
Abstract
Ancestry inference is of great interest in diverse areas of scientific researches, including the forensic biology, medical genetics and anthropology. Various methods have been published for distinguishing populations. However, few reports refer to sub-populations (like ethnic groups) within Asian populations for the limitation of markers. Several InDel loci located very tightly in physical positions were treated as one marker by us, which is multi-InDel. The multi-InDel shows potential as Ancestry Inference Marker (AIM). In this study, we performed a genome-wide scan for multi-InDels as AIM. After examining the FST distributions in the 1000 Genomes Database, 12 candidates were selected and validated for eastern Asian populations. A multiplexed assay was developed as a panel to genotype 12 multi-InDel markers simultaneously. Ancestry component analysis with STRUCTURE and principal component analysis (PCA) were employed to estimate its capability for ancestry inference. Furthermore, ancestry assignments of trial individuals were conducted. It proved to be very effective when 210 samples from Han and Tibetan individuals in China were tested. The panel consisting of multi-InDel markers exhibited considerable potency in ancestry inference, and was suggested to be applied in forensic practices and genetic population studies.
Similar content being viewed by others

Introduction
A fair amount of Ancestry Information Markers (AIMs) have been proposed for various purposes including detecting stratification in biomedical studies1,2 and determining an individual’s ancestry from a forensic context3,4,5,6,7,8,9,10,11,12,13,14. In the genotype-phenotype studies, such as the popular genome-wide association studies (GWAS), the presence of uncontrolled population structure may lead to false-positive or false-negative results15,16,17,18,19. To eliminate the adverse impact to the outcomes, AIMs are used to detect and control the potential population stratification derived from genetic ancestry. Besides, in the forensic practices, ancestry information could provide vital clues and narrow down the investigation scope, when existing profiling is unsuccessful, no DNA database matches are obtained or in the absence of reliable eyewitness testimony. In reality, samples from forensic contexts are always challenging, with limited quantity or poor quality. Therefore, a small-scale but robust panel of AIMs is preferred. In fact, it is the capability of discerning subtle differences between closely related populations, which may share many similarities in evolution or/and close residences in geography, that matters in the practical police investigations. Such researches20,21,22 have been reported for some special regions. Yet it is absent in eastern Asian. Regarding to this range, Chinese Han population is naturally the ideal study object. As the largest ethnic group in the world, it comprises about 20% of the global human population. Chinese Tibetan population also deserved to be studied for the unique genetic characteristics inherited from their ancestries, which adapt the Tibetan people to the plateau environment. Researches on ancestry analyses of these two ethnic populations are undoubtedly beneficial to clinical association studies as well as forensic identifications. No results have been published yet.
Small molecular regions comprised of two or more SNPs have been reported to convey more identity and ancestry-related information9,23. Systems consisting of such multi-allelic haplotype markers have also been defined and advocated developing for forensic use, because they could serve to identify relatives with higher probabilities than simple di-allelic SNPs24. Replacing SNPs with InDels, a novel concept termed multi-InDel has been reported to behave well in parentage tests and to be hopefully used for forensic applications in the near future25,26. Meanwhile, the prospect of this new kind of markers in the population genetic studies deserves to be explored and discovered, especially that regarding population stratification and ancestry inference. Reasons are listed as below: (1) The looselinkage9 multi-InDel markers adopted shows conservation of evolutionary lineages. They have evolved from the ancestral human haplotype without evidence of recurring recombination; (2) Different from the traditional complete linkage disequilibrium (LD), historic recombination may occurred, while the recombinants have drifted to sufficiently high frequency to become common haplotypes; (3) With high inter-population variability, InDel is perfectly suitable for population genetic researches. Furthermore, the feasibility and practicability of this research are significantly enhanced thanks to the advantages of InDels including, ubiquity throughout the genome, second only to SNPs; well characterized27,28; low mutation rates; simplicity of laboratory analysis, like routine capillary electrophoresis (CE) typing; short amplicon strategy that could be adopted for degraded DNA samples; as well as the possibility of genotyping several markers in a single PCR multiplex reaction.
Based on above, we made a bold speculation that multi-InDel might do well in the study of population stratification, even ancestry inference in admixed populations. And we proved it through an example of a multiplex assay including 12 multi-InDel markers in this paper.
Materials and Methods
Ethics Statement
Human blood samples were collected upon approval of Ethics Committee at Sichuan University, P. R. China. A written informed consent was obtained from each participant in this study. This study was approved by the Ethics Committee of Sichuan University, P. R. China.
Sample Collection and DNA Extraction
A total of 210 blood samples were collected, among which 110 samples from Chinese Tibetan individuals were collected in Tibet Autonomous Region and 100 samples from Han individuals were collected in our laboratory. All the samples were obtained with written inform content and self-declared ancestry information according to quality control procedures. Genomic DNAs were extracted from peripheral blood samples or bloodstain samples using the Chelex-100 method as described by Walsh et al.29. DNA was quantitated using the NanoDrop 1000 Spectrophotometer (Thermo Fisher, MA, USA). In addition, samples for known cryptically related individuals were removed30.
Marker Selection
Candidate Indels were chosen from the 1000 Genomes database31,32 (http://www.ncbi.nlm.nih.gov/variation/tools/) when the following principles were both met: (a) FST > 0.4 when comparison was performed between any two of the five continental populations (AFR, African; EAS, East Asian; EUR, European; SAS, South Asian; AMR, Admixed American); and (b) FST > 0.15 when comparison was performed between any two of the five sub-populations in EAS (CHS, Southern Han Chinese, China; CHB, Han Chinese in Beijing, China; JPT, Japanese in Tokyo, Japan; KHV, Kinh in Ho Chi Minh City, Vietnam; CDX, Chinese Dai in Xishuangbanna, China). Based on these core loci, additional criterions were applied to construct multi-InDel markers, including: (i) the physical distance between the InDels in one multi-InDel marker was less than 250 bp for the formation; (ii) there were at least two haplotypes for each multi-InDel marker to ensure the heterozygosity; and (iii) the amplicon length was less than 300 bp.
The scanning procedure focuses mainly on the formation of combined InDel markers and the high level of FST parameters for studied populations. Specific steps include (1) calculating FST values for subject populations as described by Weir and Cockerham33; (2) sorting FST and screening loci with standard parameters meeting the preset criteria; and (3) checking flanking regions for proper combinations of InDels with extremely short physical distances.
Flanking regions of the selected multi-InDel markers were also obtained from the University of California Santa Cruz Genome Browser (Human February 2009 Assembly; GRCh37/hg19) at http://genome.ucsc.edu/. Sequences were checked for variants and polymorphism structures, which are likely to interfere with primer design or data analysis.
Primer Design and Optimization
Primer designing was performed with the Primer Premier v5.0 (PREMIER Biosoft, Palo Alto, CA, USA), applying the following criteria: PCR product size from 70 to 300 bp; Tm values from 52 to 60 °C and GC content from 30 to 60%. The “PIGtail” or partial tails of the original GTTTCTT nucleotide sequence were added to the 5’end of the unlabeled primers to promote full adenylation34. The obtained primer pairs were examined for potential interactions with each other using AutoDimer software35. They were also checked for non-specific hybridizations in other genome regions using the National Center for Biotechnology Information (NCBI) Basic Local Alignment Search Tool (BLAST) at http://blast.ncbi.nlm.nih.gov/Blast.cgi. All the markers were then schematically organized by expected amplicon length and assigned into four different dye-labeling fluorochromes (FAM, HEX, TAMRA, and ROX) (Applied Biosystems, Foster City, CA).
Optimizations of the multiplex assay were performed on the basis of primer concentrations and peak heights. After that, 210 collected Chinese samples from Han and Tibetan individuals were genotyped with the developed PCR system. Based on the size information of each haplotype, Panel and Bin files were programmed.
PCR Setup and CE Genotyping
The PCR system was a 12.5 μL reaction volume containing 1× Qiagen multiplex PCR master mix, 1 × Q-Solution, 2 μL primer mix and 0.5–2 ng of template gDNA. Thermal cycling conditions consisted of an initial step at 95 °C for 15 min; 32 cycles at 94 °C for 30 s, 56 °C for 90 s, and 72 °C for 60 s; and a final extension at 60 °C for 45 min. For the Tm value, 54 °C, 56 °C and 58 °C were chosen for testing.
PCR products were analyzed by mixing 1 μL of each amplified product with 9 μL in a 17:1 mixture of Hi-Di formamide (Applied Biosystem, Foster City, CA) and SIZ 500 (AGCU Co, China) for CE. Fragment detection and separation were performed on ABI Prism 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA). Genotyping data was analyzed with GeneMapper v3.2.1 software (Applied Biosystems, Foster City, CA). Default setting of peak height (above 200 RFU) was applied for the analysis. The positive controls (9947A) and negative controls (DI water) were performed by the same reaction condition.
Analytical Method
Haplotype frequencies were determined by counting. Analyses of genetic parameters such as FST values were performed with vcftools_0.1.12b36. PCA analysis based on the genotypes of the 12 multi-InDel markers was operated with SPSS 16.0.
Individual ancestry components were primarily examined via model-based clustering algorithms implemented in STRUCTURE 2.3.437, which was based on a Bayesian Markov Chain Monte Carlo algorithm. Analyses were performed with five replicates from K = 1 to K = 7 using the no-admixture model and correlated allele frequencies (100,000 burn-ins iterations and 10,000 MCMC repeats) to estimate the selected 12 multi-Indel markers. Structure Harvester38 was applied to estimate the optimum K value. Results of ancestry proportions were compared to the self-identified ancestry. We ran STRUCTURE for increased K values to observe if the multi-InDel panel had the potential to discern more subtle structures in the selected samples.
Inference of ancestry affiliation was estimated using the Snipper Classify using frequencies option (at: http://mathgene.usc.es/snipper/ “Classification with the 32 STR training set or a custom Excel file of frequencies”), a direct link provided by the Snipper portal. Ten out of the 210 collected Chinese samples from Han and Tibetan individuals were randomly chosen as blind trials. The rest were developed to the training set, an Excel-based data input system using one worksheet per marker (alleles as columns, populations as rows) listing allele frequencies in each cell. Ancestry assignments were performed to test the accuracy of this inference system with the 12 multi-InDel markers.
Quality Control
The main experiments were conducted at the Forensic Genetics Laboratory of West China School of Basic Science and Forensic Medicine, Sichuan University, P.R. China, which is an accredited laboratory by ISO 17025. All the methods were carried out in accordance with the approved guidelines of Institute of Forensic Medicine, West China School of Basic Science and Forensic Medicine, Sichuan University, P.R. China.
Results and Discussion
Multi-InDel Markers Chosen for distinguishing Ethnic Populations
After data analysis and loci screening based on data resource from 1000 Genomes Database, experiment validations were additionally conducted to eliminate some loci with which we cannot obtain stable results. Among various reasons, successful multiplex PCR for all selected markers is the first to be taken into consideration. Primers for each locus were initially tested in a singleplex PCR reaction to evaluate the performance. The criteria for primer “failure” are defined as those that produce profiles that exhibit incomplete adenylation, the presence of PCR artifacts, low signal, nonspecific products, or no PCR products at all. Once the successful primers at each locus were determined, those were equally combined together for a primer mix of 0.1 μM at first. Based on the results of genotyping profiles, the optimization of each primer’s concentrations in the final primer mix was performed. Furthermore, successful PCR without stable performance in the replicated experiments are eliminated, too. Situations are filtered out such like (1) repeated sequences appear in the flanking region of candidate loci; (2) core sequences are variable; and (3) SNPs are detected in the primer binding regions. A total of 12 multi-InDel markers picked into the panel are all robust, accurate, specific and sensitive to ensure the integral efficacy of the panel as a whole. Detail information is presented in Table 1, including the rs numbers, localizations, and alleles of the contained InDels. Multi-InDel No.5 is an example of two InDels (rs568000255 and rs148177611, 65 bp apart) defining three haplotypes in the 210 collected individuals, namely 122, 126, and 127 according to their amplicon size, as illustrated in Fig 1. For each haplotype, distributions in the two sub-populations are strikingly different, which guarantees the capability of the marker to differ one population from the other. Coupled with the other 11 multi-InDel markers, especially that containing more InDels, the assay developed will be sufficiently informative for ancestry inference.

Haplotype frequency distributions of Multi-Indel No.5 in Han and Tibetan populations in China.
Blue bars denote allele122, red and green bars alleles 126 and 127, respectively.
Unlike other AIMs2,12,20,21,39, we chose InDel as the core unit of the markers. InDel is suitable for population analysis, since the allele distributions show significant differences in different geographic regions40,41. Studies based on simple InDels have proved its great potential in ancestry inference researches3,7,42,43. FST and In are commonly used to measure the ancestral information of AIMs. However, a high correlation has been found between these two parameters2. In this study, we treated FST value as the leading factor among the selecting criteria, since it concerns the power of differentiation more directly and typically. Population-specific markers were preferred. They included loci with a polymorphism detected in one population but absent in the other or those with a common allele in one population that was rare in others. In the process of selection, we arbitrarily took FST = 0.15 as the lower limit when sub-populations within EAS were compared to each other so as to balance the number of markers and the power of each marker. In this way, the total power of differentiation was guaranteed. Additionally, continental populations were compared with each other for the warranty of real difference in the allele distributions, which is essential to the aim of ancestry inference.
For multi-InDels, multiple haplotypes with heterozygosity are defined depending on the accumulation of the variants at the different sites, the occurrence of rare crossovers historically, the vagaries of random genetic drift, and/or selection23. Profiles with multiple haplotypes provide more information than that with simple di-allelic markers in ancestry inference as well as individual identification or even identifying biological relatives. Generally, increasing the number of genetic markers applied is a conventional approach to achieve more reliable results. While thanks to the sufficient heterozygosity, similar effectiveness could be obtained with a small-scale of multi-InDel markers without any sacrifice of stability or ancestry information.
Twelve Multi-InDel Markers in One Panel
The final panel was composed of 12 multi-InDel markers, which can be amplified with reproducibility in a fast and easy PCR multiplex reaction followed by electrophoresis. The primer information such as sequences, Tm values and concentrations in the final multiplex was listed in Table 2. The panel proved to be capable of amplifying all of the markers in samples containing different amounts of largely variable DNA (0.1–10 ng). In this effective detection range, full and clear genotyping profiles were obtained from all tested samples. Signal strength variation and stutters appearing in some individual samples with extremely high/low DNA concentration, albeit slightly higher than those of kit markers, did not interfere markedly with profile interpretation. Moreover, the length of a multi-InDel marker was restricted to smaller than 300 bp. Supposing a genome-wide average of ~1% recombination per mega base and no recombination hot spots within the locus, an extent of <300 bp for a multi-InDel marker is projected to bring the recombination rate down to a value comparable to the mutation rate for SNPs. Very rare historical recombination events remained to assume identity by descent within a family after the exclusion of recombination hot spot. At the same time, the small-amplicon strategy diminishes allele and locus dropout and improves the chances of successful analysis of challenging samples with degraded DNA.
Validation of the 12 Multi-InDel Markers as AIMs
Several strategies including ancestry component analysis with STRUCTURE, ancestry affiliation prediction with snipper as well as unsupervised cluster analysis with PCA were performed to estimate the developed panel.
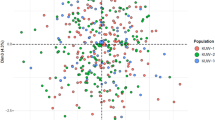
210 Chinese samples from Han and Tibetan individuals were combined to perform an ancestry component analysis. STRUCTURE runs for K = 2–4 are shown in Fig 2. Optimum K value was estimated to be K = 2 by Structure Harvester. Bar plot of K = 2 analysis reveals that all the collected samples can be primarily separated into two clusters, with one color bar representing each continental origin. The two principal components dominate in Chinese Tibetan and Han, respectively. Additionally, a slight flow representing Han component appears in the supposed Tibetan region, which may be explained by the historical features of Han, such as complex ancestral origin, long history of interaction with surrounding ethnic groups and recent migrations. Nevertheless, Chinese Tibetan displays predominantly component by itself, which is demonstrated more clearly by the triangle plot. No more subtle stratification was observed by further increasing the K value. Compared with previous researches13, we distinguish sub-populations in a further step.

Ancestry component analysis result for 210 Chinese samples from Han and Tibetan individuals with the 12 selected multi-InDel markers.
(a) Bar plot of STRUCTURE runs for K = 2–4. Chinese samples from Tibetan and Han individuals are marked by Arabic numerals 1 and 2, respectively. The bars with different colors represent different ancestry origins from the analyses. Structure Harvester estimated the optimum K value to be K = 2 (marked by blue arrows in the Ln estimated probabilities plots). (b) Triangle plot of STRUCTURE runs for K = 2. (c). 2-dimensional plot of PCA analysis. The first two principal components (PC1 vs. PC2) demonstrate the population stratification in the tested samples. PC1 is represented by the X axis, and PC2 is represented by the Y axis.
To interpret the inference results in a comprehensive and straightforward manner, we performed an assignment test using snipper. All the trial samples were accurately assigned into their self-identified populations based on the training set. No misclassification occurred during the test of ancestry inference. Snipper, a Bayesian ancestry analysis system, was designed for forensic AIM-SNPs initially. Ancestry assignments of SNP genotype profiles are derived from the ratio of the two highest likelihoods from multiple population comparisons with likelihoods calculated from allele frequencies estimated using training. Later, Snipper has been improved to handle the multiple alleles of STRs by accepting training sets of user-input allele frequencies rather than genotypes6. Results from this web-based tool are clear and easy to understand with the familiar way we adopted in the routine forensic practice.
Result of the PCA test is shown in Fig 2 too. 210 Chinese samples from Han and Tibetan individuals are spread along PC1 (the X axis), suggesting population stratification in tested samples, although not so pronounced as that in the ancestry component analysis through STRUCTURE. The main tendency clearly indicates two divergent genetic structures though a little overlap appears between them. The clustering of the collected samples was mainly explained by PC1 and PC2, while other PCs were much less informative. And no discernible structure was detected in other combinations of PCs. Compared with that of Tibetan, samples of Han individual are more dispersed in the 2-dimensional PCA plot, reflecting the complex genetic background of Han. Unlike model-based algorithm STRUCTURE implemented or Bayesian analysis snipper adopted, PCA is a classical nonparametric linear dimensionality reduction technique, extracting the fundamental structure of a dataset without the need for any modeling. It has recently been shown to be powerful for the identification of population structure and the correction of stratification in the setting of association studies44,45. Coupled with a clustering tool, it can also be used for inferring population clusters and assigning individuals to sub-populations46.
In sum, using the 12 multi-InDel markers, we obtained a perfect classification in ancestry component analysis with STRUCTURE. Besides, ancestry affiliation prediction with snipper also assigned trial samples to their correct ethnicity in our test. Results of PCA clustering showed that the separation of the two ethnic populations was clear and definite.
Conclusion
A multiplex assay with 12 multi-InDel markers as AIM was developed in this study. In validation, 210 Chinese individuals from Han and Tibetan populations were separated into two clusters in accordance with their self-declared ethnic information both in model-based analysis by STRUCTURE and in the unsupervised way, PCA. The ethnic affiliations tests using the web-based tool- snipper revealed no mismatch. By constructing multi-InDel markers as the AIMs, we provided a set of markers with improved performance in distinguishing and clustering two closely resided sub-populations. It is a helpful and beneficial exploration for applications on ancestry inference, not only in forensic practices but also in population genetics.
Additional Information
How to cite this article: Sun, K. et al. Multi-InDel Analysis for Ancestry Inference of Sub-Populations in China. Sci. Rep. 6, 39797; doi: 10.1038/srep39797 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Tang, H. et al. Genetic structure, self-identified race/ethnicity, and confounding in case-control association studies. Am J Hum Genet 76, 268–275, doi: 10.1086/427888 (2005).
Qin, P. et al. A panel of ancestry informative markers to estimate and correct potential effects of population stratification in Han Chinese. Eur J Hum Genet 22, 248–253, doi: 10.1038/ejhg.2013.111 (2014).
Romanini, C. et al. Ancestry informative markers: inference of ancestry in aged bone samples using an autosomal AIM-Indel multiplex. Forensic Sci Int Genet 16, 58–63, doi: 10.1016/j.fsigen.2014.11.025 (2015).
Phillips, C. et al. Inferring ancestral origin using a single multiplex assay of ancestry-informative marker SNPs. Forensic Science International-Genetics 1, 273–280, doi: 10.1016/j.fsigen.2007.06.008 (2007).
Phillips, C. et al. Building a forensic ancestry panel from the ground up: The EUROFORGEN Global AIM-SNP set. Forensic Sci Int Genet 11, 13–25, doi: 10.1016/j.fsigen.2014.02.012 (2014).
Phillips, C. et al. Development of a novel forensic STR multiplex for ancestry analysis and extended identity testing. Electrophoresis 34, 1151–1162, doi: 10.1002/elps.201200621 (2013).
Pereira, R. et al. Straightforward inference of ancestry and admixture proportions through ancestry-informative insertion deletion multiplexing. PLoS One 7, e29684, doi: 10.1371/journal.pone.0029684 (2012).
Paschou, P. et al. PCA-correlated SNPs for structure identification in worldwide human populations. PLoS Genet 3, 1672–1686, doi: 10.1371/journal.pgen.0030160 (2007).
Pakstis, A. J., Fang, R. X., Furtado, M. R., Kidd, J. R. & Kidd, K. K. Mini-haplotypes as lineage informative SNPs and ancestry inference SNPs. European Journal of Human Genetics 20, 1148–1154, doi: 10.1038/ejhg.2012.69 (2012).
Libiger, O. & Schork, N. J. A Method for Inferring an Individual’s Genetic Ancestry and Degree of Admixture Associated with Six Major Continental Populations. Front Genet 3, 322, doi: 10.3389/fgene.2012.00322 (2012).
Kidd, K. K. et al. Progress toward an efficient panel of SNPs for ancestry inference. Forensic Sci Int Genet 10, 23–32, doi: 10.1016/j.fsigen.2014.01.002 (2014).
Kidd, J. R. et al. Analyses of a set of 128 ancestry informative single-nucleotide polymorphisms in a global set of 119 population samples. Investig Genet 2, 1, doi: 10.1186/2041-2223-2-1 (2011).
Jia, J. et al. Developing a novel panel of genome-wide ancestry informative markers for bio-geographical ancestry estimates. Forensic Sci Int Genet 8, 187–194, doi: 10.1016/j.fsigen.2013.09.004 (2014).
Fondevila, M. et al. Revision of the SNPforID 34-plex forensic ancestry test: Assay enhancements, standard reference sample genotypes and extended population studies. Forensic Sci Int Genet 7, 63–74, doi: 10.1016/j.fsigen.2012.06.007 (2013).
Pritchard, J. K. & Rosenberg, N. A. Use of unlinked genetic markers to detect population stratification in association studies. American Journal of Human Genetics 65, 220–228, doi: 10.1086/302449 (1999).
Freedman, M. L. et al. Assessing the impact of population stratification on genetic association studies. Nat Genet 36, 388–393, doi: 10.1038/ng1333 (2004).
Marchini, J., Cardon, L. R., Phillips, M. S. & Donnelly, P. The effects of human population structure on large genetic association studies. Nat Genet 36, 512–517, doi: 10.1038/ng1337 (2004).
Platt, A., Vilhjalmsson, B. J. & Nordborg, M. Conditions Under Which Genome-Wide Association Studies Will be Positively Misleading. Genetics 186, 1045–1052, doi: 10.1534/genetics.110.121665 (2010).
Price, A. L., Zaitlen, N. A., Reich, D. & Patterson, N. New approaches to population stratification in genome-wide association studies. Nat Rev Genet 11, 459–463, doi: 10.1038/nrg2813 (2010).
Phillips, C. et al. Eurasiaplex: A forensic SNP assay for differentiating European and South Asian ancestries. Forensic Science International-Genetics 7, 359–366, doi: 10.1016/j.fsigen.2013.02.010 (2013).
Santos, C. et al. Pacifiplex: an ancestry-informative SNP panel centred on Australia and the Pacific region. Forensic Sci Int Genet 20, 71–80, doi: 10.1016/j.fsigen.2015.10.003 (2016).
Galanter, J. M. et al. Development of a panel of genome-wide ancestry informative markers to study admixture throughout the Americas. PLoS Genet 8, e1002554, doi: 10.1371/journal.pgen.1002554 (2012).
Kidd, K. K. et al. Current sequencing technology makes microhaplotypes a powerful new type of genetic marker for forensics. Forensic Sci Int Genet 12, 215–224, doi: 10.1016/j.fsigen.2014.06.014 (2014).
Ge, J., Budowle, B., Planz, J. V. & Chakraborty, R. Haplotype block: a new type of forensic DNA markers. Int J Legal Med 124, 353–361, doi: 10.1007/s00414-009-0400-5 (2010).
Huang, J., Luo, H., Wei, W. & Hou, Y. A novel method for the analysis of 20 multi-Indel polymorphisms and its forensic application. Electrophoresis 35, 487–493, doi: 10.1002/elps.201300346 (2014).
Fan G, Y. Y., Luo, H & Hou, Y. Use of multi-InDels as novel markers to analyze 13 X-chromosome haplotype loci for forensic purposes. Electrophoresis (2015).
Wang, D. G. et al. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science 280, 1077–1082 (1998).
Sachidanandam, R. et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409, 928–933, doi: 10.1038/35057149 (2001).
Walsh, P. S., Metzger, D. A. & Higuchi, R. Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10, 506–513 (1991).
Pemberton, T. J., Wang, C. L., Li, J. Z. & Rosenberg, N. A. Inference of Unexpected Genetic Relatedness among Individuals in HapMap Phase III. American Journal of Human Genetics 87, 457–464, doi: 10.1016/j.ajhg.2010.08.014 (2010).
Genomes Project, C. et al. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073, doi: 10.1038/nature09534 (2010).
Genomes Project, C. et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65, doi: 10.1038/nature11632 (2012).
Cockerham, C. C. & Weir, B. S. Covariances of relatives stemming from a population undergoing mixed self and random mating. Biometrics 40, 157–164 (1984).
Brownstein, M. J., Carpten, J. D. & Smith, J. R. Modulation of non-templated nucleotide addition by taq DNA polymerase: Primer modifications that facilitate genotyping. Biotechniques 20, 1004-+ (1996).
Vallone, P. M. & Butler, J. M. AutoDimer: a screening tool for primer-dimer and hairpin structures. Biotechniques 37, 226–231 (2004).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158, doi: 10.1093/bioinformatics/btr330 (2011).
Falush, D., Stephens, M. & Pritchard, J. K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 164, 1567–1587 (2003).
Earl, D. A. & vonHoldt, B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genetics Resources 4, 359–361, doi: 10.1007/s12686-011-9548-7 (2011).
Silva, M. C. et al. Development of two multiplex mini-sequencing panels of ancestry informative SNPs for studies in Latin Americans: an application to populations of the State of Minas Gerais (Brazil). Genet Mol Res 9, 2069–2085, doi: 10.4238/vol9-4gmr911 (2010).
Mills, R. E. et al. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res 16, 1182–1190, doi: 10.1101/gr.4565806 (2006).
Weber, J. L. et al. Human diallelic insertion/deletion polymorphisms. Am J Hum Genet 71, 854–862, doi: 10.1086/342727 (2002).
Santos, C. et al. Completion of a worldwide reference panel of samples for an ancestry informative Indel assay. Forensic Sci Int Genet 17, 75–80, doi: 10.1016/j.fsigen.2015.03.011 (2015).
Santos, N. P. C. et al. Assessing Individual Interethnic Admixture and Population Substructure Using a 48-Insertion-Deletion (INSEL) Ancestry-Informative Marker (AIM) Panel. Human Mutation 31, 184–190, doi: 10.1002/humu.21159 (2010).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38, 904–909, doi: 10.1038/ng1847 (2006).
Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. PLoS Genet 2, e190, doi: 10.1371/journal.pgen.0020190 (2006).
Liu, N. & Zhao, H. A non-parametric approach to population structure inference using multilocus genotypes. Human Genomics 2, 353, doi: 10.1186/1479-7364-2-6-353 (2006).
Acknowledgements
We are very grateful to the volunteers in our study. This work was supported by grants from the Five- thirteenth National Science and Technology Support Program of China (2016YFC0800703) and from the National Natural Science Foundation of China (No.81571854). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
K.S. wrote the manuscript, Y.Y. and K.S. collected the samples, K.S. conducted the experiment, K.S. and T.L. analyzed the results, Y.H. conceived the experiment. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Sun, K., Ye, Y., Luo, T. et al. Multi-InDel Analysis for Ancestry Inference of Sub-Populations in China. Sci Rep 6, 39797 (2016). https://doi.org/10.1038/srep39797
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep39797
This article is cited by
-
Screening Indels from the whole genome to identify the candidates and their association with economic traits in several goat breeds
Functional & Integrative Genomics (2023)
-
Ancestry informative DIP loci for dissecting genetic structure and ancestry proportions of Qinghai Tibetan and Tibet Tibetan groups
Molecular Biology Reports (2020)
-
Population genetics, diversity and forensic characteristics of Tai–Kadai-speaking Bouyei revealed by insertion/deletions markers
Molecular Genetics and Genomics (2019)
-
Fuzzy set-based generalized multifactor dimensionality reduction analysis of gene-gene interactions
BMC Medical Genomics (2018)
-
Genetic structure and polymorphisms of Gelao ethnicity residing in southwest china revealed by X-chromosomal genetic markers
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
