
Abstract
EFhd2/Swiprosin-1 is a cytoskeletal Ca2+-binding protein implicated in Ca2+-dependent cell spreading and migration in epithelial cells. EFhd2 domain architecture includes an N-terminal disordered region, a PxxP motif, two EF-hands, a ligand mimic helix and a C-terminal coiled-coil domain. We reported previously that EFhd2 displays F-actin bundling activity in the presence of Ca2+ and this activity depends on the coiled-coil domain and direct interaction of the EFhd2 core region. However, the molecular mechanism for the regulation of F-actin binding and bundling by EFhd2 is unknown. Here, the Ca2+-bound crystal structure of the EFhd2 core region is presented and structures of mutants defective for Ca2+-binding are also described. These structures and biochemical analyses reveal that the F-actin bundling activity of EFhd2 depends on the structural rigidity of F-actin binding sites conferred by binding of the EF-hands to Ca2+. In the absence of Ca2+, the EFhd2 core region exhibits local conformational flexibility around the EF-hand domain and C-terminal linker, which retains F-actin binding activity but loses the ability to bundle F-actin. In addition, we establish that dimerisation of EFhd2 via the C-terminal coiled-coil domain, which is necessary for F-actin bundling, occurs through the parallel coiled-coil interaction.
Similar content being viewed by others

Introduction
More than 100 actin-related proteins exist in eukaryotic cells, and these proteins regulate the transition of actin polymerisation and depolymerisation to form highly complex structures1,2,3. Actin-related proteins are classified according to their specific functions in actin organisation, such as bundling (crosslinking), severing and capping of the actin cytoskeleton2,4. Higher ordered actin filaments are stabilised by many actin-bundling proteins that contain coiled-coil domains (cortexillin, SCAB1, coronin-1) and rod domains (α-actinin, villin) for self-association, which organise actin filaments into bundles as homodimers arranged in a parallel or antiparallel fashion. In addition, actin organisation activity of several actin-related proteins is controlled by cellular stimuli (Ca2+) and signals5,6,7,8,9.
Intracellular Ca2+ levels affect actin organisation in various ways. Several actin-related proteins contain EF-hands or Ca2+/CaM binding domains (see Supplementary Fig. S1). For example, caldesmon contains a Ca2+/CaM binding domain that is located close to actin-binding sites. At high Ca2+ concentrations (>1 μM), Ca2+/CaM binds to caldesmon and interferes with the binding of caldesmon to actin3,10. In addition, fimbrin and non-muscle α-actinin contain multiple calponin-homology (CH) domains and EF-hands. These proteins associate with actin through CH domains, and F-actin binding or bundling activity is inhibited by Ca2+ 11. Conformational changes to EF-hands upon Ca2+ binding has been postulated to disrupt the interaction between the CH domain and actin, because EF-hands are located proximal to CH domains (see Supplementary Fig. S1)11,12,13,14. For example, the structure of Ca2+-free EF-hands of non-muscle α-actinin-1 revealed a flexible conformation around the connecting linker between the N-lobe and C-lobe, and binding of Ca2+ to EF-hands induced structural rigidification, which affected the orientation of adjacent CH domains resulting in inhibition of F-actin crosslinking activity15. In some cases, such as gelsolin, villin, fragmin and severin, Ca2+ directly affects actin-related functions through binding to multiple actin-binding sites. These proteins show F-actin bundling activity at low Ca2+ concentrations (<0.1 μM), but actin filament severing activity at high Ca2+ concentrations. For these proteins, multiple actin-binding sites bind to F-actin in an open conformation at high Ca2+ concentrations, which leads to a transition from F-actin bundling to severing activity (see Supplementary Fig. S1)3,4,11,16.
EFhd2/Swiprosin-1 (EFhd2) is a cytoskeletal Ca2+-binding protein identified in human immune, brain and mast cells17,18,19. EFhd2 is highly conserved among homologous EF-hand-containing proteins, including EFhd1/Swiprosin-2 (EFhd1) and allograft inflammatory factor-1 (AIF-1). EFhd2 and EFhd1 consist of a disordered N-terminal region followed by two EF-hands and a coiled-coil domain at the C-terminus (see Supplementary Fig. S2a). Although EFhd2 and EFhd1 have similar predicted domain compositions, except for the disordered N-terminal region, their cellular functions are different. EFhd2 is a cytoskeleton-associated protein involved in regulating immune and brain cell functions, whereas EFhd1 appears to modulate apoptosis and differentiation of neuronal and muscle cells by mitochondrial association20,21. The domain architecture of AIF-1 is different when compared with EFhd1 and EFhd2 (see Supplementary Fig. S2b). Nevertheless, AIF-1 is an F-actin bundling protein that functions, like EFhd2, to regulate the immune system20. Recently, among these homologous proteins, the role of EFhd2 in modulating actin dynamics has been examined. EFhd2 modulates cytokine expression by actin remodelling in human mast cells and functions in cancer invasion and metastasis as an actin-related regulator of membrane dynamics22,23,24,25. In our previous work, EFhd2 was found to contain multiple actin-binding sites in the core region, including the proline-rich region (PxxP motif) and two EF-hands26. We also reported previously that the EF-hands of EFhd2 are involved directly in F-actin bundling in a Ca2+-dependent manner and the coiled-coil domain is essential to the F-actin bundling activity by homodimerisation26. However, the detailed molecular mechanism describing F-actin binding and bundling by EFhd2 remains elusive because structural data are missing.
Here, we report crystal structures of the Ca2+-bound EFhd2 core domain (CDEFhd2, residues 70–184) comprising the N-terminal PxxP motif, two EF-hands, ligand mimic (LM) helix and C-terminal linker. In addition, we also report crystal structures of mutants of CDEFhd2 defective for Ca2+-binding. Furthermore, we performed chemical shift perturbation (CSP), ensemble refinement and biochemical analyses to further understand the structural basis for the Ca2+-dependent F-actin bundling function of EFhd2. Based on the experimental results, we propose that the F-actin bundling activity of EFhd2 depends on structural rigidity conferred by binding of two Ca2+ ions to the EF-hand domains. In the absence of Ca2+, EFhd2 displays local conformational flexibility around the Ca2+-binding loop of the EF-hand and C-terminal linker, supporting an explanation of the Ca2+-dependent reorganisation of actin binding sites of EFhd2 that retains F-actin binding activity but loses the ability to bundle F-actin. Additionally, we established that dimerisation of EFhd2 via the C-terminal coiled-coil domain, which is necessary for F-actin bundling, occurs through the parallel coiled-coil interaction.
Results
Crystal structure of CDEFhd2 in the Ca2+ bound state
The crystal structure of Ca2+-bound CDEFhd2 (residues 70–184) was solved using the multi-wavelength anomalous dispersion (MAD) method and refined to a Rwork = 16.7% and Rfree = 20.2% at 1.85 Å resolution (Table 1). The structure of CDEFhd2 adopts a compact and globular fold composed of the predicted PxxP motif (residues 80–90, actin-binding site 1(ABS1)) at the N-terminus followed by two EF-hands (residues 91–163, actin-binding site 2 (ABS2)), a connecting short α-helix and a C-terminal linker (residues 164–184, actin-binding site 3 (ABS3)) (Fig. 1a,b). The connecting short α-helix of CDEFhd2 resembles the ligand helix of EF-hand proteins. Thus, we named this helix the LM-helix27. In addition, two Ca2+ ions are coordinated by several negative charged residues (Asp105, Asp109 and Glu116 for EF1; Asp141, Asp143, Asp145 and Glu152 for EF2), which are well-known Ca2+-coordinating residues in EF-hand domains (Fig. 1b).

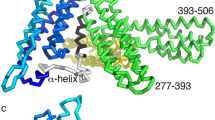
Crystal structure of human CDEFhd2.
(a) Schematic of human CDEFhd2; PxxP: proline-rich region, EF: EF-hands, LM: ligand mimic, CC: coiled-coil, ABS: actin-binding site. (b) Stereoview of the CDEFhd2 structure. The colour coding used is the same as in (a). Silver spheres indicate two Ca2+ ions located near the loops between the EF-hand motifs. (c) Hydrophobic interactions and hydrogen bonds between the PxxP motif and the EF-hand domain. Phe80, Pro82, Phe86 and Phe89 in the PxxP motif are proximal to Phe150, Leu154 and Leu155 in helix 4. Tyr83, Glu85 and Glu88 in the PxxP motif form hydrogen bonds with Lys95 in helix 1 (H1), and Arg151 and Arg158 in helix 4 (H4). (d) Intramolecular interactions between hydrophobic residues of the EF-hand domains and the LM-helix.
The PxxP motif is involved in proper intracellular localisation of target proteins through basic motifs (Arg/Lys), exposed hydrophobic residues and a pair of Pro residues28. These three conserved elements of the PxxP motif are important for phosphoinositide binding, penetration of the lipid bilayer and SH3 domain binding, respectively28. Interestingly, the PxxP motif of CDEFhd2 is not only required for association with the B-cell membrane, but was also identified as part of the multiple actin-binding sites20,26. Although 10 residues (residues 70–79) of the PxxP motif were disordered in the crystal structure, Pro80, Pro82, Phe86 and Phe89 face towards helix 4 of the EF-hands to form hydrophobic interactions, and Glu85, Glu88 and Tyr83 form hydrogen bonds with Lys95, Arg151 and Arg158 of helix 1 and 4 of the EF-hand domains. As a result, the PxxP motif (ABS1) interacts tightly with the EF-hand domains (ABS2) (Fig. 1c). Furthermore, the EF-hand domains (ABS2) not only interact tightly with the PxxP motif (ABS1) via helix 1 and 4, but also associate with the LM-helix (ABS3) through intramolecular interactions that resemble the intermolecular interactions of Ca2+-calmodulin (CaM)-peptide complexes (Fig. 1a,d).
Structural implications of EFhd2 in the absence of Ca2+
We failed to determine the structure of the apo form of EFhd2 owing to structural instability during protein purification; however, we could determine the crystal structures of CDEFhd2 mutants defective for one Ca2+-binding site (E116A for EF1, CDEFhd2EF1; E152A for EF2, CDEFhd2EF2) (see Supplementary Fig. S3). The overall structures of these two mutants in the presence of Ca2+ are similar to that of Ca2+-bound CDEFhd2 (CDEFhd2EF1, root mean square deviation (RMSD) = 0.34 Å for 102 Cα atoms; CDEFhd2EF2, RMSD = 0.61 Å for 105 Cα atoms), which implies a single Ca2+-loaded EF-hand is sufficient to maintain a stable fold (Fig. 2a). However, the Ca2+-binding loop region of EF1 of CDEFhd2EF1 (Arg106, Gly107 and Arg108) was observed to be disordered (Fig. 2b). In addition, in the structure of CDEFhd2EF2, one water molecule occupied the Ca2+ position of EF2 and forms several hydrogen bonds with Asp141, Asp143, Asp145 and Lys147. Moreover, Asp143 forms a hydrogen bond with Arg151 and the Ca2+-binding loop is slightly shifted (~2.2 Å). As a result, the water molecule is trapped in the Ca2+-binding site of EF2 (Fig. 2c). Furthermore, comparison of the crystallographic B-factors between Ca2+-bound and EF-hand mutant structures showed that the largest changes in B-factor values were for CDEFhd2EF1 (35.6 Å2), CDEFhd2 (20.4 Å2) and CDEFhd2EF2 (21.5 Å2). In particular, B-factor values for EF1 and the C-terminal linker region in the structure of CDEFhd2EF1 were increased significantly (Fig. 2d). On the other hand, only small changes of B-factor values for EF2 in the structure of CDEFhd2EF2 were observed and are probably because of small structural perturbations in the absence of Ca2+ and stabilisation by newly formed hydrogen bonds to the trapped water molecule. These results suggest that the Ca2+-binding loop of EF1 adopts a more flexible structure than EF2 in the absence of Ca2+, resulting in large conformational fluctuations to EF1 and a concomitant increase in the overall B-factor. Next, we performed CSP analysis using the conditions of Ca2+-free and bound states to identify conformational changes to CDEFhd2 in the absence Ca2+. The Ca2+-dependent conformational changes to CDEFhd2 were monitored by measuring resonance perturbations in 2D 1H-15N HSQC spectra (see Supplementary Fig. S4a). Consistent with the crystal structures of the CDEFhd2 mutant, significant CSPs were associated with residues in Ca2+ binding loop region (Phe101, Asp105, Asp109, Phe111, Ile 112 and Glu116) of EF1. Noticeably, CSPs of hydrophobic residues in the Ca2+-binding loop of EF1 are likely to be associated with the failure of structure determination caused by the instability of EFhd2 in the absence of Ca2+. Significant CSPs for the disordered region (residues 70–80, PxxP motif) of the crystal structure appears to be associated with the conformational changes to EF1 in the absence of Ca2+. On the other hand, noticeable CSPs were not detected in the LM-helix region (see Supplementary Fig. S4b,c). Thus, we expect the LM-helix region to maintain its structure in the absence of Ca2+.

Structure comparison between Ca2+-bound CDEFhd2 (green), CDEFhd2EF1 (cyan) and CDEFhd2EF2 (orange).
(a) Structural superposition of Ca2+-bound CDEFhd2 (green), CDEFhd2EF1 (cyan), and CDEFhd2EF2 (orange). (b) The Ca2+-binding loop of EF1 of Ca2+-bound CDEFhd2 (green) and CDEFhd2EF1 (cyan). (c) Ca2+-binding loop of EF2 of Ca2+-bound CDEFhd2 (green) and CDEFhd2EF2 (orange). (d) Plot of the crystallographic B-factor per residue of Ca2+-bound CDEFhd2 (20.4 Å2, green), CDEFhd2EF1 (35.6 Å2, cyan) and CDEFhd2EF2 (21.5 Å2, orange). The disordered region of the PxxP motif in the crystal structure of CDEFhd2 is shown by the blue dashed line.
Collectively, we postulate that Ca2+ depletion leads to local conformational flexibility of actin-binding sites (EF1, C-terminal linker) and this reduces the F-actin bundling ability of EFhd2 in the absence of Ca2+, as observed in previous results26.
Ensemble refinement of the CDEFhd2, CDEFhd2EF1 and CDEFhd2EF2
Based on the results of the crystal structures and CSP analysis, we hypothesise that Ca2+ depletion induces changes in local flexibility of the actin binding sites of EFhd2, which implies a Ca2+-dependent regulation of F-actin bundling activity of EFhd2 through protein dynamics. To evaluate the effect of Ca2+ on structural dynamics of EFhd2 at the atomic level, we performed ensemble refinement for CDEFhd2, CDEFhd2EF1 and CDEFhd2EF2 using Phenix.ensemble refinement29. Ensemble refinement is a useful tool to highlight functional protein dynamics through X-ray diffraction data29. Ensemble refinement of the CDEFhd2, CDEFhd2EF1 and CDEFhd2EF2 yielded a large number of models that represent structural dynamics and decreases in the Rfree value (CDEFhd2 = 5.2%, CDEFhd2EF1 = 3.7%, CDEFhd2EF1 = 3.8%) compared with the single structure. In the model structures, different degrees of mobility in CDEFhd2, CDEFhd2EF1 and CDEFhd2EF2 were observed (Fig. 3). As expected, Ca2+-bound CDEFhd2 displayed a rigid conformation and CDEFhd2EF1 showed the largest degrees of mobility, indicating that Ca2+ depletion of EF1 has a larger impact on conformational dynamics (Fig. 3a,b). In addition, CDEFhd2EF2 also showed a moderate degree of mobility (Fig. 3c). The significant increase of the root-mean-square fluctuation (RMSF) in the EF1 of CDEFhd2EF1 is consistent with the crystallographic B-factor and CSP analysis, which support an increase in local flexibility of the actin binding sites of EFhd2 in the absence of Ca2+ (Fig. 3d). Interestingly, the C-terminal linker (residues, 176–184) followed by the LM-helix also showed significant RMSF increases in CDEFhd2EF1 and CDEFhd2EF2 (Fig. 3d). These results support the postulate that the EFhd2 core domain forms local dynamic conformations (EF1, C-terminal linker) in the absence of Ca2+.

Structural comparison of ensemble models of CDEFhd2, CDEFhd2EF1 and CDEFhd2EF2.
Ensemble models of (a) Ca2+-bound CDEFhd2, (b) CDEFhd2EF1 and (c) CDEFhd2EF2. Structures coloured by B-factor. The B-factors are coloured from a blue/thin line to a red/thicker line with increasing B-factor. (d) The root-mean-square fluctuation (RMSF) of ensemble models for Ca2+-bound CDEFhd2 (green), CDEFhd2EF1 (cyan) and CDEFhd2EF2 (orange). RMSF in the EF1 of CDEFhd2EF1 is consistent with the crystallographic B-factor. The disordered region of the PxxP motif in the crystal structure of CDEFhd2 is shown by the blue dashed line.
High Ca2+ binding affinities of two EF-hand domains
In order to measure the Ca2+-binding affinity of each EF-hand domain, we performed isothermal titration calorimetry (ITC) experiments using full-length EFhd2EF1 and EFhd2EF2. We observed that each EF-hand domain possesses high Ca2+ binding (EF1, Kd = 96 ± 15 nM; EF2, Kd = 70 ± 1 nM). The ITC results for Ca2+ binding to EFhd2 mutants (EFhd2EF1, EFhd2EF2) demonstrate exothermic profiles; thus, the reaction is enthalpically driven. For the EF1 site (using EFhd2EF2), Kd = 96 ± 15 nM, stoichiometry = 1.08 ± 0.02, ΔH = −14.7 ± 0.3 kcal/mol, and ΔS = −17.00 ± 1.30 cal/mol/K. For the EF2 site (EFhd2EF1), Kd = 70 ± 1 nM, stoichiometry = 0.98 ± 0.05, ΔH = −15.7 ± 1.8 kcal/mol, and ΔS = −19.80 ± 6.12 cal/mol/K (Fig. 4a). The decrease in entropy upon Ca2+ binding indicates that the flexible conformation of the Ca2+ binding site in the absence of Ca2+ changes to a rigid conformation. The Ca2+-binding affinity of EF-hand containing proteins is related to protein stability. In other words, high affinity towards Ca2+ leads to instability in the Ca2+-free state30. To assess the effect of Ca2+ in EFhd2 stability, we measured the Ca2+-dependent EFhd2 thermostability thorough a heat aggregation test (Fig. 4b). Consistent with a previous study showing that thermostability of EFhd2 was restored by Ca2+ at a high temperature31, the half aggregation temperature for both EF-hand mutants that bind only one Ca2+ is significantly lower (CDEFhd2EF1: 62.32 ± 0.14 °C, CDEFhd2EF2: 57.90 ± 0.60 °C) than the two Ca2+-bound EFhd2 (CDEFhd2: 84.89 ± 0.01 °C) and is consistent with ref. 30 and 31.

Ca2+-binding properties of EFhd2.
(a) ITC data for EFhd2 with Ca2+. Each experiment was repeated in triplicate. (b) Light scattering intensity at 470 nm and various temperatures for thermal aggregation of CDEFhd2 (green), CDEFhd2EF1 (cyan) and CDEFhd2EF2 (orange). The protein concentration was 250 μM. Each experiment was repeated in triplicate. A reduction in intensity at high temperatures indicates precipitation of large-sized aggregates70.
Although the crystal structures of EF-hand mutants (CDEFhd2EF1 and CDEFhd2EF2) are similar to Ca2+-bound CDEFhd2, we observed structural flexibility in the Ca2+-binding loop of EF1 and the C-terminal linker (Figs 2 and 3). Based on structural and biochemical results, we hypothesise that changes in the conformation and flexibility lead to exposure of hydrophobic residues around the Ca2+-binding loop of EF-hands and the C-terminal linker, and this exposure of hydrophobic residues affects protein stability.
We previously reported that F-actin bundling activity decreases in the absence of Ca2+ 26. We further analysed the contribution of Ca2+ binding to each EF-hand domain on F-actin binding and bundling activity (Fig. 5). It is interesting to note that wild-type (Ca2+-bound and Ca2+-unbound) and each EF-hand mutant (only one Ca2+-bound) showed similar F-actin binding activity. Surprisingly, however, F-actin bundling activities were quite different and dependent on the Ca2+-bound states. Even depletion of one Ca2+ site in a two Ca2+-binding protein showed a dramatic reduction in F-actin bundling activity to a level that is similar to that of previously reported data for a two Ca2+-depleted state26. We propose that the increased structural flexibility observed in the Ca2+-binding loop and C-terminal linker, which encompass actin-binding sites, cause a reduction in F-actin bundling activity, presumably because coordination of the F-actin binding sites for F-actin bundling is disrupted.

In vitro co-sedimentation (F-actin binding) assay and low-speed F-actin bundling assay.
(a) Co-sedimentation assay and (b) low-speed actin-bundling assay of EFhd2 (full-length) and its mutants. Protein samples (5 μM) were added to pre-polymerised actin (2 μM) in the presence of 1 mM CaCl2. BSA and α-actinin were used as a negative and positive control, respectively. The percentage of actin distribution was quantified and is presented in the bar graphs. Each experiment was repeated in triplicate.
Structural comparison between CDEFhd2 and a homologous protein, allograft inflammatory factor-1 (AIF-1)
AIF-1 and EFhd1/EFhd2 are highly evolutionarily conserved proteins, because these genes are generated from common ancestral species of the Bilateria20. In particular, EFhd2 and AIF-1 exhibit the same cellular function as an actin-binding protein. Although sequence homology between AIF-1 and EFhd2 is limited to the EF-hand domains, AIF-1 exhibits F-actin binding and crosslinking activity similar to that observed for EFhd2 (see Supplementary Fig. S2b). EFhd2 exhibits F-actin bundling activity in a Ca2+-dependent manner, whereas AIF-1 does not exhibit Ca2+ dependency for F-actin binding and bundling activity26,32,33,34.
The structure of AIF-1 has been determined in the presence and absence of Ca2+ (PDB IDs: 1WY9 and 2D58)35. To investigate the molecular basis of the effect of Ca2+ on F-actin bundling activity, we compared Ca2+-bound and Ca2+-free structures of CDEFhd2 and AIF-1. The CDEFhd2 structure is similar to the structures of Ca2+-bound and the apo form of AIF-1 (1WY9, Ca2+-bound form_Z-score = 4.0, RMSD = 2.48 Å for 76 Cα atoms; 2D58, apo-form_Z-score = 5.5, RMSD = 1.98 Å for 86 Cα atoms), even though we failed to solve the Ca2+-free structure owing to protein destabilisation during the protein purification process (see Supplementary Fig. S5a). Interestingly, EF1 of AIF-1 is stabilised by hydrogen bonds between Asn60, Asn62 and Asp66 in the absence of Ca2+ (see Supplementary Fig. S5b). In addition, a water molecule is trapped in the Ca2+ binding site of EF2 in the apo structure of AIF-1 similar to that observed for CDEFhd2EF2 (see Supplementary Fig. S5c). Therefore, CDEFhd2 reveals two Ca2+-bound EF-hands, whereas Ca2+ bound to only EF2 of AIF-1, because Ca2+-binding residues (Asp/Glu) are not conserved in EF1 of AIF-1 (see Supplementary Fig. S2)35. These structural features of CDEFhd2 and AIF-1 suggest that Ca2+ is essential for the formation of a stable structure of CDEFhd2, whereas AIF-1 is capable of maintaining a stable structure in the absence of Ca2+ through hydrogen bonds involving several residues of the Ca2+-binding loop of EF1 and a water molecule located in the Ca2+-binding site of EF2. These structural differences between CDEFhd2 and AIF-1 support the hypothesis that Ca2+ is essential for the actin-bundling function of EFhd2 by maintaining a stable structure, whereas AIF-1 exhibits F-actin binding and bundling activity regardless of Ca2+ dependency32,33,34.
EFhd2 displays an actin-bundling function with the parallel coiled-coil domain at the C-terminus
We already reported that the C-terminal coiled-coil domain is essential for the dimerisation of EFhd2 because we observed EFhd2 lost F-actin bundling activity in the absence of the coiled-coil domain26. However, it was unclear whether EFhd2 dimerised by parallel or antiparallel interactions. To establish the molecular architecture of EFhd2, we engineered recombinant fragments corresponding to the predicted coiled-coil domain (residues 199–240), including a Cys residue at either the N-terminus (CC1) or C-terminus (CC2) of the coiled-coil domain36. We expected that if the coiled-coil domain assembles as a parallel interaction, formation of a disulfide bond should occur owing to the high proximity between Cys residues of each polypeptide and the dimer should be detected on a non-reducing denaturing gel. Therefore, purified recombinant proteins were resolved on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels under reducing or non-reducing conditions. The disulfide cross-linking assay revealed that SDS-PAGE analysis gave only a monomer band under reducing conditions. In contrast, both CC1 and CC2 protein bands migrated as monomers with the dimeric form observed under non-reducing conditions. These results suggest that the coiled-coil domain of EFhd2 assembles into a parallel dimer (Fig. 6).

Schematic of the actin-bundling activity of EFhd2.
(a) SDS-PAGE analysis of the two engineered Cys mutants of the coiled-coil (residues 199–240). Protein samples were analysed under reducing or non-reducing conditions. (b) Schematic of the Ca2+-dependent F-actin bundling mechanism of EFhd2 (ABS: actin-binding site).
Discussion
Ca2+ is an essential modulator of signal transduction processes required for various cellular functions such as contraction, cell differentiation and proliferation30. The presence of EF-hand domains in EFhd2 raises the possibility that EF-hand domains upon Ca2+ binding may affect its cellular function related to actin dynamics regulation. For example, we found that Ca2+ or ethylene glycoltetraacetic acid (EGTA) had little effect on EFhd2 binding to F-actin; however, the F-actin bundling activity was significantly reduced in the Ca2+-free state and these results were visualised by electron microscopy26.
In this study, we have tried to determine the structure of EFhd2 to elucidate the Ca2+-dependent F-actin bundling mechanism of this protein. In a search for structures similar to CDEFhd2 using the Dali program37, we were able to find ~100 Ca2+-bound EF-hands with similar structures (>10 for Z-scores and <3.0 Å in RMSD), in which most of the EF-hand matches were CaM and troponin C (TnC). In addition, the structure of CDEFhd2 fits well with those of Ca2+-CaM-peptide complexes (see Supplementary Fig. S6)38,39,40,41. Ca2+-CaM-peptide complexes are more compact than the peptide unbound form because of intermolecular interactions between exposed hydrophobic grooves of CaM and hydrophobic residues of the target molecule. In addition, the Ca2+ affinity of EF-hands increases with intermolecular interactions, leading to structural stabilisation of the Ca2+-bound state30,42. Many EF-hand containing proteins can change their diverse biochemical responses through changes in conformation and/or protein stability in the presence or absence of Ca2+ 30,42. For example, members of the CaM superfamily are capable of modulating numerous intracellular processes in a Ca2+-dependent manner by undergoing conformational changes represented by “close” to “open” structures. On the other hand, several EF-hand containing proteins such as sarcoplasmic Ca2+-binding proteins (CaBPs), calcium vector protein (CaVP), calerythrin, and stromal interaction molecule-1 (STIM1) remain in an unstable form in the absence of Ca2+ in vitro43,44,45,46,47. In particular, the structure of STIM1 adopts a compact conformation through a hydrophobic interaction between EF-hands and a SAM domain (sterile α motif) similar to Ca2+-peptide-CaM complexes and CDEFhd2. In addition, mutational analysis revealed that Ca2+ depletion or disruption of hydrophobic interactions between EF-hands and the SAM domain leads to destabilisation of the entire EF-SAM complex43. Taking into consideration a previous study and structural similarity between Ca2+-peptide-CaM complexes, the observations for STIM1 and CDEFhd2 support the hypothesis that high affinity for Ca2+ and intramolecular interactions of CDEFhd2 are likely to maximise stabilisation of the EFhd2 fold. In support of this hypothesis is the thermostability results of EFhd2, which showed that the protein thermal stability at high temperature was restored by Ca2+ 31. This is further emphasised by the observation that CDEFhd2 remained stable in solution, even at high temperatures in the presence of two Ca2+ ions (Fig. 4). As mentioned earlier, in the case of CaM, the core region comprising two EF-hand domains shows significant conformational change upon Ca2+ binding, which leads to structural changes in two lobes and interaction with partner proteins. However, in the case of CDEFhd2, the structural evidence in this report (including mutant structures and CSP analysis) indicates that the core structure of the EF-hand domains is retained regardless of Ca2+ binding, because two hydrophobic clusters in CDEFhd2 are maintained (see Supplementary Figure S7a). Denessiouk et al. classified EF-hand domains in five groups based on differences in the structural changes in the core region (hydrophobic cluster I and II) upon Ca2+ binding48. CDEFhd2 may belong to type I or IV, because these types have an open conformation in the Ca2+-bound form. In the apo state, type I EF-hand domains (Parvalbumin, PVALB) maintain an open conformation; however, type IV EF-hand domains (CaM and Troponin C, TnC) exhibit a closed conformation48. This structural difference between types I and IV raised the possibility that the CDEFhd2 may belong to type I, because we expect CDEFhd2 to have an open conformation in the apo state based on the mutant structures and CSP analysis. Additionally, we compared the structures of the single Ca2+-bound state in types I and IV. Intriguingly, in the case of type IV (TnC), the single Ca2+-bound intermediate state resembled the closed conformation of the apo state (Supplementary Figure S7b)49,50,51. The structure of the single Ca2+-bound state of type I (PVALB) is close to that of the two Ca2+-bound state, although the structural difference between the apo and two Ca2+-bound states is marginal (Supplementary Figure S7c)52,53,54. This again suggests that CDEFhd2 belongs to type I, because the structures of the single Ca2+-bound state of the two CDEFhd2 mutants are similar to the structure of the two Ca2+-bound state, and the core structures of CDEFhd2 may not differ even when in a complex with interacting proteins.
We failed to solve the structure of EFhd2 in the absence of Ca2+ because of protein instability; however, structures of EF-hand mutants, CSP analysis and ensemble refinement analysis showed that CDEFhd2 undergoes changes in local structure and dynamics in the absence of Ca2+. The crystal structures of the EF-hand mutants are maintained even when one EF-hand loses Ca2+ binding capacity (Fig. 2a). However, the Ca2+ binding loop region that loses Ca2+ binding activity exhibits structural flexibility (Fig. 2b,d). Furthermore, RMSF values support the premise that F-actin binding sites of EFhd2 form locally dynamic conformations (EF1, C-terminal linker) in the absence of Ca2+ and this dynamic state reduces F-actin bundling activity (Figs 3 and 5). In particular, greater flexibility of the C-terminal linker between the EF-hands and coiled-coil domain probably leads to incorrect coordination of actin binding sites in dimer formation. Based on these results, we suggest that the EFhd2 core domain comprising the multiple actin-binding sites changes to an unstable structure by changes in local conformational flexibility in the absence of Ca2+, and these structural dynamics reduce the F-actin bundling function.
Recently, a structural model for the Ca2+-dependent F-actin crosslinking mechanism by non-muscle α-actinin-1 was reported15. Non-muscle α-actinin-1 is composed of N-terminal CH domains (actin binding sites), repeated rod domains and C-terminal EF-hands (CaM-like domain; CaMD). Non-muscle α-actinin-1 forms an antiparallel dimer via the rod domain composed by 4 spectrin-like repeats11,13. NMR structures of the holo and apo form of CaMD of α-actinin-1 reveal that apo CaMD forms a flexible structure owing to the unstructured linker between N- and C-lobes; however, Ca2+ binding leads to stabilisation of the linker, resulting in structural rearrangement of CaMD. Consequently, rearrangement of CaMD inhibits proper orientation of adjacent F-actin binding sites for F-actin crosslinking15. This observation supports the concept that Ca2+-dependent local conformational flexibility of EFhd2 plays a critical role in regulation of F-actin bundling activity by induced reorganisation of actin-binding sites.
Ca2+ is essential for leading edge formation because several Ca2+-related actin-binding proteins modulate cell motility and shape by reorganisation of F-actin structures in a Ca2+-dependent manner11,55. For example, F-actin crosslinking activity of non-muscle α-actinin and villin at the leading edge of cells is drastically inhibited at high Ca2+ concentrations (micromolar levels)8,9,11. However, Ca2+ is required for F-actin bundling function of EFhd2 in contrast with what is observed for α-actinin and villin. We speculate that various Ca2+-related F-actin bundling proteins may be involved in F-actin reorganisation as suitable regulators in specific cell environments. Furthermore, in our earlier studies, EFhd2 was mainly expressed at the leading edge of cells and improved lamellipodia formation and cell migration26. Interestingly, Beerman et al. analysed Ca2+ transients of migrating immune cells through direct measurement of Ca2+ signalling using light-sheet microscopy. They demonstrated that Ca2+ fluctuations were enhanced at the leading edge and reduced at the lagging edge of migrating immune cells56. In many EF-hand-containing proteins, including calmodulin, calbindin D9k, and vitamin K-dependent protein S, Kds for Ca2+ are highly dependent on ionic strength. For these proteins, binding affinity for Ca2+ is lowered by approximately 2.5–100 fold in the presence of 0.15 M NaCl (close to physiological conditions)57,58,59. We were able to measure Kds (70–100 nM) for EFhd2 mutants only at low ionic strength (50 mM Tris-HCl, pH 8.5, 20 mM NaCl); we failed to obtain measurements at higher ionic strengths (even at 100 mM NaCl) because of the instability of EFhd2 mutants at higher ionic strengths in the absence of Ca2+. Thus, we hypothesise that the affinity of these mutants for Ca2+ is much lower than 100 nM, and that both EF hands would not be occupied by Ca2+ at resting Ca2+ levels in live cells. This result supports the mechanism of cell migration by EFhd2 because Ca2+ is essential for the F-actin bundling function of EFhd2.
In conclusion, we demonstrate that EFhd2 shows unique structural and biological features as an EF-hand containing F-actin bundling protein. For F-actin bundling activity, structural stabilisation of the EF-hand domains was found to occur in the presence of Ca2+. The core region of EFhd2 maintains its structure in the absence of Ca2+ ; however, changes in local conformational flexibility reduce F-actin bundling activity of EFhd2 by incorrect coordination of actin-binding sites in parallel dimer formation. Finally, EFhd2 acts as a cytoskeleton-associated adaptor protein that contains two functional EF-hand domains with high Ca2+-binding affinity, which might be a useful target for further research involved in its biological functions or various pathologies25,60,61.
Methods
Cloning and protein purification of full-length EFhd2 and ΔNTD
A human EFhd2 clone encoding full-length (residues 1–240) and ΔNTD (residues 70–240) were amplified using the polymerase chain reaction (PCR) from pOTB7 (RZPD German Resource Centre, Germany). Full-length EFhd2 was cloned into a modified pET28a vector (Novagen) containing an N-terminal 6×His (His6)-tobacco etch virus (TEV) tag. ΔNTD was cloned into a modified pET28a vector (Novagen) containing a His6-Nus-TEV tag. Recombinant DNA were transformed into E. coli strain BL21 (DE3) and the cells were grown in Luria–Bertani (LB) medium containing 50 μg/mL kanamycin at 37 °C until the absorbance at 600 nm was 0.7. Expression of recombinant proteins was induced by adding isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM and cells were grown for a further 5 h at 37 °C. Cells were harvested by centrifugation (4,000 g) for 20 min at 4 °C. The cell pellet was resuspended in buffer containing 50 mM HEPES-NaOH, pH 7.5, 300 mM NaCl and 5 mM imidazole, and the cells disrupted by sonication. After removal of the cell debris by centrifugation at 14,000 g for 50 min and 4 °C, the soluble supernatant was loaded onto an equilibrated gravity-flow column (Bio-Rad, Hercules, CA, USA) packed with Ni-NTA agarose resin (Peptron, Korea). The protein was eluted with a buffer containing 50 mM HEPES-NaOH, pH 7.5, 300 mM NaCl and 300 mM imidazole. After concentrating the eluate, the protein solution was incubated with TEV protease overnight at 4 °C to remove the N-terminal His6 or His6-Nus tag. To exchange the buffer for crystallisation, the final purified protein was passed through a HiLoad 16/60 Superdex 75 gel filtration column (Pharmacia Biotech) pre-equilibrated with 20 mM HEPES-NaOH, pH 7.5, 150 mM NaCl and 1 mM CaC12. Severe degradation was observed after incubation with TEV protease. Therefore, full-length EFhd2 was cloned into a modified pET-21a vector (Novagen) containing N-terminal His6 tag. The overall purification procedure was the same as described above. However, the removal process of the N-terminal His6 tag was omitted, because the modified pET-21a vector (Novagen) does not include a protease cleavage site.
EFhd2 core domain (CDEFhd2, residues 70–184) identification
We initially tried to crystallise the full-length EFhd2 with ΔNTD. However, crystallisation of this construct failed because of severe degradation during the purification process. Thus, we performed limited proteolysis experiments to identify stable domains and ΔNTD was used for this purpose. Treatment with TEV protease overnight at 4 °C gave a stable fragment, as observed by SDS-PAGE (see Supplementary Fig. S8) and blotted onto a polyvinylidene fluoride membrane to perform N-terminal sequencing analysis (Korea Basic Science Institute, Seoul, Korea). The stable core domain was identified to span residues 70–184 and corresponds to a PxxP motif and two EF-hand domains (see Supplementary Fig. S3).
Cloning and protein purification of CDEFhd2
Human CDEFhd2 (residues 70–184) was amplified using PCR from full-length EFhd2 (residues 1–240) and cloned into the modified pET-28a vector (Novagen) containing an N-terminal His6-TEV tag. The expressed recombinant protein was purified using the procedure used to purify full-length EFhd2. For seleno-L-methionine (Se-Met) incorporation, a plasmid encoding the CDEFhd2 was transformed into the methionine-auxotrophic E. coli strain B834 (DE3) (Novagen). Colonies were inoculated into LB medium containing 50 μg/mL kanamycin and incubated at 37 °C with shaking for ~24 h, and then cells were harvested by centrifugation at 4,000 g for 20 min and 4 °C. The cell pellet was resuspended in minimal medium to wash and remove the LB medium, and washed cells were harvested by centrifugation at 4,000 g for 20 min and 4 °C. After washing, the cell pellet was transferred to a fresh 2 L culture of minimal medium (M9 media) supplemented with 25 mg/mL Se-Met, 2% glucose, 0.1 M magnesium sulfate and amino acids, and grown at 37 °C. Protein expression by the cells was induced by the addition of IPTG to a final concentration of 0.5 mM. After 24 h incubation at 37 °C, the cells were harvested by centrifugation at 4,000 g for 20 min and 4 °C. The overall purification procedure of the Se-Met substituted CDEFhd2 was the same as the native CDEFhd2 protein purification procedure. The purified protein was concentrated using an Amicon Ultra-15 30 K (Millipore) and stored in a deep freezer. During purification, the presence of EFhd2 was confirmed by SDS-PAGE.
Cloning and purification of EFhd2 mutants
To investigate the structural properties of Ca2+ binding, we have mutated one acidic residue (E116A for EF1, CDEFhd2EF1; E152A for EF2, CDEFhd2EF2) of each EF-hand domain of CDEFhd2 to abolish the Ca2+ binding ability (see Supplementary Fig. S3). CDEFhd2 mutants were accomplished by PCR and site-directed mutagenesis using the CDEFhd2 cDNA. All mutants were cloned into a modified pET28a vector (Novagen) containing an N-terminal His6-TEV tag. The overall purification procedure of the CDEFhd2 mutants was the same as that used for purifying native CDEFhd2. To investigate the Ca2+-binding affinity or Ca2+-dependent actin-binding and -bundling activity, we have mutated one acidic residue of each EF-hand domain of full-length EFhd2 (E116A for EF1, EFhd2EF1; E152A for EF2, EFhd2EF2) (see Supplementary Fig. S3). Point mutations (EFhd2EF1, EFhd2EF2) were accomplished by PCR and site-directed mutagenesis using the full-length EFhd2 cDNA. All mutants were cloned into the modified pET21a vector (Novagen) containing an N-terminal His6 tag. The overall purification procedure of full-length EFhd2 mutants was the same as that used to purify native full-length EFhd2.
Crystallisation, data collection, and structure determination
To crystallise native and selenomethionine (Se-Met) substituted CDEFhd2, initial screening was performed by the sitting-drop vapour-diffusion method in a 96-well INTELLI-PLATE (Art Robbins Ins.) using the Crystal Screen, Index, SaltRx, MembFac, Natrix, Crystal Screen Lite and Crystal Screen Cryo (Hampton Research). Drops were prepared by mixing 0.5 μL of the protein and 0.5 μL reservoir solutions. Initial crystals of native CDEFhd2 were observed in 0.1 M Tris-HCl (pH 8.5), 0.2 M Na-acetate, and 30% (w/v) PEG 4000, and Se-Met substituted CDEFhd2 crystals were observed in 0.1 M Tris-HCl (pH 8.5), 0.165 M Na-acetate and 25% (w/v) PEG 4000. Final native and Se-Met substituted CDEFhd2 crystals were obtained using a well solution of 0.1 M Tris-HCl (pH 8.5), 23% (w/v) PEG 4000 and 0.16 M Na-acetate, and 0.1 M Tris-HCl (pH 8.5), 0.165 M Na-acetate, 23.5% (w/v) PEG 4000 and 3% (v/v) dioxane, respectively. In addition, crystallisation of CDEFhd2EF1 and CDEFhd2EF2 was performed by the sitting-drop vapour-diffusion method in a 96-well INTELLI-PLATE (Art Robbins Ins.) using the Crystal Screen, Index, SaltRx, MembFac, Natrix, Crystal Screen Lite and Crystal Screen Cryo (Hampton Research), and PEGs Suite (Qiagen). Drops were prepared by mixing 0.5 μL of the protein and 0.5 μL reservoir solutions. Crystals of CDEFhd2EF1 and CDEFhd2EF2 were observed in 0.1 M Tris-HCl (pH 8.5) and 32% (w/v) PEG 2000. All CDEFhd2 crystals were cryoprotected by soaking them for 10 min in mother liquor containing an additional 15% (v/v) glycerol before flash freezing in a stream of nitrogen gas at 95 K. Native and MAD data sets were collected on beamline 7A at the Pohang Accelerator Laboratory (Pohang, Korea). Raw data integration and scaling were performed with the HKL200062. Both the native and Se-Met substituted CDEFhd2 were crystallised in the orthorhombic form and space group P212121 and cell dimensions of a = 37.3, b = 50.7, c = 53.4 Å. A native data set of 1.85 Å resolution was collected and the MAD dataset of Se-Met substituted protein crystals were collected to 2.10 Å. The crystal contains one molecule in an asymmetric unit with a calculated Matthews coefficient of 1.99 Å3/Da and an estimated solvent content of 38.6%63. Four out of the expected six Se sites in the asymmetric unit were found using the program SOLVE64 using 2.10 Å resolution data yielding phases with a figure of merit of 0.51. Refinement was performed with PHENIX65 and manual rebuilding was performed using the COOT program63. Cycles of group and individual B-factor refinement were performed with PHENIX65. In the last step of the refinement, 117 water and two Ca2+ ions were added. A final crystallographic R-value of 16.7% (Rfree = 20.2%) was obtained. The N-terminus residues from 70 to 79 of the PxxP motif were poorly defined in the electron density maps owing to disorder in the crystal lattice. Therefore, we could observe the structure of the predicted PxxP motif (residues 80–90) at the N-terminus, two EF-hand domains (residues 91–163) and the connecting short LM α-helix (residues 170–177) region at the C-terminus (Fig. 1). CDEFhd2EF1 and CDEFhd2EF2 datasets were collected at beamline 5C at the Pohang Accelerator Laboratory to 1.95 Å and 1.94 Å, respectively. Both the CDEFhd2EF1 and CDEFhd2EF2 structures had the space group P212121 and cell dimensions of a = 36.3, b = 51.5, c = 53.6 Å and a = 35.6, b = 52.1, c = 55.3 Å, respectively. Raw data integration and scaling were performed with HKL200062. The Matthews coefficient for CDEFhd2EF1 and CDEFhd2EF2 was calculated as 1.84 and 1.88 Å3/Da, respectively, which corresponds to a solvent content of 33.0 and 34.7% assuming one molecule in the asymmetric unit63. Initial automatic model building was performed with AutoMR. The model was then refined in cyclic rounds of manual model building in COOT with refinement using PHENIX65,66. Refinement of CDEFhd2EF1 and CDEFhd2EF2 was performed using PHENIX to Rwork = 18.2% and Rfree = 20.7%, and Rwork = 17.9% and Rfree = 20.7%, respectively. All structures of the CDEFhd2 mutants were solved by molecular replacement using the refined native CDEFhd2 structure and molecular graphics were created using PyMol67. The refinement statistics are given in Table 1.
NMR Spectroscopy
For NMR experiments, the CDEFhd2-expressing cells were grown in M9 medium containing 15N ammonium chloride and 13C glucose as the sole nitrogen and carbon sources, respectively. The overall purification procedure followed the approach used to purify the native protein. To remove pre-bound Ca2+, proteins were treated with 25-fold excess EGTA and then dialysed extensively against buffer with or without CaCl2. During the purification process, 5 mM 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate was added to the buffer to retain protein stability. The Ca2+-dependent structural changes to CDEFhd2 were monitored by resonance perturbations in the two-dimensional (2D) 1H-15N HSQC spectra. NMR data were recorded on a Bruker Avance 800 spectrometer at 25 °C. Data were processed with NMRPipe68 and analysed with SPARKY program (Goddard TD and Kneller DG, SPARKY 3, University of California, San Francisco). The magnitude of the 1H-15N chemical shift differences (∆δ, ppm) were calculated using the equation: ∆δ = {(δH2) + 0.2x(δN2)}1/2, where δH and δN are changes to the proton (1H) and nitrogen (15N) chemical shift perturbation, respectively. CSPs for peaks that disappeared upon addition of Ca2+ are set to 1 ppm. We considered CSP to be significant if ∆δ ≥ 0.2 ppm.
Measurement of Ca2+-binding affinity using ITC
Since the full-length EFhd2 (residues 1–240) was more stable than CDEFhd2 in the absence of Ca2+, full-length EFhd2 mutants (EFhd2E116A, EFhd2E152A) were used to measure Ca2+-binding affinities of EFhd2. Protein samples were treated initially with 25-fold excess EGTA and EDTA for >20 h at 4 °C to remove pre-bound metal ions. We dialysed extensively against buffer (50 mM Tris-HCl (pH 8.5) and 20 mM NaCl) for 48 h at 4 °C and changing the buffer every 12 h. To measure the residual Ca2+ concentration after the dialysis step, we used quantitative fluorescence measurement using the Ca2+-indicator fura-2 (non acetoxymethyl ester (AM) form, Molecular Probes, Eugene, OR) (see Supplementary Fig. S9). For determining the intensity of fura-2 at various Ca2+ concentrations, we prepared standard solutions refer to the method of Kong et al.69. After the dialysis process, EFhd2 mutants (EFhd2EF1, EFhd2EF2) (5 μM) were mixed with 10 μM fura-2. Fluorescence spectra of standard solutions and EFhd2 mutants were collected using a FlexStation Ш (Molecular Devices) at room temperature (excitation wavelength: 280 nm to 460 nm, emission wavelength: 510 nm, slit: 4 nm). The residual Ca2+ concentration used for the ITC measurement was around the 1 nM range, indicating that our dialysis process was sufficient to measure Ca2+ binding affinity using the ITC experiment. The protein sample (70 and 150 μM for EFhd2EF1 and EFhd2EF2, respectively) was titrated with 30 injections of ligand (10 μL) in a VP-ITC calorimeter (MicroCal). The ligand solution (0.6 and 1.2 mM Ca2+ for EFhd2EF1 and EFhd2EF2, respectively) was prepared in the same buffer. All measurements were conducted at 25 °C, and binding isotherms were analysed using Origin software supplied with the calorimeter.
Protein stability measured using a heat aggregation assay
To measure the stability of EFhd2 in a Ca2+-dependent manner, the half aggregation temperature of native CDEFhd2, CDEFhd2EF1, and CDEFhd2EF2 were determined spectrophotometrically. The protein solution contained 5 mM Tris-HCl (pH 8.0), 1 mM CaCl2 and 250 μM of protein in a final volume of 4.0 mL. The temperature was increased at the rate of 4 °C per 90 s. Turbidity was monitored by the absorption at 470 nm and room temperature using an ultraviolet-visible spectrometer (Ultrospec 2000; Pharmacia Biotech).
Ensemble refinement of CDEFhd2, CDEFhd2EF1 and CDEFhd2EF2
To evaluate structural dynamics at the atomic level, we performed ensemble refinement using the Phenix.ensemble refinement31 for CDEFhd2, CDEFhd2EF1 and CDEFhd2EF2. Harmonic restraints were applied for all amino acids with visible electron density at a level of 1σ in the 2mFo-DFc electron density map using parameters slack = 1.0 and weight = 0.001.
Protein preparation and the crosslinking experiment of cysteine mutants within the EFhd2 coiled-coil domain (residues 199–240)
To determine whether the C-terminal coiled-coil domain formed a dimer by parallel or antiparallel coiled-coil interaction, we designed recombinant fragments of the coiled-coil domain (residues 199–240) with Cys mutations at the N-terminus (CC1) or C-terminus (CC2) of the coiled-coil domain. The sequence for CC1 starts with CysGlyGly at the N-terminus, whereas for CC2 the C-terminus ends with GlyGlyCys. CC1 and CC2 clones were PCR amplified from the cDNA of the coiled-coil domain of EFhd2. CC1 was subcloned into pGEX-4T-1 and the glutathione S-transferase (GST) tag at the N-terminus was removed by thrombin treatment during the purification process. In addition, CC2 was subcloned into a modified pET-21a vector (Novagen) containing an N-terminal His6 tag. The purification procedure was the same as those used for the other EFhd2 proteins. CC1 and CC2 proteins were analysed by SDS-PAGE under reducing and non-reducing conditions to identify the disulfide bond between Cys residues that mediate dimerisation.
In vitro actin-binding and -bundling assay
F-Actin binding (co-sedimentation) and bundling assays were performed as reported26. In brief, non-muscle actin derived from human platelets was purchased from Cytoskeleton Inc. (Denver, CO, USA). Actin was mixed in G-buffer (5 mM Tris-HCl, pH 8.0 and 0.5 mM CaCl2) to produce an actin stock solution and polymerised in actin polymerisation buffer (0.2 mM Tris-HCl, pH 8.0, 100 mM KCl, 2 mM MgCl2 and 0.5 mM ATP) at room temperature for 1 h and then incubated with EFhd2 or its mutants from 5 min to 1 h at room temperature. Actin filaments with bound proteins were pelleted by centrifugation at 100,000 g for 2 h at room temperature (for the F-actin binding assay) or 15,000 g for 10 min at room temperature (for the F-actin bundling assay). BSA and α-actinin were used as a negative and positive control, respectively. Equal amounts of pellet and supernatant were resolved by SDS-PAGE and proteins were visualised by Coomassie Blue staining. The percentage of actin in the supernatant (S) and pellet (P) was quantified by densitometry using ImageJ 1.44p.
Additional Information
Accession codes: Atomic coordinates and structure factors of CDEFhd2, CDEFhd2EF1, and CDEFhd2EF2 have been deposited in the RCSB Protein Data Bank with accession codes 5I2L, 5I2O, and 5I2Q.
How to cite this article: Park, K. R. et al. Structural implications of Ca2+-dependent actin-bundling function of human EFhd2/Swiprosin-1. Sci. Rep. 6, 39095; doi: 10.1038/srep39095 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Egelman, E. H. More insights into structural plasticity of actin binding proteins. Structure. 12, 909–910 (2004).
Matsudaira, P. Modular organization of actin crosslinking proteins. Trends Biochem Sci. 16, 87–92 (1991).
Vandekerckhove, J. Actin-binding proteins. Curr Opin Cell Biol. 2, 41–50 (1990).
Puius, Y. A., Mahoney, N. M. & Almo, S. C. The modular structure of actin-regulatory proteins. Curr Opin Cell Biol. 10, 23–34 (1998).
Stock, A. et al. Domain analysis of cortexillin I: actin-bundling, PIP(2)-binding and the rescue of cytokinesis. EMBO J. 18, 5274–5284 (1999).
Zhang, W., Zhao, Y., Guo, Y. & Ye, K. Plant actin-binding protein SCAB1 is dimeric actin cross-linker with atypical pleckstrin homology domain. J Biol Chem. 287, 11981–11990 (2012).
Oku, T. et al. Homotypic dimerization of the actin-binding protein p57/coronin-1 mediated by a leucine zipper motif in the C-terminal region. Biochem J. 387, 325–331 (2005).
Noegel, A., Witke, W. & Schleicher, M. Calcium-sensitive non-muscle alpha-actinin contains EF-hand structures and highly conserved regions. FEBS Lett. 221, 391–396 (1987).
Bartles, J. R. Parallel actin bundles and their multiple actin-bundling. Curr Opin Cell Biol. 1, 72–78 (2000).
Morgan, K. G. & Gangopadhyay, S. S. Invited review: cross-bridge regulation by thin filament-associated proteins. J Appl Physiol. 91, 953–962 (1985).
Revenu, C., Athman, R., Robine, S. & Louvard, D. The co-workers of actin filaments: from cell structures to signals. Nat Rev Mol Cell Biol. 8, 635–646 (2004).
Broderick, M. J. & Winder, S. J. Spectrin, alpha-actinin, and dystrophin. Adv Protein Chem. 70, 203–246 (2005).
Tang, J., Taylor, D. W. & Taylor, K. A. The three-dimensional structure of alpha-actinin obtained by cryoelectron microscopy suggests a model for Ca2+-dependent actin binding. J Mol Biol. 310, 845–858 (2001).
Volkmman, N., DeRosier, D., Matsudaira, P. & Hanein, D. An atomic model of actin filaments cross-linked by fimbrin and its implications for bundle assembly and function. J Cell Biol. 153, 947–956 (2001).
Dromota Prebil et al. Structure and calcium-binding studies of calmodulin-like domain of human non-muscle a-actinin-1. Sci Rep. 6, 27383 (2016).
Choe, H. et al. The calcium activation of gelsolin: insights from the 3A structure of the G4-G6/actin complex. J Mol Biol. 324, 691–702 (2001).
Vuadens, F. et al. Identification of swiprosin 1 in human lymphocytes. Proteomics. 4, 2216–2220 (2004).
Ferrer-Acosta, Y. et al. EFhd2 is a novel amyloid protein associated with pathological tau in Alzheimer's disease. J Neurochem. 125, 921–931 (2013).
Kroczek, C. et al. Swiprosin-1/EFhd2 controls B cell receptor signaling through the assembly of the B cell receptor, Syk, and phospholipase C gamma2 in membrane rafts. J Immunol. 184, 3665–3676 (2010).
Dütting, S., Brachs, S. & Mielenz, D. Fraternal twins: Swiprosin-1/EFhd2 and Swiprosin-2/EFhd1, two homologous EF-hand containing calcium binding adaptor proteins with distinct functions. Cell Commun Signal. 9, 2 (2011).
Tominaga, M. et al. Molecular characterization of mitocalcin, a novel mitochondrial Ca2+-binding protein with EF-hand and coiled-coil domains. J Neurochem. 96, 292–304 (2006).
Thylur, R. P. et al. Swiprosin-1 is expressed in mast cells and up-regulated through the protein kinase C beta I/eta pathway. J Cell Biochem. 108, 705–715 (2009).
Ramesh, T. P. et al. Swiprosin-1 Regulates Cytokine Expression of Human Mast Cell Line HMC-1 through Actin Remodeling. Immune Netw. 9, 274–284 (2009).
Huh, Y. H. et al. Swiprosin-1 modulates actin dynamics by regulating the F-actin accessibility to cofilin. Cell Mol Life Sci. 70, 4841–4854 (2013).
Huh, Y. H. et al. Swiprosin-1 stimulates cancer invasion and metastasis by increasing the Rho family of GTPase signalling. Oncotarget. 6, 13060–13071 (2015).
Kwon, M. S. et al. Swiprosin-1 is a novel actin bundling protein that regulates cell spreading and migration. PLoS One. 8, e71626 (2013).
Klosowiak, J. L. et al. Structural coupling of the EF hand and C-terminal GTPase domains in the mitochondrial protein Miro. EMBO Rep. 14, 968–974 (2013).
Sato, T. K., Overduin, M. & Emr, S. D. Location, location, location: membrane targeting directed by PxxP motifs. Science. 294, 1881–1885 (2001).
Burnley, B. T., Afonine, P. V., Adams, P. D. & Gros, P. Modeling dynamics in protein crystal structures by ensemble refinement. Elife. 1, e00311 (2012).
Gifford, J. L., Walsh, M. P. & Vogel, H. J. Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem J. 405, 199–221 (2007).
Ferrer-Acosta, Y., Rodriquez Cruz, E. N., Vaquer Adel, C. & Vega, I. E. Functional and structural analysis of the conserved EFhd2 protein. Protein Pept Lett. 5, 573–583 (2013).
Schulze, J. O. et al. Structural and functional characterization of human Iba proteins. FEBS J. 275, 4627–4640 (2008).
Sasaki, Y. et al. Iba1 is an actin-cross-linking protein in macrophages/microglia. Biochem Biophys Res Commun. 286, 292–297 (2001).
Autieri, M. V. & Chen, X. The ability of AIF-1 to activate human vascular smooth muscle cells is lost by mutations in the EF-hand calcium-binding region. Exp Cell Res. 307, 204–211 (2005).
Yamada, M. et al. X-ray structures of the microglia/macrophage-specific protein Iba1 from human and mouse demonstrate novel molecular conformation change induced by calcium binding. J Mol Biol. 364, 449–457 (2006).
Steinmetz, M. O. et al. A distinct 14 residue site triggers coiled-coil formation in cortexillin I. EMBO J. 17, 1883–1891 (1998).
Holm, L. & Rosenström, P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, 545–549 (2010).
Ye, Q. et al. Structure of calmodulin bound to a calcineurin peptide: a new way of making an old binding mode. Biochemistry. 45, 738–745 (2006).
Rellos, P. et al. Structure of the CaMKIIdelta/calmodulin complex reveals the molecular mechanism of CaMKII kinase activation. PLoS Biol. 8, e1000426 (2010).
Meador, W. E., Means, A. R. & Quiocho, F. A. Modulation of calmodulin plasticity in molecular recognition on the basis of x-ray structures. Science. 262, 1718–1721 (1993).
Ogura, K. et al. Solution structures of yeast Saccharomyces cerevisiae calmodulin in calcium- and target peptide-bound states reveal similarities and differences to vertebrate calmodulin. Genes Cells. 17, 159–172 (2012).
Yap, K. L., Ames, J. B., Swindells, M. B. & Ikura, M. Diversity of conformational states and changes within the EF-hand protein superfamily. Proteins. 37, 499–507 (1999).
Stathopulos, P. B., Zheng, L., Li, G. Y., Plevin, M. J. & lkura, M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell. 135, 110–122 (2008).
Prêcheur, B., Cox, J. A., Petrova, T., Mispelter, J. & Craescu, C. T. Nereis sarcoplasmic Ca2+ binding protein has a highly unstructured apo state which is switched to the native state upon binding of the first Ca2+ ion. FEBS Lett. 395, 89–94 (1996).
Christova, P., Cox, J. A. & Craescu, C. T. Ion-induced conformational and stability changes in Nereis sarcoplasmic calcium binding protein: evidence that the APO state is a molten globule. Proteins. 40, 177–184 (2000).
Théret, I., Baladi, S., Cox, J. A., Sakamoto, H. & Craescu, C. T. Sequential calcium binding to the regulatory domain of calcium vector protein reveals functional asymmetry and a novel mode of structural rearrangement. Biochemistry. 39, 7920–7926 (2000).
Aitio, H. et al. Characterization of apo and partially saturated states of calerythrin, an EF-hand protein from S. erythraea: a molten globule when deprived of Ca2+. Protein Sci. 10, 74–82 (2001).
Denessiouk, K., Permyakov, S., Denesyuk, A., Permyakov, E. & Johnson, M. S. Two structural motifs within canonical EF-hand calcium-binding domains identify five different classes of calcium buffers and sensors. PLoS One. 9, e109287 (2014).
Strynadka, N. C. et al. Structural details of a calcium-induced molecular switch: X-ray crystallographic analysis of the calcium-saturated N-terminal domain of troponin C at 1.75 A resolution. J Mol Biol. 273, 238–255 (1997).
Gagné, S. M., Li, M. X. & Sykes, B. D. Mechanism of direct coupling between binding and induced structural change in regulatory calcium binding proteins. Biochemistry. 36, 4386–4392 (1997).
Tsuda, S. et al. Low-temperature-induced structural changes in the Apo regulatory domain of skeletal muscle troponin C. Biochemistry. 38, 5693–5700 (1999).
Bottoms, C. A., Schuermann, J. P., Agah, S., Henzl, M. T. & Tanner, J. J. Crystal structure of rat alpha-parvalbumin at 1.05 Angstrom resolution. Protein Sci. 7, 1724–1734 (2004).
Henzl, M. T. & Tanner, J. J. Solution structure of Ca2+-free rat-parvalbumin. Protein Sci. 3, 431–438 (2008).
Cates, M. S. et al. Metal-ion affinity and specificity in EF-hand proteins: coordination geometry and domain plasticity in parvalbumin. Structure. 7, 1269–1278 (1999).
Evans, H. & Falke, J. J. Ca2+ influx is an essential component of the positive-feedback loop that maintains leading-edge structure and activity in macrophages. Proc. Natl. Acad. Sci. USA 104, 16176–16181 (2007).
Beerman, R. W. et al. Direct in vivo manipulation and imaging of calcium transients in neutrophils identify a critical role for leading-edge calcium flux. Cell Rep. 13, 2107–2117 (2015).
Lines, S. et al. Electrostatic contributions to the binding of Ca2+ in calbindin D9k. Biochemistry. 30, 154–162 (1991).
Dahlbäck, B., Hildebrand, B. & Linse, S. Novel type of very high affinity calcium-binding sites in beta-hydroxyasparagine-containing epidermal growth factor-like domains in vitamin K-dependent protein S. J Biol Chem. 265, 18481–18489 (1990).
Linse, S., Helmersson, A. & Forsén, S. Calcium binding to calmodulin and its globular domains. J Biol Chem. 266, 8050–8054 (1991).
Hagen, S. et al. The B cell receptor-induced calcium flux involves a calcium mediated positive feedback loop. Cell Calcium. 51, 411–417 (2012).
Vega, I. E. et al. A novel calcium-binding protein is associated with tau proteins in taupathy. J Neurochem, 106, 96–106 (2008).
Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997).
Matthews, B. W. Solvent content of protein crystals. J. Mol. Biol. 33, 491–497 (1968).
Terwilliger, T. C. & Berendsen, J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 55, 849–861 (1999).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 66, 213–221 (2010).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC Available at www.pymol.org.
Delaglio, F. et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 3, 277–293 (1995).
Kong, S. K. & Lee, C. Y. The use of fura 2 for measurement of free calcium concentration. Biochemical Education 23, 0307-4412(95) 00003-8 (2010).
Eronina, T. et al. A protein aggregation based test for screening of the agents affecting thermostability of porteins. PLoS One. 6, e22154 (2011).
Acknowledgements
We thank Dr. Roberto Dominguez (Department of Physiology, University of Pennsylvania) for helpful discussions. We also thank the staff at beamlines BL-5C and 7A of the Pohang Accelerator Laboratory (Pohang, South Korea) for their kind help with data collection. This work was supported by grants from the National Research Foundation (NRF) (Grants: 2013M3A9A7046297, 2013R1A2A2A01068440, 2015M2A2A4A03044653).
Author information
Authors and Affiliations
Contributions
K.R.P., M.S.K., C.D.J. and S.H.E. designed the experiments. K.R.P. and J.Y.K. performed gene cloning. K.R.P. performed purification, crystallisation and biochemical assays. K.R.P., J.Y.A., J.G.L., H.S.Y., Y.L., T.G.K., and J.J.L. performed data collection and structure determination. M.S.K. and J.Y.A performed biochemical assays. J.S.P. and H.K.C. performed chemical shift perturbation analysis. All authors contributed to data analyses. K.R.P. and S.H.E. wrote the manuscript with critical editorial input from S.H.L., W.K.S., H.K.C. and C.D.J.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Park, K., Kwon, MS., An, J. et al. Structural implications of Ca2+-dependent actin-bundling function of human EFhd2/Swiprosin-1. Sci Rep 6, 39095 (2016). https://doi.org/10.1038/srep39095
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep39095
This article is cited by
-
A diagnostic circulating miRNA signature as orchestrator of cell invasion via TKS4/TKS5/EFHD2 modulation in human gliomas
Journal of Experimental & Clinical Cancer Research (2023)
-
Cargo-specific recruitment in clathrin- and dynamin-independent endocytosis
Nature Cell Biology (2021)
-
EFHD2 promotes epithelial-to-mesenchymal transition and correlates with postsurgical recurrence of stage I lung adenocarcinoma
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
