
Abstract
Pseudomonas aeruginosa, an opportunistic, but serious multidrug-resistant pathogen, secretes a ceramidase capable of cleaving the N-acyl linkage of ceramide to generate fatty acids and sphingosine. We previously reported that the secretion of P. aeruginosa ceramidase was induced by host-derived sphingolipids, through which phospholipase C-induced hemolysis was significantly enhanced. We herein investigated the gene(s) regulating sphingolipid-induced ceramidase expression and identified SphR, which encodes a putative AraC family transcriptional regulator. Disruption of the sphR gene in P. aeruginosa markedly decreased the sphingomyelin-induced secretion of ceramidase, reduced hemolytic activity, and resulted in the loss of sphingomyelin-induced ceramidase expression. A microarray analysis confirmed that sphingomyelin significantly induced ceramidase expression in P. aeruginosa. Furthermore, an electrophoretic mobility shift assay revealed that SphR specifically bound free sphingoid bases such as sphingosine, dihydrosphingosine, and phytosphingosine, but not sphingomyelin or ceramide. A β-galactosidase-assisted promoter assay showed that sphingosine activated ceramidase expression through SphR at a concentration of 100 nM. Collectively, these results demonstrated that sphingosine induces the secretion of ceramidase by promoting the mRNA expression of ceramidase through SphR, thereby enhancing hemolytic phospholipase C-induced cytotoxicity. These results facilitate understanding of the physiological role of bacterial ceramidase in host cells.
Similar content being viewed by others


Introduction
Sphingolipids are ubiquitous components of eukaryotic cell membranes; however, these sphingoid base-containing lipids are not present in prokaryotes, except for specific bacteria such as Sphingomonas and Sphingobacterium1. In mammals, sphingolipids are well-known lipid mediators that participate in a member of cellular events such as cell growth, differentiation, and apoptosis2,3. In the sphingolipid-mediated signaling cascade, sphingolipid metabolites exert different biological activities, and, thus, enzymes involved in the sphingolipid metabolic pathway have been intensively studied4. Accumulating evidence has indicated that sphingomyelin (SM) is enzymatically converted to ceramide (Cer), sphingosine (Sph), and then Sph 1-phosphate (S1P) by sphingomyelinase (SMase), ceramidase (CDase) and Sph kinase, respectively2. S1P is degraded by S1P lyase to form hexadecenal and phosphoethanolamine, which are further metabolized to fatty acids and glycerophospholipids, respectively5.
CDase (EC 3.5.1.23) is an amidohydrolase capable of cleaving the N-acyl linkage between Sph and the fatty acids of Cer6. CDase has been classified into three groups, i.e., acid, neutral, and alkaline CDases depending on optimal pH and primary structures. Acid CDase, composed of α- and β-subunits, is involved in the catabolism of Cer in lysosomes7. Neutral CDase, a single polypeptide enzyme, is localized at the plasma membrane as a type II integral membrane protein or is present as a soluble form8. Alkaline CDase, the smallest polypeptide among the three CDases, is localized at the ER or Golgi apparatus, at which they participate in controlling intracellular Cer levels9. The three CDases are not related to each other based on their primary structures and may have evolved from different ancestor genes. The distribution of the three CDases is somewhat different; however, mammals possess the three types of CDase, i.e., acid CDase is found in vertebrates only, whereas alkaline CDase has been detected in yeasts and mammals, but not in prokaryotes. On the other hand, the genetic information on neutral CDase is conserved from bacteria to humans. We previously identified the neutral CDase in Pseudomonas aeruginosa (formerly designated as alkaline CDase due to an optimum pH at 8.5, but was re-classified into neutral CDase after real alkaline CDases showing an optimum pH at 9–10 were found) and cloned its gene8,10,11. Furthermore, we elucidated the crystal structure of P. aeruginosa CDase, and demonstrated that it cleaved the N-acyl linkage of Cer using a similar mechanism to a zinc-dependent carboxypeptidase12.
The Gram-negative bacterium P. aeruginosa is a widespread environmental bacillus that causes a wide range of severe opportunistic infections in individuals with weakened immune systems13,14. However, healthy individuals may also develop mild illnesses caused by this bacterium. Furthermore, an increase in multidrug-resistant strains of P. aeruginosa is a serious issue for hospital-acquired infections. P. aeruginosa is known to produce multiple virulence factors including proteases, lipases, phospholipases, and exotoxins. These virulence factors play important roles when P. aeruginosa invades host cells and causes severe cellular damage15,16. Among these virulence factors, hemolytic phospholipase C (PlcH) hydrolyzes phosphatidylcholine (PC) and SM to generate diacylglycerol (DG) and Cer, respectively, and phosphorylcholine. Since PC and SM are ubiquitous lipid components in mammalian membranes, PlcH lyses human and sheep erythrocytes17,18 and causes severe damage to host cells19,20. We also demonstrated that PlcH-induced hemolysis was significantly attenuated in CDase-deficient mutants of P. aeruginosa, indicating that CDase is also a virulence factor of P. aeruginosa that enhances the cytotoxicity of PlcH21.
P. aeruginosa possesses metabolic pathways for various lipids in order to utilize them as nutrients, and, thus, this microbe has the ability to survive under physiologically severe conditions22,23,24. For example, PC is initially hydrolyzed to phosphorylcholine and DG and is then further catabolized to phosphate, choline, glycerol, and fatty acids, which are utilized by P. aeruginosa as sources of phosphorus, nitrogen, and carbon23,25.
Although P. aeruginosa possesses PlcH and neutral CDase (CerN, neutral CDase of P. aeruginosa is registered as CerN in the P. aeruginosa genome database), this pathogen does not possess sphingolipids such as SM, Cer, and Sph. PlcH and CerN were found to be secreted into the culture medium of P. aeruginosa when SM was added21. The expression of PlcH in P. aeruginosa is transcriptionally controlled by the concentration of phosphorus ions; however, the mechanism by which sphingolipids induce the expression of CerN currently remain unclear. In this study, we identified the transcriptional regulator SphR, which mediates the gene expression of CerN in response to sphingolipids, and intensively investigated the specificity of SphR for sphingolipids. It is important to note that the specificity of SphR toward sphingolipids is strict, namely, it responds to free sphingoid bases such as sphingenine, sphinganine (dihydro-Sph), and 4-hydroxysphinganine (phyto-Sph), but not to SM, Cer, 1-deoxy-Sph, or S1P.
This study provides the molecular basis for understanding the mechanisms responsible for the transcriptional regulation of CerN in bacteria that do not possess sphingolipids and facilitates understanding on how pathogens incorporate sphingolipids as a nutrient to survive under severe conditions including the host environment.
Results
Identification of gene(s) involved in sphingolipid-mediated CerN induction in P. aeruginosa
We previously reported that the release of CerN into a culture medium of P. aeruginosa was strongly enhanced by the addition of sphingolipids such as SM, Cer, and Sph21. In order to elucidate the molecular mechanism of underlying sphingolipid-mediated CerN induction, we adopted a transposome-based gene disruption strategy in the present study. We initially generated a P. aeruginosa mutant (AN17-BGT), in which the cerN gene was replaced with the E. coli β-galactosidase gene (Fig. 1). Using this mutant, the induction of CerN with sphingolipids was easily monitored by β-galactosidase activity, which may be assessed using chromogenic substrates such as pNP-β-galactose. We then introduced EZ-Tn5 <KAN-2> Tnp Transposome into AN17-BGT and selected mutants lacking β-galactosidase activity in the presence of SM. After the screening of 1,700 mutants, we identified PA5324 as a candidate gene involved in sphingolipid-mediated CerN induction. In the genome database of P. aeruginosa PAO126,27, PA5324 was assigned as the AraC-type transcriptional regulator, SphR28.

Genomic organization of PA0842-PA0847 in the wild type (WT) and mutant (AN17-BGT) of P. aeruginosa.
Arrows represent open reading frames and indicate their orientations and sizes.
Functions of SphR in hemolysis caused by P. aeruginosa
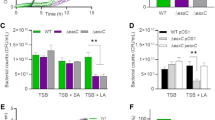
We generated SphR-deficient mutants of P. aeruginosa using the tetracycline resistance (Tcr) cassette as described in the “Methods” section. The CerN activities of wild-type P. aeruginosa (WT) and SphR-deficient mutants were measured in the presence or absence of SM. As shown in Fig. 2a, CerN activity increased in WT when SM was added to the culture, while its activity was retained at the basal level in SphR-deficient mutants (KO) even in the presence of SM. Similarly, the deletion of SphR in P. aeruginosa resulted in hemolytic activity being significantly lower than that in WT, and this was restored in part by the complementation of SphR to the SphR-deficient mutant, indicating that hemolytic activity is also regulated by SphR (Fig. 2b). Since CerN enhances the hemolytic activity of PlcH21, these results strongly suggest that SphR plays an important role in hemolysis caused by P. aeruginosa through the regulation of CerN.

CerN activity, hemolytic activity, cerN expression, and cell growth of the wild-type (WT) and SphR-null mutant (KO) of P. aeruginosa in the presence or absence of SM.
(a) CerN activity of the WT and SphR-null mutant of P. aeruginosa PAO1. Strains were cultured at 30 °C for 10 h with PY medium in the presence (200 μM) or absence of SM. The CerN activity of the culture supernatant was measured as described in the Methods section. Values are expressed as the mean ± S.D. (n = 3). (b) Time course for the hemolysis of sheep erythrocytes by WT, KO, KO complemented with SphR, and the mock transfectant. Hemolytic activity was measured by the method described in the “Methods” section. Values are expressed as the mean ± S.D. (n = 4). ●, WT; ○, KO; □, KO complemented with SphR; ■, mock. Significance is shown as follows: *p < 0.05; **p < 0.01; ***p < 0.001; significantly different from WT for KO or KO complemented with SphR for the mock. (c) Expression of cerN in the presence or absence of SM. The mRNA levels of cerN and PA3001 were analyzed by real-time RT-PCR. Open and black bars represent the WT and KO, respectively. Values are expressed as the mean ± S.D. (n = 3). Significance is shown as follows: ***p < 0.001 significantly different from WT. (d and e) Cell growth of the WT and SphR-null mutant of P. aeruginosa. (d) Strains were cultured at 30 °C for 4 h with 190 μl of PY medium in the absence (open bars) or presence (gray bars) of 200 μM SM. After cultivation, 10 μl of the detection reagent was added to the wells of a 96-well plate and incubated at 30 °C for 2 h. (e) Strains were cultured at 30 °C for 20 h with 190 μl of SM synthetic medium (0.05% NH4Cl, 0.05% K2HPO4, 0.5% NaCl, 0.05% TDC, and 0.05% SM). After cultivation, 10 μl of the detection reagent was added to the wells of a 96-well plate and incubated at 30 °C for 1 h. Values are expressed as the mean ± S.D. (n = 4). Significance is shown as follows: ***p < 0.001 significantly different from WT. Statistical analyses were performed by Welch’s t-test.
Roles of SphR in the transcriptional regulation of CerN
We then examined the roles of SphR in the transcription of the cerN gene in P. aeruginosa using real-time PCR. The addition of SM resulted in cerN transcription levels being more than 80-fold higher in WT than in the absence of SM; however, this sphingolipid-mediated increase in cerN transcription was not observed in SphR-deficient mutants (Fig. 2c). The transcription of PA3001 (housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase) did not increase in WT with the addition of SM. These results indicate that the sphingolipid-mediated induction of CerN occurs at the transcriptional level and is regulated by SphR.
Effects of the deletion of SphR on P. aeruginosa growth
We compared the cell growth of WT and SphR-deficient mutants in nutrition-rich medium (PY medium) and synthetic medium containing SM (SM synthetic medium) as the sole source of carbon. No significant differences were observed in cell growth between WT and SphR-deficient mutants in PY medium in the presence or absence of SM (Fig. 2d). On the other hand, the growth of SphR-deficient mutants was markedly slower than that of WT in SM synthetic medium (Fig. 2e), suggesting that SM is not utilized by SphR-deficient mutants as a source of carbon because CerN was not expressed in P. aeruginosa without SphR (Fig. 2a,c).
DNA microarray analysis
SM is an abundant lipid in the plasma membranes of host cells and may influence the expression of genes involved in SM metabolism in P. aeruginosa during the infectious process and colonization. Therefore, we performed a DNA microarray analysis in order to identify the genes induced in P. aeruginosa when SM was added to the culture. The mRNA quantity of P. aeruginosa was measured using cells cultured in the presence or absence of SM (control). The expression of 94 genes was significantly stronger by more than 1.5-fold (P < 0.05), in the presence of SM than in the control (Table 1). Of those, the expression of 27 genes increased by more than 2-fold, and that of 5 genes by more than 6-fold. SM added to the culture of P. aeruginosa may be sequentially converted to phosphorylcholine and Cer by PlcH (SMase), choline and phosphate by phosphatase, and fatty acids and Sph by CerN. Thus, the results of the DNA microarray may reflect gene expression induced not only by SM, but also by SM metabolites such as Cer, Sph, fatty acids, phosphorylcholine, and choline. As expected, the transcript of the cerN gene (PA0845) showed the greatest increase in P. aeruginosa in the presence of SM than in the control (~19-fold, P = 4.91E-6). This result is consistent with those shown in Fig. 2c. The expression of plcH (PA0844) and its accessory protein gene (plcR, PA0843) also increased in the presence of SM, however, the degree of this increase was lower than cerN (2-fold, P < 0.001). The transcriptional level of SphR did not change, whereas those of PA5325 - PA5328, which are adjacent to sphR (PA5324) in the P. aeruginosa genome, increased by 6~44-fold in the presence of SM; however, the biological relevance of these four genes remains to be clarified. Although transcripts of genes related to sulfur metabolism (PA0280-PA0283, PA1837-PA1838, and PA4442-PA4443) moderately increased (2~3-fold) in the presence of SM, the mechanism underlying the regulation of these genes by SM is unknown. The expression of PA5373 (betB) and PA5374 (betI), which form an operon for choline catabolism with PA5372 (betA), increased 2-fold in the presence of SM. Furthermore, PA5375 (betT1), a putative choline transporter, exhibited a significant increase (1.2-fold, P = 0.0138). The increase observed in the transcription of choline catabolism-related genes (bet genes) appeared to be mediated by choline, but not SM25. Similarly, the transcription of Fad (fatty acid degradation) genes (PA3014/FaoA/FadB5, PA3013/FaoB/FadA5, PA4994/probable acyl CoA dehydrogenase, and PA4995/probable acyl CoA dehydrogenase) was found to moderately increase in the presence of SM, suggesting that the expression of Fad genes in P. aeruginosa is regulated by fatty acids released from SM through the corporative reactions of PlcH (SMase) and CerN.
Lipids inducing cerN expression
As previously reported, not only SM, but also its metabolites such as Cer and Sph induce CerN activity in P. aeruginosa21. However, we found that β-galactosidase activity was only strongly induced when P. aeruginosa AN17-BGT was cultured in the presence of Sph, but not SM, Cer, or palmitic acid (Pal) (Fig. 3a). In AN17-BGT, SM may be hydrolyzed to Cer by PlcH, whereas Cer is not converted to Sph because the cerN gene was replaced with the β-galactosidase gene in this mutant strain. This result suggests that the transcription of the cerN gene is induced by Sph, but not SM in P. aeruginosa through SphR. In order to elucidate the relationship between the Sph concentrations and extent of the transcription of the cerN gene, AN17-BGT was cultured with Sph at various concentrations and β-galactosidase activity was measured. As shown in Fig. 3b, β-galactosidase activity was detected in the presence of no less than 100 nM Sph and activity increased up to 100 μM Sph. These results indicate that the cerN gene is transcriptionally activated by Sph in a concentration-dependent manner.

Effects of various lipids on the activation of the cerN promoter.
(a) Effects of SM and its metabolites on the activation of the cerN promoter. A 15-μl aliquot of the strain AN17-BGT cell suspension (A600 = 2.0) was inoculated into 1.5 ml of PY medium containing 0.05% sodium taurodeoxycholate in the presence or absence of lipids and cultured at 30 °C for 8 h. After cultivation, promoter activities were measured by the method described in the “Methods” section. -; Control experiment without lipids, SM; sphingomyelin, Cer; ceramide, Pal; palmitic acid, Sph; sphingosine. Values are expressed as the mean ± S.D. (n = 3). **p < 0.01; ***p < 0.001; significantly different from the control experiment without lipids. (b) Sph dependence of the cerN promoter. A 15-μl aliquot of the AN17-BGT cell suspension (A600 = 2.0) was inoculated into 1.5 ml of PY medium containing 0.05% sodium taurodeoxycholate in the presence of Sph and cultured at 30 °C for 8 h. After cultivation, promoter activities were measured by the method described in the “Methods” section. Values are expressed as the mean ± S.D. (n = 3). **p < 0.01; ***p < 0.001; significantly different from the control experiment without Sph. Statistical analyses were performed by Welch’s t-test.
Analysis of the cerN promoter
By using 5′-RACE, we showed that the cerN transcription start point was located 80 nucleotides upstream of ATG (Fig. 4a). In order to identify the minimal promoter region required for cerN transcription, we generated plasmid constructs containing different sized promoter regions driven by LacZ. P. aeruginosa PAO1 was transformed with the constructs (deletion mutants) and the promoter activities of the mutants were evaluated by measuring β-galactosidase activity in the presence of SM. As shown in Fig. 4b and c, β-galactosidase activity was markedly weaker in the mutant possessing 100-bp, but not 200-bp nucleotides from the transcription start point than in the control possessing the putative full promoter sequence, indicating that an integral region for Sph-inducible transcription is present between −200 and −100 bp upstream of the cerN gene. Further investigations using several deletion mutants possessing 135- to 200-bp nucleotides identified the critical region in the cerN promoter, which was present between −155 and −135 bp upstream of the cerN gene (Fig. 4b and d).

Promoter analysis for the cerN gene.
(a) The transcription initiation site was identified by 5′-RACE. The ATG start codon (PA0845, CerN), stop codon (PA0846), and cerN transcriptional start site (indicated by the bent arrow) are indicated in bold typeface. Underlined nucleotides indicate the putative −35 and −10 promoter sequences and Shine-Dalgarno (SD) sequence. (b) A map of the cerN promoter region. The ATG start codon and cerN transcriptional start site are indicated by arrows. (c and d) Schematic map of the truncation constructs for the cerN promoter and the promoter activity of each construct. A 15-μl aliquot of the cell suspension (A600 = 2.0) was inoculated into 1.5 ml of PY medium containing 0.05% sodium taurodeoxycholate in the presence of 200 μM SM and cultured at 30 °C for 9 h. After cultivation, promoter activities were measured by the method described in the “Methods” section. Promoter activities are expressed in Miller units. +1 denotes the cerN ATG start codon. Values are expressed as the mean ± S.D. (n = 3). Significance is shown as follows: *p < 0.05; **p < 0.01; ***p < 0.001; significantly different from Full in the presence of SM for (c) and −200 in the presence of SM for (d). Statistical analyses were performed by Welch’s t-test.
In order to elucidate the nucleotide sequence required for binding to Sph in the cerN promoter, the sequence of PA0845 (cerN) was aligned with that of the PA5325 and a PA5328-PA5326 operon, all of which may be regulated by SphR (Table 1). The conserved nucleotides among these regions were asterisked (Fig. 5a) and the putative consensus sequences were underlined (Fig. 5b). The estimated consensus region appeared to consist of two repeating 15-bp nucleotides separated by a 7-bp spacer when SphR formed a dimer similar to other bacterial transcriptional regulators such as AraC. We generated several mutants (M1–M4), in which a few nucleotides were replaced with A in the conserved sequence and evaluated the promoter activities of these 4 mutants in the presence of SM. As a result, we found that the mutants M1, M2, and M4 completely lost their promoter activities, while the activity of the mutant M3 decreased to one fourth that of WT (Fig. 5c). This result indicates that the conserved sequence is critical for SM-inducible promoter activity.

Identification of the SphR-binding sequence in the cerN promoter.
(a) Alignment of the PA0845 (cerN) promoter region and 5′-UTR of PA5325 and PA5328. Alignment was performed using the CLUSTAL algorithm58. Identical nucleotides in all three sequences are indicated by asterisks and identical nucleotides in two sequences are indicated by dots. (b) Alignment of conserved sequences in the promoter regions of the PA0845 (cerN) and 5′-UTR of PA5325 and PA5328. Upper, identical nucleotides in all three sequences and those that are conserved in two sequences are indicated in the consensus sequence by uppercase and lowercase letters, respectively. Putative SphR-binding sequence motifs are underlined. Lower, point mutations (M1~M4) in the conserved region are underlined. (c) Promoter activity of each construct. Promoter activities are expressed in Miller units. Values are expressed as the mean ± S.D. (n = 3). Significance is shown as follows: ***p < 0.001; significantly different from WT in the presence of SM. Statistical analyses were performed by Welch’s t-test.
Specificity of SphR toward various lipids
The transcription of the cerN gene was induced in P. aeruginosa by Sph, but not SM, Cer, or fatty acids (Fig. 3a). We examined whether the Sph-inducible transcriptional regulation of CerN depends on SphR by evaluating the specificity of SphR toward various lipids. Recombinant MBP-fused SphR (MBP-SphR, 84 k) was used in this experiment because free SphR was not soluble under the conditions used. The binding of MBP-SphR to the cerN promoter was examined using an electrophoretic mobility shift assay (EMSA), in which the biotin-labeled cerN promoter (−200~−81) was used as a probe. As shown in Fig. 6a, MBP-SphR formed a complex with the probe in order to shift up on the gel in the presence of Sph; however, this mobility shift of the probe was not observed in the absence of Sph. The binding of MBP-SphR to the cerN promoter attenuated competitively with the addition of the unlabeled probe. No complex was formed with MBP per se and the cerN promoter. These results indicate that SphR binds to the cerN promoter in the presence of Sph. We then evaluated the specificity of SphR to various lipids by EMSA and found that the specificity of SphR was very strict, i.e., SphR bound to Sph, but not SM, Cer, Pal, DG, or PC under the conditions used (Fig. 6b). Among the long-chain bases tested, SphR bound to Sph, dihydroSph, and phytoSph, but not to S1P or 1-deoxy-Sph. Furthermore, SphR recognized the stereoisomers of Sph, i.e., it bound to D-erythro-Sph more strongly than L-threo-Sph (Fig 6c). These results clearly indicate that SphR recognizes the OH group at C1 and stereochemical configuration at C3 of Sph; however, the double bond between C4 and C5, or hydroxy group at C4 of Sph is not critical for binding to SphR. An acyl chain length of more than 14 in Sph is sufficient for binding to SphR.

Specificity of MBP-SphR for sphingoid bases.
(a–c) An electrophoretic mobility shift assay (EMSA) showing the DNA-binding activity of MBP-SphR. (a) A biotin-labeled probe (0.5 nM) was incubated with MBP-SphR (1.5 μM) or MBP (1.5 μM) as indicated for 30 min, and this was followed by electrophoresis, membrane transfer, and avidin-HRP detection. Ligand specificity was analyzed using various lipids (b) and Sph derivatives (c). (b) -; Control experiment without lipids, Sph; sphingosine, Cer; ceramide, Pal; palmitic acid, SM; sphingomyelin, DG; diacylglycerol, PC; phosphatidylcholine. (c) -; Control experiment without lipids, Sph; sphingosine, DHS, dihydrosphingosine, PHS; phytosphingosine, L-t-Sph; L-threo-sphingosine, 1-de-Sph, 1-deoxy-sphingosine, S1P; sphingosine-1 phosphate, C16Sph; C16-sphingosine, C14Sph; C14-sphingosine. The unbound probe and shift product are indicated by black and white arrows, respectively. (d) Binding of MBP-SphR to biotin-Sph. The binding of MBP-SphR to Sph was assessed using a pull-down assay with biotin-Sph. Values are expressed as the mean ± S.D. (n = 3). Significance is shown as follows: ***p < 0.001; significantly different from the control experiment without biotin-Sph or with MBP. Statistical analyses were performed by Welch’s t-test.
In order to examine whether Sph directly binds to purified MBP-SphR, we performed a pull-down assay, in which a complex of MBP-SphR with biotin-labeled Sph was pulled down by avidin-labeled magnetic beads and detected on SDS-PAGE after staining with Coomassie Brilliant Blue. As shown in Fig. 6d, MBP-SphR was only detected in the presence of biotin-labeled Sph and MBP per se did not bind to biotin-Sph, indicating that SphR directly binds to Sph.
In conclusion, we demonstrated that Sph specifically binds to SphR, through which the transcription of the cerN gene is initiated in P. aeruginosa, i.e., the Sph-inducible transcription of CerN entirely depends on SphR as a transcriptional regulator.
Discussion
In an attempt to elucidate the mechanism underlying the sphingolipid-mediated induction of CerN in P. aeruginosa, we examined gene(s) that regulate CerN transcription in response to sphingolipids. As a result, we identified the Sph-specific AraC-type transcriptional regulator, SphR, which is involved in Sph-mediated cerN gene expression. AraC-type transcriptional regulators are known to control a number of cellular processes such as carbon metabolism, stress responses, and virulence in Gram-positive and -negative bacteria29,30. Sixty-one putative AraC-type transcriptional regulators were identified in the P. aeruginosa genome when analyzed using a Pfam database31. Some AraC-type transcriptional regulators such as VqsM is related to quorum sensing32, PchR is involved in pyochelin (siderophore) biosynthesis33 and GbdR is involved in phosphorylcholine catabolism25,34. These transcriptional regulators were previously reported to be important for P. aeruginosa infections. AraC-type transcriptional regulators are generally composed of an N-terminal ligand binding domain that is responsible for dimerization and a C-terminal helix-turn-helix (HTH) DNA-binding domain. The binding of a ligand to the N-terminal domain of the transcriptional regulator induces a conformational change, by which it binds to the promoter region of the target gene via a C-terminal domain. We speculated that SphR may be converted to an active form when Sph binds to the N-terminal region, and this active form may then bind to the promoter region of the cerN gene via the C-terminal HTH motif, thereby initiating cerN mRNA transcription.
LaBauve and Wargo also recently identified SphR in P. aeruginosa using a different approach. They performed a DNA microarray in the presence or absence of pulmonary surfactants and found that the expression of several genes including PA5325 and PA5328-PA5326 (these three genes are regulated as an operon) increased under the conditions employed. They ascertained that these genes were transcriptionally regulated by Sph in pulmonary surfactants through SphR as a transcriptional regulator28. Since Sph is toxic for bacteria and the deletion of SphR was found to decrease the tolerance of P. aeruginosa toward Sph, they speculated that PA5325 (sphA) and PA5328-PA5326 (sphB, C, D) are genes related to the import and metabolism of Sph in P. aeruginosa, respectively.
P. aeruginosa synthesizes a small amount of PC (~4% of all phospholipids)35,36 and appears to possess all the enzymes required to metabolize this phospholipid. Thus, host-derived PC may be utilized by this bacterium as a nutrient for its growth and survival. Host PC is degraded by phospholipase C and DG lipases to phosphocholine, glycerol, and fatty acids in P. aeruginosa23. Fatty acids, phosphocholine, and glycerol are further metabolized by the β-oxidation pathway, Bet pathway, and Glp pathway in the bacterium, respectively. Son et al. found that these genes, which are involved in PC metabolism were up-regulated in P. aeruginosa by the addition of PC to the culture23. Sun et al. also reported that P. aeruginosa mutants lacking double or triple genes involved in PC metabolism exhibited a severe growth defect on synthetic medium containing PC as the sole carbon source as well as reduced survival rates in the lungs of mice37. These findings indicate that host PC is an integral nutrient for P. aeruginosa to infect and survive in the lungs of mice.
SM also appears to be a nutrient for P. aeruginosa. SM is a phosphocholine-possessing phospholipid, similar to PC; however, the lipid portion of SM is Cer instead of DG in PC. Host SM has been suggested to be hydrolyzed by two SM hydrolases (PlcB and PlcH) of P. aeruginosa in order to generate Cer and phosphocholine38. Cer is further decomposed by CerN into Sph and fatty acids. In the present study, we found that not only PlcH and its accessory protein (PlcR), but also the Fad (β-oxidation) and Bet (choline metabolism) genes were up-regulated in P. aeruginosa either in the presence of PC or SM. On the other hand, the DG-lipase and Glp (glycerol metabolism) genes were induced by PC, but not SM, while SphA and SphBCD were induced by SM, but not PC in P. aeruginosa. These results clearly indicate that SM is metabolized in part by the same pathway as PC; however, the metabolic pathways of Cer (Sph moiety) and DG (glycerol moiety) are completely different from each other in P. aeruginosa (see Supplementary Fig. S1).
Several genes were up-regulated in P. aeruginosa in the presence of SM, but not PC. Of these, PA0845 is the gene encoding neutral CDase (CerN) and PA5325 (sphA) is the gene encoding a homologue of porin, which makes a channel in the plasma membrane as a transporter. PA5328-PA5326 (sphBCD), three genes that form an operon, showed homology to cytochrome C oxidase (cbb3-type, subunit III), D-arabinono-1,4-lactone oxidase, and alanine racemase (N-terminal domain), respectively. These proteins may be involved in Sph transport and metabolism in P. aeruginosa; however, the physiological functions of these proteins have not yet been elucidated.
CerN may be induced in P. aeruginosa in the presence of not only Sph, but also SM or Cer21. However, we found that the promoter of cerN was activated by Sph, but not by SM or Cer when the strain ANT17-BGT was used instead of P. aeruginosa WT (Fig. 3a). This contradiction may be consistently explained, i.e., SM and Cer are converted to Sph by PlcH (SMase) and CerN in P. aeruginosa WT, but are not converted to Sph in the strain AN17-BGT because the CerN gene was replaced with the E. coli β-galactosidase gene and, thus, SM/Cer was not converted to Sph. On the other hand, the promoter of cerN in the strain AN17-BGT was activated by SM when we screened β-galactosidase-deficient strains (Fig. 1). We found that the egg yolk SM used in this experiment was contaminated with Sph (~0.5% of SM), and, thus, this experiment may be regarded as being performed in the presence of Sph. Thereafter, we used Sph-free SM in the experiments described in Figs 3a and 6b.
Figure 7 shows a working model for the functions of SphR in P. aeruginosa. The presence of a positive feedback loop has been suggested between SphR and CerN, i.e., 1) Host Sph is incorporated into P. aeruginosa and induces the expression of CerN through the activation of SphR, 2) CerN is secreted into the host environment, in which host-cell Cer is hydrolyzed to generate Sph, 3) the Sph generated is then incorporated into P. aeruginosa and increases CerN expression through the activation of SphR. This positive feedback loop amplifies the generation of Sph in host environments and may exacerbate damage to host cells. In this model, PlcH also plays an important role in the production of Cer from host-derived SM. PlcH is induced by the PhoBR regulon under phosphate starvation conditions and by the GbdR regulon with the catabolism of choline from phospholipids such as PC and SM. On the other hand, SphR regulates intracellular Sph levels in P. aeruginosa through the expression of sphABCD, which may be involved in the transport and metabolism of Sph28. Since Sph exhibited the antibacterial activity toward many bacteria including P. aeruginosa39,40, the regulation of intracellular Sph levels is integral for bacteria exposed to Sph. According to this point of view, the study by Pewzner-Jung et al. is of interest. They reported that tracheal and bronchial epithelial Sph levels play important roles in the prevention of P. aeruginosa in healthy individuals and Sph levels are reduced in patients with cystic fibrosis due to reductions in acid CDase activity41. Collectively, SphR exerts two different physiological functions in P. aeruginosa, i.e., attacking host cells through the generation of Sph by the expression of CerN and defending against Sph through a decrease in intracellular Sph levels by the expression of SphABCD.

Proposed mechanisms of SphR in P. aeruginosa.
The model shows Sph-induced cerN gene expression through SphR. The generation of Sph is amplified by a positive feedback loop between SphR and CerN. This figure also represents the regulation of intracellular Sph levels in the bacterium by SphR28. CerN; neutral CDase, Cer; ceramide, FA; fatty acids, PlcH; hemolytic phospholipase C, RNA Pol; RNA polymerase, SM; sphingomyelin, Sph, sphingosine.
We found that Sph at a concentration of no less than 100 nM induced the transcription of cerN in P. aeruginosa (Fig. 3b). This concentration of Sph was markedly lower than its physiological concentration in human serum (~250 nM)42, and, thus, we speculate that the cerN gene is expressed in P. aeruginosa in some host environments. We isolated a CDase-producing bacterium, P. aeruginosa AN17, from the skin of patients with atopic dermatitis (AD)10 and we demonstrated that the skin of these patients is significantly colonized by CDase-secreting bacteria, including P. aeruginosa, compared to that of healthy individuals43. The human epidermis contains a certain amount of Sph and dihydroSph44, which may induce the expression of cerN in P. aeruginosa-infected skin through SphR. We found that CerN hydrolyzes Cer in the human skin efficiently in the presence of anionic phospholipids derived from Staphylococcus aureus, which is found in atopic skin frequently, in place of detergents45. Recently, Oizumi et al. demonstrated that Sph produced by the action of CerN is converted to S1P and induces production of inflammatory mediators in human keratinocytes46. In addition, exogenous Sph, dihydroSph, and phytoSph are found to be modulators of differentiation and lipid metabolism in keratinocytes47,48. These observations indicate that CerN disturbs the skin barrier function by decreasing the levels of skin Cer and by increasing the long-chain bases. This study also suggests the important role of CerN as an exacerbating factor in P. aeruginosa-infected skin in conditions such as AD and related dermatitis. From another point of view, P. aeruginosa is an important opportunistic human pathogen, but is also found in diverse environmental conditions, such as soil and water. Because all eukaryotes and some bacteria possess sphingolipids as constituents of biological membranes, the bacterium may produce CerN through SphR, for decomposing SM or Cer from sphingolipid-producing organisms to obtain fatty acids as a nutrient source.
In order to obtain a better understanding of the relationship between SphR and CerN in bacteria, we performed a blast search using the amino acid sequences of SphR and CerN. Among 56 bacteria with SphR homologues (E-value < e-30), we found that 12 bacteria (including P. aeruginosa) had highly homologous sequences of CerN (see Supplemental Fig. S2). Furthermore, their distribution was found in Gram-negative and Gram-positive bacteria, and frequently in the genus Mycobacterium. Mycobacterium are well-known pathogens towards humans and animals; however, other bacteria, except for Collimonas pratensis and C. fungivorans, which have both SphR and CerN, have also been identified as pathogenic bacteria towards humans and animals. Although the functions of SphR homologues currently remain unknown, the distribution of both proteins enables us to verify the importance of SphR in CerN production in pathogenic bacteria.
In the present study, we clarified the mechanism underlying the transcriptional regulation of cerN in P. aeruginosa, in which SphR functions as an Sph-inducible transcriptional regulator. Furthermore, we present detailed biochemical results on the interaction between Sph and SphR. The results described here may contribute to developing SphR inhibitors that have the potential to become anti-microbial drugs for SphR-possessing pathogens including P. aeruginosa. The AN17-BGT strain developed in this study is a useful tool for the high throughput (HST) screening of inhibitors of the interaction between SphR and the cerN promoter or SphR and Sph, which is easily performed by detecting β-galactosidase activity using artificial substrates.
Methods
Materials
SM, sodium taurodeoxycholate, S1P, palmitic acid, DG, and Triton X-100 were purchased from Sigma-Aldrich Corp. (St. Louis, MO, USA). D-erythro-Sph, N-palmitoyl-Sph, and dihydro-Sph were purchased from Enzo Life Sciences, Inc. (Farmingdale, NY, USA). Phyto-Sph was purchased from Olbracht Serdary Research Laboratories (Toronto, Canada). Omega-biotinyl D-erythro-sphingosine (biotin-Sph), L-threo-Sph, and 1-deoxy-Sph were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL, USA). C14-Sph and C16-Sph were purchased from Matreya, LLC (State College, PA, USA). Chlorophenol red-β-D-galactopyranoside (CPRG) was purchased from Roche Diagnostics (Mannheim, Germany). Precoated Silica gel 60 TLC plates were purchased from Merck (Darmstadt, Germany). The pHP45Ωaac vector49 was donated by Dr. H. Niki, National Institute of Genetics, Japan. All other reagents were of the highest purity available.
Bacterial strains and media
A type strain of P. aeruginosa PAO1 (JCM14847) was provided by the Japan Collection of Microorganisms, RIKEN BRC, which is participating in the National BioResource Project of MEXT, Japan. P. aeruginosa strains were cultivated in PY medium (0.5% polypeptone, 0.1% yeast extract, and 0.5% NaCl, pH 7.2) at 30 °C. E. coli strains were grown in Luria- Bertani (LB) medium at 37 °C. Media were supplemented with antibiotics such as ampicillin (100 μg/ml for E. coli), carbenicillin (100 μg/ml for E. coli), kanamycin (50 μg/ml for E. coli and 1 mg/ml for P. aeruginosa), gentamycin (20 μg/ml for E. coli and 30 μg/ml for P. aeruginosa), and tetracycline (15 μg/ml for E. coli and 200 μg/ml for P. aeruginosa) where necessary.
Construction of AN17-BGT
All plasmids were constructed using PCR with PrimeSTAR MAX DNA polymerase (Takara Bio, Kusatsu, Japan) and the In-Fusion PCR cloning kit (Takara Bio). A 5-kbp fragment, which contained the 5′-flanking region, open reading frame, and 3′-flanking region of the cerN gene, was amplified by PCR using P. aeruginosa AN17 genome DNA as a template and the primers PaCD-5k-Infu1 and PaCD-5k-Infu2, and was then cloned into the BamHI site of pGEM-3Zf(+). The resulting plasmid was named pGEM-PaCD5k. In order to replace the cerN gene with the E. coli β-galactosidase gene, the cerN gene was removed by PCR using pGEM-PaCD5k as a template and the primers PaCD-5′UTR and PaCD-3′UTR. The fragment obtained was ligated with the β-galactosidase gene amplified from pGEM-LacZ using the primers PaCD-5′UTR-LacZ-5′ and PaCD-3′UTR-LacZ-3′. The resulting plasmid was named pGEM-PaCD5k-LacZ. pGEM-PaCD5k-LacZ was linearized by PCR using the primers PaCD3′UTR-3′U and PaCD3′UTR-5′L to introduce the Tcr cassette into the 3′UTR region of the cerN gene in pGEM-PaCD5k-LacZ. The Tcr cassette was amplified by PCR using pBR322 as a template and the primers pBR322-U1-PaCD3′ and pBR322-L1-PaCD3′, and was then ligated to linearized pGEM-PaCD5k-LacZ. The resulting plasmid was named pGEM-PaCD5k-LacZ-Tet. The cassette in pGEM-PaCD5k-LacZ-Tet was amplified by PCR using pGEM-PaCD5k-LacZ-Tet as a template and the primers PaCD Pro-5k-Infu1 and PaCD Pro-5k-Infu2, and was then ligated to the BamHI site of pK19mobsacB50. The sequences of the primers used in this study are summarized in Supplementary Table S1. The resulting plasmid was named pK19-PaCD5k-LacZ-Tet. The plasmid, which contains a mutant cerN disrupted with the β-galactosidase and Tcr genes, was transferred from the broad host range-mobilizing strain E. coli S17-151 to P. aeruginosa AN17 by biparental filter matings. Tcr plasmid integrants were selected on NAC medium containing Tc. Tcr colonies were then plated on LB agar containing 5% sucrose to identify strains that had lost the vector-associated sacB gene (resistant to sucrose). Gene replacement was ascertained by PCR of the cerN gene. The resulting strain was named AN17-BGT.
Construction of the AN17-BGT mutant library and screening of β-galactosidase activity-deficient cells
An EZ-Tn5 <R6Kγori/KAN-2> Tnp Transposome kit (Epicentre, Madison, WI, USA) was used to make the AN17-BGT mutant library according to the manufacturer’s instructions. In order to obtain mutants, a 100-μl aliquot of electrocompetent cells was mixed with 1 μl of the EZ-Tn5TM <R6Kγori/KAN-2> Tnp TransposomeTM (Epicentre), and electroporation was then performed with a Gene Pulser II electroporator (Bio-Rad Laboratories, Hercules, CA, USA) in 0.2-cm cuvettes at 2.5 kV, 25 μF, and 200 Ω. Cells were transferred to a 10-ml tube containing 2 ml of PY medium and incubated at 37 °C for 2 h with shaking. Transformants were selected on NAC agar containing 1 mg/ml kanamycin. The colonies obtained were added to the wells of a 96-well microplate containing 100 μl of PY medium. On the next day, a 10-μl aliquot of the culture was transferred to a new 96-well microplate containing 100 μl of PY medium with 200 μM SM from chicken egg yolk and incubated at 25 °C for 1 day. The β-galactosidase activity of each mutant was measured using CPRG. In order to permeabilize cells, 10 μl of 0.1% SDS and 1 drop of CHCl3 were added to each well and mixed well. The β-galactosidase assay was started by adding 50 μl of 2x Z-buffer (120 mM Na2HPO4·7H2O, 80 mM NaH2PO4·H2O, 20 mM KCl, 2 mM MgSO4·7H2O, and 100 mM β-mercaptoethanol) and 10 μl of 1 mM CPRG. β-galactosidase activity was measured using a spectrophotometer at 570 nm.
Construction of the SphR mutant and complementation strains
The upstream and downstream sequence fragments (1,000 bp) flanking the sphR gene were amplified by PCR using AN17 genome DNA as a template and the primers SphR-1000-Infu1 and SphR-1000-Infu2. The amplified product was cloned into the BamHI site of pK19mobsacB and the resulting plasmid was named pK19-SphR. In order to replace the sphR gene with the Tcr cassette, the sphR gene was removed by PCR using pK19-SphR as a template and the primers SphR-KO-InfuU1 and SphR-KO-InfuL1. The fragment obtained was ligated with the Tcr cassette amplified from pBR322 by using the primers pBR322U1new and pBR322L1331new. The primers used are summarized in Supplementary Table S1. The resulting plasmid was named pK19-SphR-Tet. The plasmid containing a mutant SphR disrupted with the Tcr gene was transferred from the broad host range-mobilizing strain E. coli S17-1 to P. aeruginosa PAO1 by biparental filter matings. Gene replacement was ascertained by PCR of the sphR gene. The resulting strain was named ΔSphR. In order to construct the SphR complementation vector, we first introduce the gentamicin resistance (Gmr) cassette into the EcoRI site of pMMB66HE52. pMMB66HE was linearized by PCR using the primers pMMB66HE-U1 and pMMB66HE-L1. The Gmr cassette was amplified by PCR using pHP45Ωaac as a template and the primers aacC4-1-Infu1 and aacC4-1-Infu2, and was then ligated to linearized pMMB66HE. The resulting plasmid was named pMMB66HE-Gen. The sphR gene was amplified by PCR using pK19-SphR as a template and the primers SphR-U1-Infu1 and SphR-L1-Infu2, and was then ligated to the BamHI site of pMMB66HE-Gen, which was linearized by PCR using pMMB66HE-Gen as a template and the primers pMMB66HE-aacC4-U1 and pMMB66HE-aacC4-L1. The resulting plasmid was named pMMB66HE-Gen-SphR. pMMB66HE-Gen-SphR was transferred from the broad host range-mobilizing strain E. coli S17-1 to the strain ΔSphR by biparental filter matings. The primers used are listed in Supplementary Table S1.
Assay of CDase
CDase (CerN) activity was measured using C12-4-nitrobenzo-2-oxa-1,3-diazole (NBD)-Cer as a substrate as previously described21.
Assay of hemolysis
Fifty microliters of the P. aeruginosa cell suspension (A600 = 0.4) in saline was mixed with 50 μl of sheep erythrocytes in saline and incubated at 30 °C for an appropriate time. After being incubated, the reaction mixtures were centrifuged at 1,000 × g for 3 min, the supernatant was diluted with distilled water (3 volumes), and the release of hemoglobin was then measured using the Multiskan FC Microplate Photometer (Thermo Fisher Scientific, Inc., Waltham, MA, USA) at 492 nm.
Assay of cell viability
The cell viabilities of the wild-type and SphR-deficient mutant were evaluated by Microbial Viability Assay Kit-WST (DOJINDO LABORATORIES, Kumamoto, Japan). This colorimetric microbial viability assay is based on the reduction of WST-8 reagent by the reducing ability of viable cells53,54. Strains were grown at 30 °C overnight on NAC agar. Bacteria were then scraped from the agar plate and suspended in saline to an A550 of 0.125. A 10-μl aliquot of the cell suspension (A550 = 0.125) was inoculated into 180 μl of PY medium containing 0.05% sodium taurodeoxycholate in the presence and absence of 200 μM of SM and then incubated at 30 °C for an appropriate time. After the incubation, cell viability was analyzed by measuring the metabolic activity of cells using Microbial Viability Assay Kit-WST according to the manufacturer’s instructions.
Real-time PCR
The WT and SphR-null mutant of P. aeruginosa PAO1 were cultured in PY medium with and without 200 μM SM at 30 °C. After 8 h, cells were stabilized by RNAlater (Thermo Fisher Scientific) and harvested. Total cellular RNA was isolated from the cells using TRI reagent (Molecular Research Center, Inc., Cincinnati, OH, USA), and the SV Total RNA Isolation System (Promega Corporation, Madison, WI, USA). Reverse transcription was performed using the Prime Script RT reagent kit (Takara Bio) in accordance with the manufacturer’s protocol. Real-time PCR was performed using the SYBR Premix ExTaq kit (Takara Bio) in an Mx3000P QPCR System (Agilent Technologies, Santa Clara, CA, USA). The data obtained were normalized against the expression levels of PA3001, which were not affected by the SM treatment, as shown in the microarray analysis. Relative changes in gene expression were analyzed using the ∆∆CT method55. The sequences of the primer pairs are described in Supplementary Table S1.
Microarray analysis
In microarray experiments, P. aeruginosa strain PAO1 was cultured in 2 ml of sterilized PY medium, and incubated at 30 °C for 1 day with vigorous shaking. An aliquot of the cell suspension (OD600 = 0.01) was inoculated into 2 ml of PY medium containing 0.05% sodium taurodeoxycholate and 200 μM of SM, and then shaken at 30 °C for 8 h. Cells were stabilized by RNAlater and harvested. Total cellular RNA was isolated from cells using TRI reagent and the SV Total RNA Isolation System. The integrity of total RNA samples was assessed by the Experion Automated Electrophoresis System (Bio-Rad). Labeled cDNA probes were prepared according to the protocol supplied by the manufacturer (Affymetrix, Santa Clara, CA, USA) and hybridized to a GeneChip P. aeruginosa Genome Array (Affymetrix) according to the manufacturer’s instructions. Data analysis was initially performed with Affymetrix Expression Console™ Software. Microarray experiments and data analyses were supported by Cell Innovator Inc. (Fukuoka, Japan). Gene expression changes with significance were identified by Linear Models for Microarray Analysis (limma) package56 of Bioconductor software. Fold changes were calculated as the average ratios between the signal in the absence (control) and presence of SM. Genes induced by 1.5-fold or more and a P value of <0.05 were considered to be significant. Our results have been uploaded to the Gene Expression Omnibus database (accession number GSE83078).
Identification of the transcriptional start site using 5′-RACE
P. aeruginosa strain PAO1 was cultured in 2 ml of sterilized PY medium, and incubated at 30 °C for 1 day with vigorous shaking. An aliquot of the cell suspension (OD600 = 0.02) was inoculated into 2 ml of PY medium containing 0.05% sodium taurodeoxycholate and 100 μM of SM, and shaken at 30 °C for 8 h. Cells were stabilized by RNAlater and harvested. Total cellular RNA was isolated from cells using TRI reagent and the SV Total RNA Isolation System. Purified total RNA was used for transcription start site identification of the cerN gene using the 5′-Full RACE Core Set (Takara Bio) according to the manufacturer’s instructions. First-strand cDNA was synthesized by the gene-specific reverse primer PaCDProRT, and first PCR was performed by first-strand cDNA as the template with the primers PaCDPro-A1 and PaCDProS1. Second PCR was performed using the first PCR reactant as the template with the primers PaCDPro-A2 and PaCDProS2, and the product was cloned into the pGEM T-easy vector (Promega) and sequenced. The primers used are listed in Supplementary Table S1. The first nucleotide following the PaCDProRT sequence was taken as the transcriptional start.
Promoter analysis
The promoter region of the cerN gene, β-galactosidase gene, 3′-UTR region of the cerN gene, and Tcr cassette were amplified by PCR using pK19-PaCD5k-LacZ-Tet as a template and the primers PaCDPro2-Infu1 and PaCDPro2-Infu2-1. The amplified product was inserted into the linearized pMMB66EH vector, which was amplified by PCR using pMMB66EH as a template and the primers pMMBdel-U1-BamHI and pMMBdel-L1-BamHI. The recombinant plasmid was designated pMMBProCD2. Point and deletion mutations were introduced in pMMBProCD2 using the PrimeSTAR mutagenesis basal kit (Takara Bio) according to the instructions provided by the manufacturer. The primers used for these constructs are listed in Supplementary Table S1. The resulting plasmids were transferred from the broad host range-mobilizing strain E. coli S17-1 to P. aeruginosa PAO1 by biparental filter matings. β-Galactosidase activities were measured by the method of Miller57 using cells permeabilized with chloroform.
Expression and purification of MBP-SphR
The sphR gene was amplified by PCR using pUC-SphR as a template and the primers SphR-pMAL-Infu1 and SphR-pMAL-Infu2 (see Supplementary Table S1). The amplified product was inserted into EcoRI- and NotI-digested pMAL-c5X (New England Biolabs, Ipswich, MA, USA) to generate MBP-SphR, N-terminal fusion to the maltose-binding protein (MBP). The recombinant plasmid was designated pMAL-SphR. E. coli strain BL21(DE3) cells were transformed with pMAL-SphR and grown at 25 °C overnight with shaking in 3 ml of Luria-Bertani medium supplemented with 100 μg/ml of carbenicillin. The culture was then transferred to a 500-ml flask containing 200 ml of Luria-Bertani medium supplemented with 0.2% glucose and 100 μg/ml of carbenicillin incubated at 25 °C with shaking to an OD600 of approximately 0.5. Isopropyl 1-thio-D-galactopyranoside (IPTG) was then added to the culture to a final concentration of 1 mM to induce transcription. After an additional 5-h culture at 25 °C, cells were harvested by centrifugation and suspended in 5 ml TBS containing 0.1% Triton X-100. After sonication for 3 min, cell debris was removed by centrifugation (15,000 × g for 30 min). The supernatant was loaded onto a column of amylose resin (NEB), and the column was washed with TBS. MBP-SphR was eluted from the column with TBS containing 10 mM maltose. The fractions containing MBP-SphR were pooled and loaded onto a Superdex 200 HR column (GE Healthcare) equilibrated with 25 mM MES buffer, pH 6.0, containing 100 mM NaCl. Since MBP-SphR has the ability to restore CerN activity in the SphR mutant by a plasmid expressing MBP-SphR fusion (data not shown), we used MBP-SphR in subsequent biochemical experiments.
Electrophoretic mobility shift assay
A 120-bp DNA fragment containing the −200 to −81 region of the cerN promoter was amplified by PCR using pPaCDPro400 as a template and the primers PaCDPro-200U and PaCDPro-81L (see Supplementary Table S1). The 120-bp DNA fragment obtained was labeled with the Biotin 3′ End DNA Labeling Kit (Thermo Fisher Scientific). Biotin-labeled DNA probes were detected by chemiluminescence using a LightShift Chemiluminescent EMSA Kit (Thermo Fisher Scientific). All experimental procedures were performed according to the manufacturer’s instructions, with the exception that the binding buffer condition was changed. Gel mobility shift assays were performed by incubating 10 fmol of the labeled probe with 30 pmol of proteins (MBP-SphR or MBP) in binding buffer (10 mM Tris-HCl, pH 7.5 containing 50 mM KCl, 1 mM dithiothreitol, 1 mM EDTA, 3 mM MgCl2, 50 μg/ml bovine serum albumin (BSA), 4% glycerol, 0.05% NP-40, and 50 ng/μl poly(dI-dC)) at 30 °C for 30 min. Five microliters of 5x Loading Buffer was added to the 20-μl reaction before being loaded onto a 5% native polyacrylamide gel in 0.5X TBE. After electrophoreses at 100 V for 70 min, samples were transferred onto a positively charged nylon membrane (Biodyne B, Pall Corporation, Port Washington, NY, USA) in 0.5x TBE at 380 mA for 30 min. Transferred DNAs were cross-linked to the membrane at 120 mJ/cm2 and detected using horseradish peroxidase-conjugated streptavidin according to the manufacturer’s instructions. The DNA band signal was visualized using Ez-Capture (ATTO CORPORATION, Tokyo, Japan).
Biotin-Sph binding assay
Biotin-Sph (1 nmol) and MBP-SphR (0.2 nmol) were mixed in 100 μl of binding buffer (20 mM Tris-HCl, pH 7.5 containing 100 mM KCl, 2 mM DTT, 8% glycerol, 6 mM MgCl2, 100 μg/ml BSA, 2 mM EDTA, and 1% NP-40) and incubated at room temperature for 30 min. Dynabeads MyOne Streptavidin T1 (Thermo Fisher Scientific) was added to the mixture and incubated at room temperature for 30 min. Beads were separated using a magnet, the supernatant containing non-bound MBP-SphR was removed, and the beads were washed three times with 0.1% NP-40 in TBS using magnetic separation. The beads obtained were suspended in 45 μl of SDS sample buffer. After boiling at 100 °C for 5 min, the sample (20 μl) was subjected to SDS-PAGE and stained by EzStain Aqua (ATTO). The stained gel was scanned and band intensities were analyzed using Multi Gauge software version 3.1 (FUJIFILM Corporation, Tokyo, Japan).
Additional Information
How to cite this article: Okino, N. and Ito, M. Molecular mechanism for sphingosine-induced Pseudomonas ceramidase expression through the transcriptional regulator SphR. Sci. Rep. 6, 38797; doi: 10.1038/srep38797 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Olsen, I. & Jantzen, E. Sphingolipids in bacteria and fungi. Anaerobe. 7, 103–112 (2001).
Hannun, Y. A. & Obeid, L. M. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 9, 139–150 (2008).
Hannun, Y. A. & Obeid, L. M. Many ceramides. J. Biol. Chem. 286, 27855–27862 (2011).
Futerman, A. H. & Hannun, Y. A. The complex life of simple sphingolipids. EMBO reports 5, 777–782 (2004).
Kihara, A. Sphingosine 1-phosphate is a key metabolite linking sphingolipids to glycerophospholipids. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 1841, 766–772 (2014).
Hassler, D. F. & Bell, R. M. Ceramidases: enzymology and metabolic roles. Adv. Lipid Res. 26, 49–57 (1993).
Park, J. H. & Schuchman, E. H. Acid ceramidase and human disease. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 1758, 2133–2138 (2006).
Ito, M., Okino, N. & Tani, M. New insight into the structure, reaction mechanism, and biological functions of neutral ceramidase. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 1841, 882–691 (2013).
Mao, C. & Obeid, L. M. Ceramidases: regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 1781, 424–434 (2008).
Okino, N., Tani, M., Imayama, S. & Ito, M. Purification and characterization of a novel ceramidase from Pseudomonas aeruginosa. J. Biol. Chem. 273, 14368–14373 (1998).
Okino, N. et al. Molecular cloning, sequencing, and expression of the gene encoding alkaline ceramidase from Pseudomonas aeruginosa. Cloning of a ceramidase homologue from Mycobacterium tuberculosis. J. Biol. Chem. 274, 36616–36622 (1999).
Inoue, T. et al. Mechanistic insights into the hydrolysis and synthesis of ceramide by neutral ceramidase. J. Biol. Chem. 284, 9566–9577 (2009).
Driscoll, J. A., Brody, S. L. & Kollef, M. H. The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs 67, 351–368 (2007).
Williams, B. J., Dehnbostel, J. & Blackwell, T. S. Pseudomonas aeruginosa: host defence in lung diseases. Respirology 15, 1037–1056 (2010).
Lyczak, J. B., Cannon, C. L. & Pier, G. B. Establishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist. Microbes and Infection 2, 1051–1060 (2000).
Gellatly, S. L. & Hancock, R. E. Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathogens and Disease 67, 159–173 (2013).
Ostroff, R. M. & Vasil, M. L. Identification of a new phospholipase C activity by analysis of an insertional mutation in the hemolytic phospholipase C structural gene of Pseudomonas aeruginosa. J. Bacteriol. 169, 4597–4601 (1987).
Stonehouse, M. J. et al. A novel class of microbial phosphocholine-specific phospholipases C. Mol. Microbiol. 46, 661–676 (2002).
Meyers, D. J. et al. In vivo and in vitro toxicity of phospholipase C from Pseudomonas aeruginosa. Toxicon 30, 161–169 (1992).
Vasil, M. L. et al. A complex extracellular sphingomyelinase of Pseudomonas aeruginosa inhibits angiogenesis by selective cytotoxicity to endothelial cells. PLoS Pathog. 5, e1000420 (2009).
Okino, N. & Ito, M. Ceramidase enhances phospholipase C-induced hemolysis by Pseudomonas aeruginosa. J. Biol. Chem. 282, 6021–6030 (2007).
Lamont, I. L., Beare, P. A., Ochsner, U., Vasil, A. I. & Vasil, M. L. Siderophore-mediated signaling regulates virulence factor production in Pseudomonas aeruginosa. Proceedings of the National Academy of Sciences, USA 99, 7072–7077 (2002).
Son, M. S., Matthews, W. J., Jr., Kang, Y., Nguyen, D. T. & Hoang, T. T. In vivo evidence of Pseudomonas aeruginosa nutrient acquisition and pathogenesis in the lungs of cystic fibrosis patients. Infect. Immun. 75, 5313–5324 (2007).
Weir, T. L. et al. Global gene expression profiles suggest an important role for nutrient acquisition in early pathogenesis in a plant model of Pseudomonas aeruginosa infection. Appl. Environ. Microbiol. 74, 5784–5791 (2008).
Wargo, M. J., Szwergold, B. S. & Hogan, D. A. Identification of two gene clusters and a transcriptional regulator required for Pseudomonas aeruginosa glycine betaine catabolism. J. Bacteriol. 190, 2690–2699 (2008).
Stover, C. K. et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406, 959–964 (2000).
Winsor, G. L. et al. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 39, D596–600 (2011).
LaBauve, A. E. & Wargo, M. J. Detection of host-derived sphingosine by Pseudomonas aeruginosa is important for survival in the murine lung. PLoS Pathog. 10, e1003889 (2014).
Martin, R. G. & Rosner, J. L. The AraC transcriptional activators. Curr. Opin. Microbiol. 4, 132–137 (2001).
Yang, J., Tauschek, M. & Robins-Browne, R. M. Control of bacterial virulence by AraC-like regulators that respond to chemical signals. Trends Microbiol. 19, 128–135 (2011).
Finn, R. D. et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–285 (2016).
Dong, Y. H., Zhang, X. F., Xu, J. L., Tan, A. T. & Zhang, L. H. VqsM, a novel AraC-type global regulator of quorum-sensing signalling and virulence in Pseudomonas aeruginosa. Mol. Microbiol. 58, 552–564 (2005).
Michel, L., Gonzalez, N., Jagdeep, S., Nguyen-Ngoc, T. & Reimmann, C. PchR-box recognition by the AraC-type regulator PchR of Pseudomonas aeruginosa requires the siderophore pyochelin as an effector. Mol. Microbiol. 58, 495–509 (2005).
Wargo, M. J., Ho, T. C., Gross, M. J., Whittaker, L. A. & Hogan, D. A. GbdR regulates Pseudomonas aeruginosa plcH and pchP transcription in response to choline catabolites. Infect. Immun. 77, 1103–1111 (2009).
Albelo, S. T. & Domenech, C. E. Carbons from choline present in the phospholipids of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 156, 271–274 (1997).
Baysse, C. et al. Modulation of quorum sensing in Pseudomonas aeruginosa through alteration of membrane properties. Microbiology 151, 2529–2542 (2005).
Sun, Z. et al. Blocking phosphatidylcholine utilization in Pseudomonas aeruginosa, via mutagenesis of fatty acid, glycerol and choline degradation pathways, confirms the importance of this nutrient source in vivo. PLoS One 9, e103778 (2014).
Barker, A. P. et al. A novel extracellular phospholipase C of Pseudomonas aeruginosa is required for phospholipid chemotaxis. Mol. Microbiol. 53, 1089–1098 (2004).
Bibel, D. J., Aly, R. & Shinefield, H. R. Antimicrobial activity of sphingosines. J. Invest. Dermatol. 98, 269–273 (1992).
Fischer, C. L. et al. Antibacterial activity of sphingoid bases and fatty acids against Gram-positive and Gram-negative bacteria. Antimicrob. Agents Chemother. 56, 1157–1161 (2012).
Pewzner-Jung, Y. et al. Sphingoid long chain bases prevent lung infection by Pseudomonas aeruginosa. EMBO Mol. Med. 6, 1205–1214 (2014).
Mistry, P. K. et al. Glucocerebrosidase 2 gene deletion rescues type 1 Gaucher disease. Proceedings of the National Academy of Sciences, USA 111, 4934–4939 (2014).
Ohnishi, Y., Okino, N., Ito, M. & Imayama, S. Ceramidase activity in bacterial skin flora as a possible cause of ceramide deficiency in atopic dermatitis. Clin. Diagn. Lab. Immunol. 6, 101–104 (1999).
Wertz, P. W. & Downing, D. T. Free Sphingosine in Human Epidermis. J. Invest. Dermatol. 94, 159–161 (1990).
Kita, K. et al. Activation of bacterial ceramidase by anionic glycerophospholipids: Possible involvement in ceramide hydrolysis on atopic skin by Pseudomonas ceramidase. Biochem. J. 362, 619–626 (2002).
Oizumi, A. et al. Pseudomonas-Derived Ceramidase Induces Production of Inflammatory Mediators from Human Keratinocytes via Sphingosine-1-Phosphate. PLoS One 9, e89402 (2014).
Paragh, G. et al. Novel sphingolipid derivatives promote keratinocyte differentiation. Exp. Dermatol. 17, 1004–1016 (2008).
Sigruener, A. et al. Effects of sphingoid bases on the sphingolipidome in early keratinocyte differentiation. Exp. Dermatol. 22, 677–679 (2013).
Blondelet-Rouault, Marie-Hrl6ne, W., J., Lebrihi, Ahmed, Branny, Pavel & Pernodet, J.-L. Antibiotic resistance gene cassettes derived from the f2 interposon for use in E. coli and Streptomyces. Gene. 190, 315–317 (1997).
Schafer, A. et al. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene. 145, 69–73 (1994).
Simon, R., Priefer, U. & Puhler, A. A broad host range mobilization system for in vivo genetic engineering: Transposon mutagenesis in gram negative bacteria. Biotechnology. (N. Y.) 1, 784–791 (1983).
Fürste, J. P. et al. Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene. 48, 119–131 (1986).
Tsukatani, T. et al. Colorimetric cell proliferation assay for microorganisms in microtiter plate using water-soluble tetrazolium salts. J. Microbiol. Methods 75, 109–116 (2008).
Tsukatani, T. et al. Colorimetric microbial viability assay based on reduction of water-soluble tetrazolium salts for antimicrobial susceptibility testing and screening of antimicrobial substances. Anal. Biochem. 393, 117–125 (2009).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
Smyth, G. K. In Bioinformatics and Computational Biology Solutions using R and Bioconductor (ed Gentleman, R., C., V., Dudoit, S., Irizarry, R., Huber, W. ) 397–420 (Springer, 2005).
Miller, J. H. In A Short Course in Bacterial Genetics – A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. (ed Miller, J. H. ) 71–80 (Cold Spring Harbor Laboratory Press, 1992).
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 (1994).
Acknowledgements
This work was supported by JSPS KAKENHI Grant Number 23570142 (NO) and 15K06976 (NO), and a Research Grant for Young Investigators of the Faculty of Agriculture, Kyushu University, Japan (NO). We thank the National BioResource Project (NIG, Japan) for the gift of the plasmid pHP45Ωaac.
Author information
Authors and Affiliations
Contributions
N.O. designed this study. N.O. performed all experiments and analyzed data. M.I. supervised the study. N.O. and M.I. wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Okino, N., Ito, M. Molecular mechanism for sphingosine-induced Pseudomonas ceramidase expression through the transcriptional regulator SphR. Sci Rep 6, 38797 (2016). https://doi.org/10.1038/srep38797
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep38797
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
