
Abstract
Orofacial clefts are among the most common birth defects in humans worldwide. A large-scale, genome-wide association study (GWAS) in the Chinese population recently identified several genetic risk variants for nonsyndromic cleft lip with or without cleft palate (NSCL/P). We selected 16 significant SNPs from the GWAS I stage (P < 1.00E-5) that had not been replicated to validate their association with NSCL/P in 1931 NSCL/P cases and 2258 controls. Ultimately, we identified a NSCL/P susceptibility loci (rs17095681 at 10q25.3, intron of SHTN1 and 27.2 kb downstream of VAX1, Pmeta = 3.80E-9, OR = 0.64) in Chinese Han and Hui populations. This locus was not high LD with the reported loci in 10q25.3. It was a newly identified independent locus in 10q25.3 associated with NSCL/P. These results imply that SHTIN1 may involve in the pathogenesis of NSCL/P advance our understanding of the genetic susceptibility to NSCL/P.
Similar content being viewed by others
Introduction
Nonsyndromic cleft lip with or without cleft palate (NSCL/P) is one of the most common birth defects with heterogeneous etiologies, including both genetic and environmental factors and their joint effects1. Linkage and association analyses have identified a number of candidate genes and chromosomal loci that may be associated with the risk for NSCL/P1,2. However, mutations and/or polymorphisms in these genes can only explain a small fraction of the genetic contribution to the pathogenesis of this structural abnormality due to genetic heterogeneity and gene-environment interactions. The only gene with a confirmed role in CL/P etiology across multiple populations is IRF6. The protein encoded by IRF6 is a key determinant of the keratinocyte proliferation-differentiation switch and the formation of oral periderm3,4. A functional variant of IRF6 (rs642961), located within the promoter sequence and disrupting the binding site for the transcription factor AP2a, also significantly increases the risk for NSCL/P5.
With recent advances in high-density single nucleotide polymorphism (SNP) genotyping arrays and statistical methodology, genome-wide association studies (GWAS) have heralded a new era of gene discovery for complex diseases. To date, five GWASs and one genome-wide meta-analysis on NSCL/P have been performed, which identified 13 loci or genes (8q24, IRF6, MAFB, ABCA4, NOG, VAX1, PAX7, EPHA3, THADA, SPRY2, TPM1, 8q21.3 and CREBBP)6,7,8,9,10,11 that exceeded genome-wide significant levels. It has been shown that the 8q24 region harbors remote cis-acting enhancers that control Myc expression in the developing face. Deletion of this regulating interval in mice results in mild alteration of facial morphology, including CL/P12. Most of these GWASs used samples of European origin, with the exception of Beaty et al. in 2010 and Sun et al. in 2013, which used Asian samples including Chinese samples.
The GWAS performed by Sun et al. in a large scale of Chinese population identified several risk genetic variants for NSCL/P11. In this project, Two GWASs were performed, validating 30 loci that were significant in both GWAS studies. However, some loci that exceeded the significance threshold (P < 1E-5) in the first GWA study but were not significant or imputed successfully in the second GWA study have yet to be validated. In this paper, we selected 16 such SNPs and validated them in Chinese population to further identify susceptibility loci/genes for NSCL/P. We identified one locus showing significant association with NSCL/P risk.
Results
Validation results
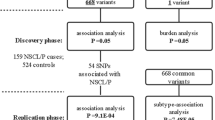
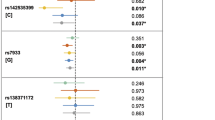
Sixteen SNPs were selected for validation based on a stepwise series of criteria (see Methods), none of which were in high LD with each other. We then performed the validation of these 16 SNPs in 1668 Chinese Han cases and 1924 Chinese Han controls from multiple hospitals in China. Three SNPs showed evidence of association with NSCL/P (Pmeta < 0.05, Supplementary Table S1), but the association direction of two of these SNPs was not in concordance with the GWAS stage. One SNPs, rs17095681 at 10q25.3 (Pmeta = 8.50E-05, OR = 0.70) displayed consistent association with NSCL/P, and the significant direction was in concordance with the GWAS stage (Table 1; Supplementary Table S1). We then validated rs17095681 in Chinese Hui population, which is a minority of China, including 263 case and 334 control from General Hospital of Ningxia Medical University. The validation result was close to significance (P = 6.42E-2) and the association direction was in concordance with the GWAS stage (OR = 0.64). The Meta analysis P-values of Han and Hui population validations was 1.50E-5 (Table 1). In the combined analysis of GWAS and validation stages, rs17095681 showed strong evidence of association (Pmeta = 3.80E-9, OR = 0.64), which reached the genome-wide significance level among the Chinese Han and Hui samples (Pmeta < 5.00E-8, Table 1). Another reported significant locus in 10q25.3, rs7078160, was also validated in these validation samples11. To test the independence of these two SNPs, We fix one SNP as conditional factor and analysis the association between the other SNP and NSCL/P. The conditional analysis results indicated that the effect of rs17095681 was not correlated with rs7078160 (P value of rs17095681 was Pmeta = 7.82E-4 conditioned on rs7078160, Supplementary Table S2). The LD analysis indicated that rs17095681 was not linkage with rs7078160 in four validation cohort (r2 < 0.1, Supplementary Table S3). These results indicated that rs17095681 may be an independent locus associated with NSCL/P at 10q25.3 in Chinese Han and Hui populations.
Analysis of different genetic models
For the significant SNP rs17095681, we used other genetic models, the additive model, allelic model and genotypic model for further analysis. We observed that this locus significant under the dominant model achieved similar results under the additive model (Pmeta = 8.96E-5), allelic model (Pmeta = 2.56E-5) and genotypic model (Pmeta = 9.85E-6) in validation stage (Table 2). In the combined analysis of the two stages, this locus achieved similar results with dominant model under the genotypic (het) model (Pmeta = 3.40E-9). It also achieved near genome-wide significance in additive (Pmeta = 6.50E-8) and allelic model (Pmeta = 5.65E-8). In Summary, the SNP rs17095681 was associated with NSCL/P significantly in additive model, allelic model and genotypic model.
Discussion
In this validation study of NSCL/P, we identified one SNP at 10q25.3, rs17095681, which were significantly associated with NSCL/P risk in Chinese Han and Hui populations. It reached the genome-wide significance threshold (Pmeta = 3.40E-9) in the combined analysis. The SNP rs17095681 is located in the intron of SHTN1 (also known as KIAA15598) and 27.2 kb downstream of VAX1 (ventral anterior homeobox 1, Fig. 1). It has been reported that four SNPs with PGWAS < E-4 are located in a 30-kb region that is 50 kb downstream of VAX18. The SNP rs17095681 is not in high LD (r2 > 0.8) with these SNPs. Rs7078160 has been successfully validated in a Chinese population11. The SNPs rs17095681 and rs7078160 are 33 kb apart from each other, but the effect of rs17095681 is not correlated with rs7078160 (Supplementary Table S2), indicating that rs17095681 is in an independent block associated with NSCL/P.

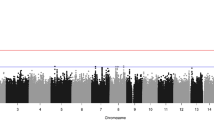
Regional plots of the susceptibility locus rs17095681.
Regional plots of newly discovered locus rs17095681 associated with the risk for NSCL/P in a Chinese population in the GWAS discovery stage. The results (−log10 P, P-value of GWAS) are shown for SNPs in the region 400 kb upstream or downstream of the marker SNP. The marker SNP is shown as a purple diamond in the GWAS stage and as a purple circle in the combined stage. The LD values (r2) between the SNPs and the most strongly associated SNP (diamond), rs17095681 are indicated by the heat scale. The genes within the region of interest are annotated, and the directions of transcripts are shown in arrows.
SHTN1 code a linker molecule shootin 1 that couples F-actin retrograde flow and the cell adhesion molecule (CAM) L1-CAM13 at neuronal growth cones to promote neuronal polarization and axon outgrowth. The attractive axon guidance molecule netrin-114,15 induces Pak1-mediated shootin1 phosphorylation in axonal growth cones16 which in turn enhances the coupling between F-actins and shootin1, thereby promoting the traction forces for axon outgrowth. It has been reported that netrin-1 gene NTN1 is associated with NSCL/P9,11,17. NTN1 encodes the protein NETRIN 1, which plays a role in the developing the nervous system by promoting both axonal outgrowth and axonal guidance in pathfinding18,19,20,21. In addition, NTN1 was up regulated in dental pulp stem cell cultures from NSCL/P patients11. This information suggested that SHTIN1 and NTN1 play important roles in the development of NSCL/P.
The other gene near SNP rs17095681, VAX1 was also been reported associated with NSCL/P. Mice with homozygous Vax1 mutations display craniofacial malformations including cleft palate22. Two individuals with a 10q terminal deletion syndrome with breakpoints in 10q25 have been reported, one with a submucous cleft palate23 and the other with a cleft lip24.
In summary, in this validation of a NSCL/P GWAS in Chinese populations, we identified a susceptibility locus at 10q25.3 that reached genome-wide significance. The rs1709568 SNP at 10q25.3 is located in an independent block and therefore is not related to any previously reported associated loci. Genes near this locus participate in the processes of neuronal axon outgrowth and cell migration. It is known that cell migration are crucial for oralfacial development. Further studies with larger sample sizes are warranted to replicate our findings. Fine mapping around this locus and related functional studies should also be performed to elucidate the molecular mechanisms underlying the observed associations.
Methods
Study populations
We performed the validation study using samples from four regions in China, including 1931 NSCL/P cases and 2258 controls. A summary of all cases and controls in the study is provided in Table 3. All of the samples were unrelated individuals of Chinese descent, obtained from Guangdong Maternal and Child Health Care Hospital (validation a, 497 Chinese Han cases and 497 Chinese Han controls for validation), Western China Hospital of Stomatology Sichuan University (validation b, 480 Chinese Han cases and 482 Chinese Han controls, independent with GWAS samples), the Institute of Stomatology, Nanjing Medical University (validation c, 446 Chinese Han cases, 522 Chinese Han controls, independent with GWAS samples), and General Hospital of Ningxia Medical University (validation d, 245 Chinese Han cases, 423 Chinese Han controls; validation e, 263 Chinese Hui cases and 334 Chinese Hui control). All cases were recruited in local hospitals and independently confirmed as NSCL/P by two gynecologic pathologists during routine diagnosis. Syndromic cleft lip or palate patients and cleft palate-only patients were excluded. Controls were recruited in local hospitals for individuals receiving routine physical examinations or healthy newborns whose parents volunteered to donate their umbilical cord blood. All controls were clinically assessed to be without cleft lip or palate or family history of cleft lip or cleft palate (including first, second, and third degree relatives). The cases and controls were frequency-matched for age and gender. At recruitment, informed consent was obtained from each subject. This study was approved by the ethics committees of Guangdong Maternal and Child Health Care Hospital, Western China Hospital of Stomatology Sichuan University, the Institute of Stomatology, Nanjing Medical University and General Hospital of Ningxia Medical University and the methods were carried out in accordance with the approved guidelines.
SNP selection and genotyping
SNPs for the replication stage were selected using the following criteria: (i) SNPs with P < 1.00E-5 in the first GWA study but were not significant or imputed successfully in the second GWA study; (ii) only the SNP with the lowest P-value was selected when multiple SNPs were observed but in strong linkage disequilibrium (LD) (r2 > = 0.8); (iii) primers could be successfully designed using Sequenom primer design software; and (iv) SNPs had not previously been validated. A total of 16 SNPs that matched these criteria were included in the replication stage. Genotyping of replicates was conducted by the Sequenom MassARRAY system at Beijing CapitalBio Technology Company, Beijing, China.
Quality control at the replication stage
We excluded SNPs with a call rate < 90% or a deviation from Hardy-Weinberg equilibrium (P < 0.05) in the controls. All 16 SNPs exceed quality control and were used for further analysis.
Association analysis in the replication and combined stage
For the replication studies, associations between SNP genotypes and disease status were assessed in a dominant model in PLINK v1.07 (http://pngu.mgh.harvard.edu/Bpurcell/plink/) using logistic regression modeling with gender as a covariate. Joint analyses of all combined samples at the validation stage and GWAS stage were conducted by using either the random effects model (I2 > 25%) or by using the fixed-effect model (I2 < 25%). Another genetic model (the additive, allelic and genotypic model) was also calculated for the associated SNPs. The chromosome regions of significant loci were plotted using an online tool, LocusZoom 1.1 (http://csg.sph.umich.edu/locuszoom/).
Additional Information
How to cite this article: Wang, Y. et al. Validation of a genome-wide association study implied that SHTIN1 may involve in the pathogenesis of NSCL/P in Chinese population. Sci. Rep. 6, 38872; doi: 10.1038/srep38872 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Dixon, M. J., Marazita, M. L., Beaty, T. H. & Murray, J. C. Cleft lip and palate: synthesizing genetic and environmental influences. Nat Rev Genet. 12, 167–178 (2011).
Jugessur, A. et al. Genetic determinants of facial clefting: analysis of 357 candidate genes using two national cleft studies from Scandinavia. PLoS ONE. 4, e5385. doi: 10.1371/journal.pone.0005385 (2009).
Zucchero, T. M. et al. Interferon regulatory factor 6 (IRF6) gene variants and the risk of isolated cleft lip or palate. N Engl J Med. 351, 769–780 (2004).
Richardson, R. J., Dixon, J., Jiang, R. & Dixon, M. J. Integration of IRF6 and Jagged2 signalling is essential for controlling palatal adhesion and fusion competence. Hum Mol Genet. 15, 2632–2642 (2009).
Rahimov, F. et al. Disruption of an AP-2α binding site in an IRF6 enhancer is strongly asso ated with cleft lip. Nat Genet. 40, 1341–1347 (2008).
Birnbaum, S. et al. Key susceptibility locus for nonsyndromic cleft lip with or without cleft palate on chromosome 8q24. Nat Genet. 41, 473–477 (2009).
Grant, S. F. et al. A Genome-wide association study identifies a locus for nonsyndromic cleft lip with or without cleft palate on 8q24. J Pediatr. 155, 909–913 (2009).
Mangold, E. et al. Genome-wide association study identifies two susceptibility loci for nonsyndromic cleft lip with or without cleft palate. Nat Genet. 42, 24–26 (2010).
Beaty, T. H. et al. A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat Genet. 42, 525–529 (2010).
Ludwig, K. U. et al. Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat Genet. 44, 968–971 (2012).
Sun, Y. et al. Genome-wide association study identifies a new susceptibility locus for cleft lip with or without a cleft palate. Nat Commun. 6, 6414. doi: 10.1038/ncomms7414 (2015).
Uslu, V. V. et al. Long-range enhancers regulating Myc expression are required for normal facial morphogenesis. Nat Genet. 46, 753–758 (2014).
Kamiguchi, H., Hlavin, M. L., Yamasaki, M. & Lemmon, V. Adhesion molecules and inherited diseases of the human nervous system. Annu Rev Neurosci. 21, 97–125 (1998).
Serafini, T. et al. The netrins define a family of axon outgrowth-promoting proteins homologous to C. elegans UNC-6. Cell. 78, 409–424 (1994).
Li, X. et al. Netrin signal transduction and the guanine nucleotide exchange factor DOCK180 in attractive signaling. Nat. Neurosci. 11, 28–35 (2008).
Toriyama, M., Kozawa, S., Sakumura, Y. & Inagaki, N. Conversion of a signal into forces for axon outgrowth through Pak1-mediated shootin1 phosphorylation. Curr. Biol. 23, 529–534 (2013).
Beaty, T. H. et al. Confirming genes influencing risk to cleft lip with/without cleft palate in a case-parent trio study. Hum. Genet. 132, 771–781 (2013).
Liu, G. et al. Netrin requires focal adhesion kinase and Src family kinases for axon outgrowth and attraction. Nat. Neurosci. 7, 1222–1232 (2004).
Li, W. et al. Activation of FAK and Src are receptor-proximal events required for netrin signaling. Nat. Neurosci. 7, 1213–1221 (2004).
Ren, X. R. et al. Focal adhesion kinase in netrin-1 signaling. Nat. Neurosci. 7, 1204–1212 (2004).
Masuda, T., Yaginuma, H., Sakuma, C. & Ono, K. Netrin-1 signaling for sensory axons: Involvement in sensory axonal development and regeneration. Cell Adh. Migr. 3, 171–173 (2009).
Hallonet, M., Hollemann, T., Pieler, T. & Gruss, P. Vax1, a novel homeobox- containing gene, directs development of the basal forebrain and visual system. Genes Dev. 1, 3106–3114 (1999).
Petersen, B., Strassburg, H. M., Feichtinger, W., Kress, W. & Schmid, M. Terminal deletion of the long arm of chromosome 10: a new case with breakpoint in q25.3. Am J Med Genet. 77, 60–62 (1998).
Mulcahy, M. T., Pemberton, P. J., Thompson, E. & Watson, M. Is there a monosomy 10qter syndrome? Clin Genet. 21, 33–35 (1982).
Acknowledgements
This work is funded by the China National High-Tech Research and Development Program Grant (2012AA020101) and partly funded by the National Key Basic Research Program Grant (2012CB720703), the Key Project of the National Natural Science Foundation of China (81230022, 81170981, and 81200808), the National Natural Science Foundation of China (81160131), Ph.D. Programs Foundation of Ministry of Education of China (20113234110003), the Key Project of the Ningxia Province Natural Science Foundation (NZ141116), and The Natural Science Foundation of Jiangsu Province (BK2012447). The authors thank all participants who donated biological samples and questionnaire information to this study. We thank all staff and students who have worked on this project.
Author information
Authors and Affiliations
Contributions
J.C., Y.Y., L.W. and A.Y. directed the study, obtained financial support and wereresponsible for the study design, interpretation of results and manuscript writing. Y.W., performed overall project management with Y.S., Y.H. and Y.P., performed statistical analyses with L.Z. and X.X. and drafted the initial manuscript. B.S., M.J. and H.J. directed each participating study and jointly organized this study. X.D. and L.M. were responsible for sample processing and managed the genotyping data. J.S., X.D. and Z.Z. were responsible for subject recruitment and sample preparation of Huaxi and Ningxia samples. H.J. and L.M. were responsible for subject recruitment and sample preparation of Nanjing samples. F.L. were responsible for subject recruitment and sample preparation of Guangzhou samples. All authors approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wang, Y., Sun, Y., Huang, Y. et al. Validation of a genome-wide association study implied that SHTIN1 may involve in the pathogenesis of NSCL/P in Chinese population. Sci Rep 6, 38872 (2016). https://doi.org/10.1038/srep38872
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep38872
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
