaid on 8% SDS-PAGE gels for electrophoresis. The separated proteins were either stained or transferred to polyvinylidene difluoride (PVDF) membranes.
After proteins had been transferred to PVDF membranes, the membranes were blocked in PBST buffer (135 mM NaCl, 2 mM KCl, 10 mM Na2HPO4, 1.7 mM KH2PO4, 0.1% Tween-20, pH 7.5) containing 5% (w/v) skim milk for 2 h, then incubated with 0.3 μg/ml activated Cry2Aa (EnviroLogix Inc., Portland, ME, USA) in blocking buffer for 2 h at room temperature. The membranes were washed in PBST buffer three times, then incubated for 2 h with a polyclonal antibody against Cry2Aa (diluted 1:3,500; Genscript Biology Company, Nanjing, China). After washing as above, the membranes were incubated with a goat anti-rabbit IgG horseradish peroxidase (HRP)-linked antibody (diluted 1:5,000). After final washes, the membranes were developed with an ECL chemiluminescence detection kit (Fermentas/Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s recommendations.
Mass spectrometry
After blotting, areas on the gel were excised according to the PVDF membrane signals and destained with destaining solution (30% acetonitrile/100 mM NH4HCO3). Each gel sample was then subject to a series of processes, including incubation with 100 mM DTT at 56 °C for 30 minutes, treatment with 200 mM indole-3-acetic acid (IAA) after removal of the supernatant, and incubation with 100 mM NH4HCO3. The liquid was removed, and 100% acetonitrile was added for 5 minutes. The samples were freeze-dried before being subject to trypsin digestion for 24 hours at 37 °C, then analyzed using liquid chromatography-electrospray ionization tandem mass spectrometry (LC-ESI-MS/MS) at the Shanghai Life Science Research Institute, Chinese Academy of Sciences, Shanghai, China. The amino acid sequence results were compared with those in the NCBI database using Mascot2.2 software.
Gene cloning and sequence analysis
Total RNA was isolated from the midguts of actively feeding 4th instar S. exigua larvae using the RNAiso reagent (TaKaRa, Dalian, China) after contaminating genomic DNA had first been eliminated with RNase-free DNase. The RNA preparation was subject to reverse transcription with the PrimeScriptTM RT reagent Kit (TaKaRa, China), according to the manufacturer’s instructions. Partial Se-polycalin, Se-V-ATPase subunit A, Se-V-ATPase subunit B and Se-4-hydroxybutyrate CoA-transferase cDNA sequences were obtained from Prof. Fei Li (Zhejiang University). A smart RACE (rapid amplification of cDNA ends) cDNA amplification kit (Clontech, TaKaRa Bio Inc., Dalian, China) was used to amplify full-length target genes from S. exigua larvae for which pairs of gene-specific primers were designed using Primer 5.0 software based on the partial sequences (Supplementary Tables S1–S5). The PCR products were subcloned into the PMD (18)-T vector (Takara, Dalian, China) and sequenced by the Nanjing Genscript Company, China. The resultant sequences were submitted to GenBank.
Full-length cDNAs were subjected to bioinformatic analysis using an ORF finder tool (http://www.ncbi.nlm.nih.gov/gorf/gorf.html). Sequence alignment was performed using DNAMAN software, and phylogenetic analysis was performed using MEGA4.051,52. Amino acid sequences from other species were used to construct a phylogenetic tree (Supplementary Table S8). Deduced protein sequences were obtained using the ExPASy translate tool Translate (http://web.expasy.org/translate/) from the Swiss Institute of Bioinformatics. N-terminal signal peptides were predicted using the SignalP 4.0 server (http://www.cbs.dtu.dk/services/SignalP/). The GPI modification site prediction server (big-PI Predictor: http://mendel.imp.ac.at/sat/gpi/gpi_server.html was used to predict GPI-anchor signal sequences and GPI anchoring sites. The presence of N- and O-glycosylation sites in predicted protein sequences was assessed using NetNGlyc 1.0 (http://www.cbs.dtu.dk/services/NetNGlyc/) and NetOGlyc 4.0 (http://www.cbs.dtu.dk/services/NetOGlyc/), respectively.
Production of recombinant S. exigua proteins and microtiter plate binding assays
A method similar to a recently described protocol23 was used to produce recombinant proteins. Briefly, PCR products were purified with Wizard PCR Preps DNA Purification System (Promega, Madison, WI, USA) and double digested with FastDigest restriction enzymes (Fermentas, Thermo Fisher Scientific, USA) for 10 min at 37 °C. The products were ligated into the previously digested pET-30a (+) vector to generate pET-30a/Se-peptide plasmids. However, we failed to obtain a Se-polycalin fragment using this expression system, and the pGEX-6P-1 vector was therefore used to construct the recombinant plasmid pGEX-6P-1/Se-polycalin containing a His tag (see primers in Supplementary Table S1). Se-polycalin was divided into three fragments corresponding to bases 1–1,113, 1,114–2,226 and 2,227–3,339 of the Se-polycalin coding sequence, which were named peptide1, peptide2 and peptide3. Insert sequence and orientation were confirmed by sequencing by the Genscript Biology Company, Nanjing, China.
For expression, 200 ng of each recombinant plasmid was transformed into Escherichia coli strain BL21 (DE3) (TransGen Biotech, Inc, Beijing, China) and positive clones were cultured overnight at 37 °C in LB medium containing 50 mg/ml kanamycin. When the absorbance was 0.5 to 0.8 at 600 nm, protein expression was induced with 0.1 mM isopropyl-D-thiogalactoside (IPTG) for 6 h at 37 °C, with horizontal shaking at 200 rpm. The E. coli cells were harvested by centrifugation at 10,000× g for 10 min at 4 °C, after which pellets were resuspended in a solution of 20 mM sodium phosphate, 500 mM sodium chloride, 30 mM imidazole and 5 M urea (pH 7.4), containing 1 mM phenylmethanesulfonyl fluoride (PMSF). Cells were lysed by sonication for 15 min on ice. The protein fragments with a His tag were purified using a nickel-nitrilotriacetic acid (Ni-NTA) affinity column (HisTrap HP column, GE Healthcare Life Sciences, Piscataway, NJ, USA) and eluted with eluting buffer (20 mM sodium phosphate, 500 mM sodium chloride, 500 mM imidazole and 5 M urea, pH 7.4). The cleaved proteins were refolded by dialysis using a gradient of decreasing urea concentration. The purified proteins were analyzed using SDS-PAGE gels, and protein concentrations determined using the Bradford50 method described above.
An ELISA for measuring the binding of Se-polycalin, Se-V-ATPase subunit A, Se-V-ATPase subunit B, Se-actin, Se-Rack, and Se-4-HB-CoAT, peptides to Cry2Aa in microplates was developed using a protocol described by Chen et al.53. In brief, 96-well plates containing 0.5 μg Cry2Aa per well were incubated for 12 h at 4 °C, followed by three washes with PBST buffer and treatment with blocking buffer (PBS, 0.1% Tween 20, 0.5% gelatine) for 1 h at 37 °C. The ELISA plates were washed, then incubated with 0–1,000 nM S. exigua recombinant peptides for 2 h at 37 °C. After washing, an anti-His antibody (1:5,000) (Genscript Biology Company, Nanjing, China) was added and incubated overnight at 4 °C. After additional washes, followed by incubation with a secondary goat-anti-mouse-alkaline phosphatase (1:2,000) antibody for 2 h at 37 °C, freshly prepared substrate (3 mM nitrophenyl phosphate) was added. The reaction was stopped with 0.02 M NaOH, and the absorbance at 405 nm was measured using a Bio-Rad microplate reader.
Competitive binding dot blot assay
Activated Cry2Aa protein was biotinylated using the EZ-Link sulpho-N-hydroxysuccinimide (NHS) liquid chromatography (LC) biotinylation kit (Pierce, FL, USA) according to the manufacturer’s instructions. A homologous competitive binding assay for Cry2Aa by S. exigua recombinant peptides was then conducted. In total, 2 μg of purified peptides bound to a nitrocellulose membrane was blocked in PBST containing 3% BSA and incubated for 3 h with 0.2 μg/ml biotinylated Cry2Aa and unlabelled toxin at weight ratios of 1:0, 1:50 and 1:500. Following washing, streptavidin-HRP was used to detect biotinylated toxin using an ECL chemiluminescence detection kit (Fermentas/Thermo Fisher Scientific, Waltham, MA USA), as described previously23.
RNA interference knockdown of S. exigua target genes
A method adapted from Ren et al.54 was used to produce a dsRNA-expressing vector. Target gene fragments were amplified from S. exigua midgut cDNA using PrimeSTAR HS DNA polymerase (TaKaRa Bio Inc., Dalian, China). The products were individually cloned into the plasmid pET-2P to generate recombinant pET2P/Se-target-gene plasmids. pET2P/EGFP recombinant plasmid production of EGFP dsRNA was used to generate the control EGFP dsRNA23. Recombinant plasmids were transferred into competent E. coli HT115 (DE3) cells. Individual colonies were cultured at 37 °C in 500 ml LB medium containing 50 μg/ml kanamycin. After reaching an OD600 of 1.0, the production of dsRNA was induced by the addition of 0.4 mM IPTG, and the cultures were allowed to grow for an additional 5 h at 37 °C. The bacteria were precipitated by centrifugation at 5,000 rpm for 10 min, and dsRNA was extracted according to the method described by Timmons et al.55 and Dong et al.56. Nucleic acids were analyzed for appropriate size by 1% agarose gel electrophoresis.
Newly hatched larvae were allowed to feed for 48 h at 27 °C on an artificial diet to which either 50 μg/cm2 Se-target genes, EGFP dsRNA, or water, had been added. The larvae were then transferred to the wells of a 6-well plate where they were allowed to continue feeding for 7 days at 27 °C. Each well contained 5 ml of artificial diet plus either 2.6 μg/cm2 activated Cry2Aa toxin (equivalent to the LC70 value determined by a pilot study), or water (the control). Five replicates were conducted with a total of 120 larvae in each treatment.
qPCR assay
Three groups of samples were prepared, including different tissues, developmental stages and dsRNA treatment groups (three replicates for each treatment). Quantitative real-time PCR (qPCR) was used to measure differences in gene expression between tissues, developmental stages and dsRNA treatment groups. qPCR primers were designed using the NCBI profile server (http://www.ncbi.nlm.nih.gov/tools/primer-blast) (Supplementary Tables S1–S5). S. exigua RpL10 and GAPDH were used as internal reference genes (Supplementary Table S6)54,57. The following standard qPCR protocol was used: denaturation at 95 °C for 30 s, followed by 40 cycles of 95 °C for 10 s and 59 °C for 30 s. All qPCR samples were run in triplicate using SYBR Premix ExTaq™ (TaKaRa) and a Bio-Rad Detection iQ2 System. Melting curve analysis from 55 to 95 °C was performed to determine the specificity of the qPCR primers. To determine the efficiency of qRT-PCR primers, a 5-fold dilution series of second-instar larval cDNA corresponding to 1 μg total RNA was used to produce a standard curve (cDNA concentration vs. Ct) with efficiencies calculated from the slope using linear regression. The corresponding qRT-PCR efficiencies were calculated according to the equation: E = (10[1/slope] − 1)*10058,59 (Supplementary Table S7).
Data analysis
Quantitative expression data were analyzed using the 2−∆∆Ct method58. Corrected larval mortalities were calculated using Abbott’s formula60. Means and variances of treatments were analyzed with one-way ANOVA implemented in SPSS for Windows (SPSS 18.0, Chicago, IL, USA).
Additional Information
How to cite this article: Qiu, L. et al. Proteomic analysis of Cry2Aa-binding proteins and their receptor function in Spodoptera exigua. Sci. Rep. 7, 40222; doi: 10.1038/srep40222 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Bravo, A., Likitvivatanavong, S., Gill, S. S. & Soberon, M. Bacillus thuringiensis: A story of a successful bioinsecticide. Insect Biochem Mol Biol 41, 423–431 (2011).
Chen, H. et al. Transgenic indica rice plants harboring a synthetic cry2A* gene of Bacillus thuringiensis exhibit enhanced resistance against lepidopteran rice pests. Theor Appl Genet 111, 1330–1337 (2005).
Adamczyk, J. J. et al. Evaluations of bollgardR, bollgard IIR, and widestrikeR technologies against beet and fall armyworm larvae (lepidoptera: noctuidae). Fla Entomol 91, 531–536 (2008).
James, C. Global Status of Commercialized Biotech/GM Crops: 2015. ISAAA: Ithaca, NY (2015).
Wu, K. M., Lu, Y. H., Feng, H. Q., Jiang, Y. Y. & Zhao, J. Z. Suppression of cotton bollworm in multiple crops in China in areas with Bt toxin–containing cotton. Science 321, 1676–1678 (2008).
Wan, P. et al. Increased frequency of pink bollworm resistance to Bt toxin Cry1Ac in China. PLoS ONE 7, e29975 (2012).
Zhang, H. et al. Diverse genetic basis of field-evolved resistance to Bt cotton in cotton bollworm from China. Proc Natl Acad Sci USA 109, 10275–10280 (2012).
Lu, Y. et al. Mirid bug outbreaks in multiple crops correlated with wide-scale adoption of Bt cotton in China. Science 328, 1151–1154 (2010).
Liu, Y. & Jiang, X. Biological control of Spodoptera exigua (Hb.). Plant Prot 28, 54–56 (2002).
Wan, P., Wu, K., Huang, M., Yu, D. & Wu, J. Population dynamics of Spodoptera litura (Lepidoptera: Noctuidae) on Bt cotton in the Yangtze River valley of China. Environ Entomol 37, 1043–1048 (2008).
Lu, X. Y., Zhang, L. M., Luo, J. Y., Wang, C. Y. & Cui, J. J. The effectiveness of Cry2Ab protein against early-instar larvae of Spodoptera exigua and S. litura . J Biosafety 22, 109–14 (2013).
Ferré, J. & Van Rie, J. Biochemistry and genetics of insect resistance to Bacillus thuringiensis . Annu Rev Entomol 47, 501–533 (2002).
Zhao, J. Z. et al. Concurrent use of transgenic plants expressing a single and two Bacillus thuringiensis genes speeds insect adaptation to pyramided plants. Proc Natl Acad Sci USA 102, 8426–8430 (2005).
Sohail, M. N. et al. Development of broad-spectrum insect-resistant tobacco by expression of synthetic cry1Ac and cry2Ab genes. Biotechnol Lett 34, 1553–1560 (2012).
Stewart, S. D., Adamczyk, J. J., Knighten, K. S. & Davis, F. M. Impact of Bt cottons expressing one or two insecticidal proteins of Bacillus thuringiensis Berliner on growth and survival of noctuid (Lepidoptera) larvae. J Econ Entomol 94, 752–760 (2001).
Avisar, D. et al. The Bacillus thuringiensis delta-endotoxin Cry1C as a potential bioinsecticide in plants. Plant Sci 176, 315–324 (2009).
Zheng, X., Wang, P., Wang, X. & Lei, C. Damage, occurrence and control of Spodoptera exigua on transgenic cottons. Plant Prot 36, 34–38 (2010).
Hernandez-Martinez, P., Ferre, J. & Escriche, B. Susceptibility of Spodoptera exigua to 9 toxins from Bacillus thuringiensis . J Invertebr Pathol 97, 245–250 (2008).
Lu, Q. et al. A fragment of cadherin-like protein enhances Bacillus thuringiensis Cry1B and Cry1C toxicity to Spodoptera exigua (Lepidoptera: Noctuidae). J Integr Agric 11, 628–638 (2012).
Kota, M. et al. Overexpression of the Bacillus thuringiensis (Bt) Cry2Aa2 protein in chloroplasts confers resistance to plants against susceptible and Bt-resistant insects. Proc Natl Acad Sci USA 96, 1840–1845 (1999).
De Cosa, B., Moar, W., Lee, S. B., Miller, M. & Daniell, H. Overexpression of the Bt cry2Aa2 operon in chloroplasts leads to formation of insecticidal crystals. Nat Biotechnol 19, 71–74 (2001).
Chitkowski, R. L., Turnipseed, S. G., Sullivan, M. J. & Bridges, W. C., Jr. Field and laboratory evaluations of transgenic cottons expressing one or two Bacillus thuringiensis var. kurstaki Berliner proteins for management of noctuid (Lepidoptera) pests. J Econ Entomol 96, 755–762 (2003).
Qiu, L. et al. Cadherin is involved in the action of Bacillus thuringiensis toxins Cry1Ac and Cry2Aa in the beet armyworm, Spodoptera exigua . J Invertebr Pathol 127, 47–53 (2015).
Kumar, S. & Udayasuriyan, V. Cloning of cry2Aa and cry2Ab genes from new isolates of Bacillus thuringiensis and their expression in recombinant Bacillus thuringiensis and Escherichia coli strains. World J Microbiol Biotechnol 20, 11–17 (2004).
McNeil, B. C. & Dean, D. H. Bacillus thuringiensis Cry2Ab is active on Anopheles mosquitoes: single D block exchanges reveal critical residues involved in activity. FEMS Microbiol Lett 325, 16–21 (2011).
Kakinuma, Y., Ohsumi, Y. & Anraku, Y. Properties of H+-translocating adenosine triphosphatase in vacuolar membranes of Saccharomyces cerevisiae . J Biol Chem 256, 10859–10863 (1981).
Nishi, T. & Forgac, M. The vacuolar (H+)-atpases - Nature’s most versatile proton pumps. Nat Rev Mol Cell Biol 3, 94–103 (2002).
Beyenbach, K. W. & Wieczorek, H. The V-type H+ ATPase: molecular structure and function, physiological roles and regulation. J Exp Biol 209, 577–589 (2006).
Wieczorek, H. et al. Structure and regulation of insect plasma membrane H(+)V-ATPase. J Exp Biol 203, 127–135 (2000).
Candas, M., Loseva, O., Oppert, B., Kosaraju, P. & Bulla, L. A., Jr. Insect resistance to Bacillus thuringiensis: alterations in the indianmeal moth larval gut proteome. Mol Cell Proteomics 2, 19–28 (2003).
Yuan, C. et al. Proteomic analysis of BBMV in Helicoverpa armigera midgut with and without Cry1Ac toxin treatment. Biocontrol sci technol 21, 139–151 (2011).
Krishnamoorthy, M., Jurat-Fuentes, J. L., McNall, R. J., Andacht, T. & Adang, M. J. Identification of novel Cry1Ac binding proteins in midgut membranes from Heliothis virescens using proteomic analyses. Insect Biochem Mol Biol 37, 189–201 (2007).
Chen, L. Z. et al. Proteomic analysis of novel Cry1Ac binding proteins in Helicoverpa armigera (Hübner). Arch Insect Biochem Physiol 73, 61–73 (2010).
Xu, L. et al. A proteomic approach to study the mechanism of tolerance to Bt toxins in Ostrinia furnacalis larvae selected for resistance to Cry1Ab. Transgenic Res 22, 1155–1166 (2013).
Cancino-Rodezno, A. et al. Comparative proteomic analysis of Aedes aegypti larval midgut after intoxication with Cry11Aa toxin from Bacillus thuringiensis . PLoS ONE 7, e37034 (2012).
Jurat-Fuentes, J. L. & Adang, M. J. Cry toxin mode of action in susceptible and resistant Heliothis virescens larvae. J Invertebr Pathol 92, 166–171 (2006).
Knowles, B. H. Mechanism of action of Bacillus thuringiensis insecticidal δ-endotoxins. Adv In Insect Phys 24, 275–308 (1994).
English, L. H. & Cantley, L. C. Delta endotoxin is a potent inhibitor of the (Na, K)-ATPase. J Biol Chem 261, 1170–1173 (1986).
Saier, M. H. Jr. A functional-phylogenetic classification system for transmembrane solute transporters. Microbiol Mol Biol Rev 64, 354–411 (2000).
Contreras, E., Schoppmeier, M., Real, M. D. & Rausell, C. Sodium solute symporter and cadherin proteins act as Bacillus thuringiensis Cry3Ba toxin functional receptors in Tribolium castaneum . J Biol Chem 288, 18013–18021 (2013).
Gahan, L. J., Pauchet, Y., Vogel, H. & Heckel, D. G. An ABC transporter mutation is correlated with insect resistance to Bacillus thuringiensis Cry1Ac toxin. PLoS Genet 6, e1001248 (2010).
Tanaka, S. et al. The ATP-binding cassette transporter subfamily C member 2 in Bombyx mori larvae is a functional receptor for Cry toxins from Bacillus thuringiensis . FEBS J 280, 1782–1794 (2013).
Park, Y. et al. ABCC transporters mediate insect resistance to multiple Bt toxins revealed by bulk segregant analysis. BMC Biol 12, 15 (2014).
Campbell, P. M., Cao, A. T., Hines, E. R., East, P. D. & Gordon, K. H. J. Proteomic analysis of the peritrophic matrix from the gut of the caterpillar, Helicoverpa armigera . Insect Biochem Mol Biol 38, 950–958 (2008).
Macieira, S., Zhang, J., Velarde, M., Buckel, W. & Messerschmidt, A. Crystal structure of 4-hydroxybutyrate CoA-transferase from Clostridium aminobutyricum . Biol Chem 390, 1251–1263 (2009).
Ron, D. et al. Cloning of an intracellular receptor for protein kinase C: a homolog of the beta subunit of G proteins. Proc Natl Acad Sci USA 91, 839–843 (1994).
Han, L. et al. Binding site concentration explains the differential susceptibility of Chilo suppressalis and Sesamia inferens to Cry1A-producing rice. Appl Environ Microbiol 80, 5134–5140 (2014).
Garbis, S., Lubec, G. & Fountoulakis, M. Limitations of current proteomics technologies. J Chromatogr A 1077, 1–18 (2005).
Wolfersberger, M. et al. Preparation and partial characterization of amino acid transporting brush border membrane vesicles from the larval midgut of the cabbage butterfly (Pieris brassicae). Comp Biochem Physiol A Mol Integr Physiol 86, 301–308 (1987).
Bradford, M. M. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72, 248–253 (1976).
Saitou, N. & Nei, M. The neighbor-joining method: a new method for recons tructing phylogenetic trees. Mol Biol Evol 4, 406–425 (1987).
Tamura, K., Dudley, J., Nei, M. & Kumar, S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24, 1596–1599 (2007).
Chen, J., Likitvivatanavong, S., Aimanova, K. G. & Gill, S. S. A 104 kDa Aedes aegypti aminopeptidase N is a putative receptor for the Cry11Aa toxin from Bacillus thuringiensis subsp. israelensis . Insect Biochem Mol Biol 43, 1201–1208 (2013).
Ren, X. L. et al. A Spodoptera exigua cadherin serves as a putative receptor for Bacillus thuringiensis Cry1Ca toxin and shows differential enhancement of Cry1Ca and Cry1Ac toxicity. Appl Environ Microbiol 79, 5576–5583 (2013).
Timmons, L., Court, D. L. & Fire, A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans . Gene 263, 103–112 (2001).
Dong, X., Li, Q. & Zhang, H. The noa gene is functionally linked to the activation of the Toll/Imd signaling pathways in Bactrocera dorsalis (Hendel). Dev Comp Immunol 55, 233–240 (2016).
Zhu, X. et al. Selection and evaluation of reference genes for expression analysis using qRT-PCR in the beet armyworm Spodoptera exigua (Hubner) (Lepidoptera: Noctuidae). PLoS ONE 9, e84730 (2014).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29, e45 (2001).
Radonić, A. et al. Guideline to reference gene selection for quantitative real-time PCR. Biochem Biophys Res Commun 313, 856–862 (2004).
Abbott, W. S. A method of computing the effectiveness of an insecticide. 1925. J Am Mosq Control Assoc 3, 302–303 (1987).
Acknowledgements
This work was funded by grants from Ministry of Agriculture of China (Grant No. 2016ZX08011002) and National Natural Science Foundation of China (grant no. 31101445).
Author information
Authors and Affiliations
Contributions
L.Q., B.Z. and L.L. performed the experiments. W.M., C.L., X.W. and L.C. conceived and designed the experiments. L.Q. and L.C. analyzed the data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Qiu, L., Zhang, B., Liu, L. et al. Proteomic analysis of Cry2Aa-binding proteins and their receptor function in Spodoptera exigua. Sci Rep 7, 40222 (2017). https://doi.org/10.1038/srep40222
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep40222
This article is cited by
-
Identifying the Epitopes of Bacillus thuringiensis Cry2Aa Toxin Involved in Cadherin Interaction by a Monoclonal Antibody
Applied Biochemistry and Biotechnology (2023)
-
Functional validation of DvABCB1 as a receptor of Cry3 toxins in western corn rootworm, Diabrotica virgifera virgifera
Scientific Reports (2020)
-
Synergism of the Bacillus thuringiensis Cry1, Cry2, and Vip3 Proteins in Spodoptera frugiperda Control
Applied Biochemistry and Biotechnology (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
