
Abstract
Reactive oxygenation species (ROS) generated from reperfusion results in cardiac injury through apoptosis and inflammation, while PKR has the ability to promote apoptosis and inflammation. The aim of the study was to investigate whether PKR is involved in hydrogen peroxide (H2O2) induced neonatal cardiac myocytes (NCM) injury. In our study, NCM, when exposed to H2O2, resulted in persistent activation of PKR due to NCM endogenous RNA. Inhibition of PKR by 2-aminopurine (2-AP) or siRNA protected against H2O2 induced apoptosis and injury. To elucidate the mechanism, we revealed that inhibition of PKR alleviated H2O2 induced apoptosis companied by decreased caspase3/7 activity, BAX and caspase-3 expression. We also revealed that inhibition of PKR suppressed H2O2 induced NFκB pathway and NLRP3 activation. Finally, we found ADAR1 mRNA and protein expression were both induced after H2O2 treatment through STAT-2 dependent pathway. By gain and loss of ADAR1 expression, we confirmed ADAR1 modulated PKR activity. Therefore, we concluded inhibition of PKR protected against H2O2-induced injury by attenuating apoptosis and inflammation. A self-preservation mechanism existed in NCM that ADAR1 expression is induced by H2O2 to limit PKR activation simultaneously. These findings identify a novel role for PKR/ADAR1 in myocardial reperfusion injury.
Similar content being viewed by others


Introduction
Myocardial ischemia reperfusion injury is a major mechanism leading to cell damage and organ dysfunction after myocardial infarction. A great part of cardiomyocytes cell death occurs via the process of acute reperfusion phase due to the development of oxidative stress induced by the generation of reactive oxygen species (ROS), such as H2O21,2. H2O2 is highly diffusible and triggers subsequent inflammation, thus causing severe cardiac cell apoptosis and injury3.
Inflammatory injury limits the benefits of reperfusion during acute myocardial infarction. Therefore, protecting cardiomyocyte from inflammatory damage could be a rational method for ameliorating myocardial reperfusion injury4,5. NFκB and NLRP3 inflammasome pathway are both important inflammatory signaling, and new targeted therapeutic strategies such as anti-NFκB and anti-NLRP3 inflammasome were explored6,7,8. However, it is unclear how these inflammatory signaling molecules are coordinately regulated in myocardial reperfusion injury process. Myocardial apoptosis is one of major damage pattern which contributes to myocardial reperfusion injury9,10. Therefore, various ways to alleviate apoptosis were also extensively investigated.
Protein kinase PKR is double-stranded RNA (dsRNA)-activated serine/threonine protein kinase inducible by interferon(IFN). It was first identified as a mediator of the antiproliferative and antiviral actions of interferon. It is an ubiquitously expressed protein in mammalian cells and carry the potential ability for direct recognition of pathogens and activation of specific cellular responses to external stimuli. Encountering infections, PKR can regulate or act in conjunction with major inflammatory signaling pathways that are implicated in myocardial ischemia reperfusion injury, including NFκB and inflammasome NLRP311,12,13,14. In addition to regulating inflammatory signaling, PKR also promotes apoptosis through interactions with FAS-associated death domain protein and upregulation of the proapoptotic factor BAX15,16. Taken together, PKR is a potential target for treatment of myocardial reperfusion injury. Accumulating evidences also revealed that inflammatory stressors such as TNF-α, lipopolysaccharide (LPS), viral dsRNA and its mimetic compound poly (I:C), all activate the RNA editor ADAR117,18,19. By interacting with PKR and inhibiting its kinase activity, ADAR1 presents a role in buffering inflammatory stress effects18. Therefore, it is possible that ADAR1 also modulate PKR activity in cardiac myocytes.
In the present study, we examined the role of PKR in H2O2-induced injury on NCM. We observed that PKR was activated by endogenous RNA from H2O2 treated NCM, and inhibition of PKR activation significantly decreased H2O2 induced injury. The mechanisms involve the suppression of NFκB, NLRP3 inflammasome activation, and alleviation of apoptosis. We also confirmed ADAR1 was up-regulated after H2O2 treatment, and ADAR1 modulated PKR activity.
Results
H2O2 induces PKR phosphorylation in NCM
We first examined whether H2O2 was able to activate PKR in NCM. As shown in Fig. 1A, 12 h exposure of NCM to H2O2 resulted in the increased PKR phosphorylation. H2O2-mediated increase in PKR phosphorylation is in a dose-dependent manner with a peak at approximately 50 μM. Because the maximum induction of PKR phosphorylation occurred at the concentration of 50 μM, all subsequent experiments were conducted according to this concentration. Due to the key role of oxidative stress in PKR activation, we also determined the levels of ROS after 50 μM H2O2 stimulation. We found H2O2 treatment resulted in maximum elevation of ROS after 2 h, and sustained 24 h (Supple Fig. 1). Next, NCM were stimulated with H2O2 (50 μM) for 0, 2, 12, 24 hours and PKR phosphorylation was evaluated. PKR was found to be activated by H2O2 after 2 h, reaching maximum phosphorylation levels at 12 h (Fig. 1B). 2-AP was extensively used as a PKR activation inhibitor. We confirmed 4 mM is the minimum concentration to inhibit PKR activation in H2O2 treated NCM (Supple Fig. 2). Pretreatment of NCM with the 2-AP for 30 minutes before stimulation with the H2O2 significantly suppressed PKR phosphorylation (Fig. 1C). PKR is a key component of the cytoplasmic RNA sensors involved in the recognition of viral double-stranded RNA (dsRNA), and bacterial RNAs can directly binds to and activate PKR in human adult cardiac myocytes20. Therefore, we tested whether endogenous RNA released from injured NCM can activate PKR. We found RNase abolished H2O2 induced PKR activation completely (Fig. 1D). It reveals endogenous RNA results in phosphorylation of PKR. Then, we performed the PKR binding assay to elucidate the mechanism. We found RNA from untreated NCM was unable to activate PKR, however, RNA from H2O2 treated NCM resulted in phosphorylation of PKR. As a positive control, synthetic dsRNA (poly I:C) activated PKR distinctly (Fig. 1E).

H2O2 induces PKR phosphorylation in NCM.
(A) Cultured NCM were treated with H2O2 (0–100 μM) for 12 h. (B) Cultured NCM were treated with H2O2 (50 μM) for 24 h. (C) Cultured NCM were left untreated or stimulated with 4 mM 2-AP for 30 minutes, followed by stimulation with H2O2 (50 μM) for 12 h. (D) Cultured NCM were left untreated or stimulated with 100 μg/ml RNase for 30 minutes, followed by stimulation with H2O2 (50 μM) for 12 h. (A,B,C,D) The expression of p-PKR, PKR protein were measured by Western blotting, densitometric analyses of p-PKR, PKR/GAPDH were calculated. (E) PKR was from NCM and subjected to PKR binding assay. Purified PKR was incubated without or with 1 μg/ml of total RNA from NCM, NCM treated with H2O2 (50 μM) for 12 h, or Poly I:C. The data are representative of three independent experiments. (**P < 0.01; *P < 0.05). Full length blots were shown in Supple Fig. 4.
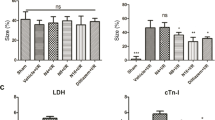
Inhibition of PKR alleviates H2O2 induced NCM injury
The integrity of cell membranes or necrosis is estimated by LDH release into the media in response to oxidant burden. Figure 2A shows H2O2 induced LDH activity in NCM significantly reduced by 2-AP (1483.77 ± 27.60 vs 489.06 ± 16.74, P = 0.03). Figure 2B shows H2O2 markedly decreased cell viability, compared with untreated cells (93.43 ± 3.10 vs 43.96 ± 6.26, P = 0.002). However, H2O2 induced decrease of cell viability was significantly attenuated by 2-AP pretreatment (43.96 ± 6.26 vs 70.45 ± 7.08, P = 0.02). To exclude the possibility that 2-AP may affect the NCM survival independent to its effect of PKR inhibition, and further confirm the role of PKR in alleviating H2O2 induced NCM injury, NCM were transfected with siRNA for PKR. First, we confirmed the knockdown efficiency of PKR by Western blotting (Supple Fig. 3). Figure 2C shows H2O2 induced LDH activity in NCM obviously reduced after down regulation of PKR (1306.95 ± 36.31 vs 520.43 ± 25.28, P = 0.03). Figure 2D reveals H2O2 induced decrease of cell viability was significantly attenuated by PKR siRNA (41.45 ± 4.74 vs 73.11 ± 7.27, P = 0.02).

Inhibition of PKR alleviates H2O2 induced NCM injury.
(A,B) Cultured NCM were left untreated or stimulated with 4 mM 2-AP for 30 minutes, followed by stimulation with H2O2 (50 μM) for 24 h. (C,D) Cultured NCM were left untreated or transfected with siRNA, followed by stimulation with H2O2 (50 μM) for 24 h. (A,C) Cultured NCM media were collected for LDH determination. (B,D) Cultured NCM were collected for cell viability determination. The data are representative of three independent experiments. (**P < 0.01; *P < 0.05).
H2O2 induced NCM apoptosis is PKR dependent
PKR mediates transverse aortic constriction (TAC) induced cardiac myocytes apoptosis21. Therefore, NCM apoptosis was assessed after H2O2 treatment with (without) 2-AP pretreatment. We observed NCM apoptosis obviously increased when exposed to H2O2 (2.01 ± 0.12 vs 22.13 ± 2.86, P = 0.004). However, this increase attenuated significantly after 2-AP pretreatment (22.13 ± 2.86 vs 11.24 ± 1.43, P = 0.02) (Fig. 3A). Next, we examined the effect of 2-AP on H2O2 induced caspase-3/7 activity on NCM. Figure 3B shows that H2O2 induced caspase3/7 activity. However, 2-AP markedly suppressed H2O2-induced caspase-3/7 activity. To investigate the mechanism, we detected the proapoptotic factor Bax and caspase-3 expression. We found H2O2 induced Bax and caspase-3 expression, but this increase was significantly buffered after 2-AP pretreatment (Fig. 3C). These data suggest that inhibition of PKR reduced H2O2 induced apoptosis by restricting Bax and Caspase-3 expression. We also evaluated the apoptosis after PKR siRNA transfection. We demonstrated NCM apoptosis obviously increased when exposed to H2O2 (2.14 ± 0.13 vs 21.79 ± 3.23, P = 0.004), and this increase attenuated significantly after PKR siRNA pretreatment (21.79 ± 3.23 vs 12.07 ± 1.39, P = 0.02) (Fig. 3D). It revealed 2-AP didn’t affect the NCM apoptosis.

H2O2 induced NCM apoptosis is PKR dependent.
(A,B,C) Cultured NCM were left untreated or stimulated with 4 mM 2-AP for 30 minutes, followed by stimulation with H2O2 (50 μM) for 12 h. (A) Analysis of NCM apoptosis by flow cytometry after staining cells with Annexin V and PI. Percentage of Annexin V positive cells were calculated. (B) Related enzyme activity of NCM apoptosis. (C) The expression of BAX, caspase-3 protein were measured by Western blotting, and densitometric analyses of BAX, caspase-3/GAPDH were calculated. The data are representative of three independent experiments. (D) Cultured NCM were left untreated or transfected with siRNA, followed by stimulation with H2O2 (50 μM) for 12 h. Analysis of NCM apoptosis by flow cytometry after staining cells with Annexin V and PI. Percentage of Annexin V positive cells were calculated. (**P < 0.01; *P < 0.05). Full length blots were shown in Supple Fig. 4.
PKR mediates NFκB activation signaling in H2O2 treated NCM
We first conducted EMSA to assess whether the activation of NFκB was PKR phosphorylation dependent. We found NFκB activation increased after H2O2 treatment compared to that in untreated group. However, this increase was inhibited after pretreatment with 2-AP (Fig. 4A). Activation of the p65 subunit of NFκB was also determined with a NFκB p65 ELISA-based assay. P65 activation induced by H2O2 and was found to be significantly reduced after 2-AP pretreatment (Fig. 4B). Next, we determined the downstream inflammatory cytokines, such as TNF-α and IL-6, mRNA expression and protein release in medium. As shown in Fig. 4C,D,E,F, H2O2 induced TNF-α and IL-6 mRNA expression and protein release in medium. 2-AP pretreated NCM displayed a significantly reduced H2O2 induced TNF-α and IL-6 mRNA expression and protein release in medium. These data indicate PKR phosphorylation is essential for H2O2 induced NFκB activation signaling.

PKR mediates NFκB activation signaling in H2O2 treated NCM.
Cultured NCM were left untreated or stimulated with 4 mM 2-AP for 30 minutes, followed by stimulation with H2O2 (50 μM) for 12 h. (A) Analysis of NFκB activation by EMSA; (B) Analysis of NFκB activation by TransAM p65 NFκB assay kit. (C) Real-time PCR analysis of TNF-α mRNA expression. (D) ELISA assay for detection the TNF-α release in medium. (E) Real-time PCR analysis of IL-6 mRNA expression. (F) ELISA assay for detection the IL-6 release in medium. The data are representative of three independent experiments. (**P < 0.01; *P < 0.05). Full length blots were shown in Supple Fig. 4.
PKR is involved in H2O2 induced inflammasome activation in NCM
PKR mediates inflammasome activation by physically interacting with NLRP3 in mice macrophages12. In this study, we found NLRP3 expression increased obviously after H2O2 treatment. However, the expression of NLRP3 did not differ significantly after 2-AP pretreatment in NCM (Fig. 5A). Next, we determined caspase-1 activation and IL-1β cleavage in NCM. Though the expressions of pro-IL-1β and pro-CASP1 were stable, caspase-1 activation and IL-1β cleavage in NCM both increased after exposed to H2O2 and inhibited after 2-AP pretreatment (Fig. 5B). We also measured IL-1β release in medium. As expected, IL-1β release increased after H2O2 treatment, and attenuated after 2-AP pretreatment (Fig. 5C).

PKR is involved in H2O2 induced inflammasome activation in NCM.
Cultured NCM were left untreated or stimulated with 4 mM 2-AP for 30 minutes, followed by stimulation with H2O2 (50 μM) for 12 h. (A) The expression of NLRP3 was measured by Western blotting, and densitometric analysis of NLRP3/GAPDH was calculated. (B) The expression of CASP1 p10, IL-1β p17, Pro-CASP1 and Pro-IL-1β proteins were measured by Western blotting, and densitometric analyses of CASP1 p10, IL-1β p17, Pro-CASP1 and Pro-IL-1β/GAPDH were calculated. (C) ELISA assay for detection the IL-1β release in medium. (**P < 0.01; *P < 0.05). Full length blots were shown in Supple Fig. 4.
ADAR1 is induced by H2O2 and modulates PKR activation
Previous reports revealed ADAR1 might affect PKR autophosphorylation18,22 We first examined changes of ADAR1 expression in NCM after H2O2 treatment. Both ADAR1 mRNA and protein levels were significantly increased with a peak in 12 hours (Fig. 6A,B). Next, we tested how ADAR1 expression was manipulated. After downregulation of STAT-2 by siRNA, we found ADAR1 expression was suppressed (Fig. 6C). Finally, we tested the effect of ADAR1 on the status of PKR phosphorylation in NCM. We observed though the total PKR Levels were unchanged, the phosphorylated PKR was downregulated while overexpression of ADAR1. We also confirmed that ADAR1 knockdown in NCM exposed to H2O2 resulted in significantly increased PKR activation with unaffected total PKR levels (Fig. 6D). These data consistently indicated that ADAR1 modulated PKR activation in NCM.

ADAR1 is induced by H2O2 and modulates PKR activation.
(A) Cultured NCM were treated with H2O2 (50 μM) for 24 h. Real-time PCR analysis of p150, p110 mRNA expression. (B) Cultured NCM were treated with H2O2 (50 μM) for 24 h. The expression of ADAR1 protein was measured by Western blotting, and densitometric analysis of ADAR1/GAPDH was calculated. (C) Cultured NCM were transfected with STAT-2 siRNA, followed by H2O2 (50 μM) for 12 h. The expression of STAT-2, ADAR1 protein were measured by Western blotting, and densitometric analyses of STAT-2, ADAR1/GAPDH were calculated. The data are representative of three independent experiments. (D) Cultured NCM were transfected with adenovirus containingADAR1-specific shRNA or ADAR1 cDNA for the purpose of knocking down or over expressing ADAR1, followed by H2O2 (50 μM) for 12 h. The expression of ADAR1, p-PKR and PKR protein were measured by Western blotting, and densitometric analyses of ADAR1, p-PKR and PKR/GAPDH were calculated. The data are representative of three independent experiments. (**P < 0.01; *P < 0.05). Full length blots were shown in Supple Fig. 4.
Discussion
In addition to its antiviral function, it is noteworthy that PKR responds to various cellular stresses23,24 and regulates cell inflammation, proliferation25, and apoptosis. However, most of the previous studies revealed the roles of PKR in confronting the chronic cellular stresses, including regulating metabolic homeostasis23, mediating TNF-α induced osteoclast formation26 and systolic overload-induced congestive heart failure21, et al. In this study, we investigated in the face of H2O2 treatment on NCM, an acute and drastic cellular stress, whether PKR is involved. We found consistently activation of PKR after H2O2 treatment, and we also revealed PKR inhibition protected against H2O2-induced injury by attenuating apoptosis and inflammation.
PKR is a key component of the cytoplasmic RNA sensors involved in the recognition of viral double-stranded RNA (dsRNA). Accumulating evidences suggested specific nucleoside modifications and structural elements are differentially represented in either microbial or mammalian RNA and therefore provide a molecular mechanism to discriminate foreign from self RNA27,28. However, under some situations, self cellular primary transcripts undergo some modifications and are recognized. The RNA released from necrotic cells may act as an endogenous TLR3 ligand for the stimulation of proinflammatory gene expression in rheumatoid arthritis synovial fibroblasts29. It has been described that endogenous RNA with a 5′-triphosphate-dependent manner can interact with PKR after induction of metabolic stress by palmitic acid (PA). This study by Youssef OA et al. indicated snoRNAs were enriched in response to PA and a subset of identified snoRNAs could bind and activate PKR in vitro30. In this study, we found that RNA from normal NCM was unable to activate PKR. However, RNA from H2O2 treated NCM resulted in PKR phosphorylation directly. The following study was warranted to identify the particular types of endogenous RNA released from H2O2 treated NCM.
NFκB, known as the major transcriptional factor of a wide range of inflammatory cytokines, triggered inflammatory response via modulating inflammatory transcription and in turn maintained NFκB activation and establish a positive autoregulatory loop to sustain the inflammatory status31. The role of NFκB activation and their downstream cytokines, such as TNF-α, in myocardial reperfusion injury has been previously reported, that myocardial ischemia reperfusion injury was alleviated after NFκB inhibition7,32. PKR has the property to mediate inflammatory signaling through NFκB activation. As expected, in our study, inhibition of PKR activation suppressed NFκB activation and downstream inflammatory cytokines expression. One of the recently identified proinflammatory signaling pathways involved in myocardial ischemia reperfusion injury is NLRP3 inflammasome, that inhibition of NLRP3 activation limited inflammatory injury following myocardial ischemia reperfusion6. PKR physically interacts with NLRP3 and mediates cleavage of inactive pro- IL-1β to their active form in mouse macrophage12. In our study, though NLRP3 expression was induced after H2O2 treatment, NLRP3 expression was stable after 2-AP pretreatment. It indicates NLRP3 expression is not PKR activation dependent. However, IL-1β cleavage and caspase-1 activation were significantly inhibited in NCM by exposure to H2O2 after 2-AP treatment, indicating that inhibition of PKR activation abrogates NLRP3 inflammasome activation. Taken together, inhibition of PKR alleviated the inflammation by suppressing the NFκB pathway and NLRP3 inflammasome activation, which were both involved in reperfusion induced injury. We deduce alleviation of inflammation after inhibition of PKR activation provided the cardiac protection against H2O2.
Loss of cardiac myocytes by apoptosis is a serious and frequent complication from reperfusion injury33. It is well known that PKR can mediate different forms of stress-induced apoptosis through eIF2α phosphorylation34 and through a Fas-associated death domain protein/caspase-8/caspase-3 signaling pathway15. NFκB activation by PKR has been suggested to mediate apoptosis35,36. The expressions of Bax, p53 and Bcl-2 were all regulated by PKR16. In present study, cardiomyocytes apoptosis induced by H2O2 attenuated after PKR inhibition, accompanied by low BAX and caspase-3 expression. Though the definitive mechanism is still unknown, we found cardiomyocytes apoptosis was suppressed after PKR inhibition.
ADAR1 expression has been broadly implicated in the other inflammatory conditions17,18,19,37. In present study, we observed ADAR1 expression was induced after H2O2 treatment in NCM. Previous reports revealed a STAT-2 dependent process of transcriptional activation of IFN-induced ADAR1 expression in mouse embryo fibroblast cells38. In our study, we confirmed H2O2 induced ADAR1 expression in NCM was also STAT-2 dependent. The expression of the mouse ADAR1 gene found on chromosome 3F2 likewise involves the utilization of multiple promoters39. The alternative signaling pathways which were involved in cardiac reperfusion injury include p38, STAT3 and interferon regulatory factor 340,41. Therefore, we can’t exclude the possibility that other signaling pathway was also involved in H2O2 induced ADAR1 expression process. In this study, we also observed over-expression of ADAR1 inhibited PKR activation. It revealed the self-preservation mechanism existed in NCM to prevent an excessive apoptosis and inflammation. Though PKR activation contributed to cardiac myocytes injury, ADAR1 expression was induced to limit PKR activation simultaneously. A similar pattern for ADAR1 in buffering inflammatory stress response in murine myoblasts was described previously18.
In conclusion, we observed PKR activation was induced by H2O2 in NCM and inhibition of PKR protects against H2O2-induced injury by attenuating apoptosis and inflammation. A self-preservation mechanism existed in cardiac myocytes that ADAR1 expression is induced by H2O2 to limit PKR activation simultaneously. The underlying mechanism is summarized in Fig. 7. These findings identify a novel role for PKR/ADAR1 in cardiac reperfusion injury.

Diagram of underlying mechanism.
Materials and Methods
NCM isolation and culture
Standard principles of laboratory animal care were followed. Male C57BL⁄6 mice (aged 6–8 wk, 18–21 g) were purchased from Shanghai Laboratory Animal Center (Shanghai, China). The present study was performed in accordance with the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences, published by the National Institutes of Health. All animal procedures carried out in this study were reviewed, approved, and supervised by the Institutional Animal Care and Use Committee of Shanghai Jiao Tong University. A breeding program was performed to produce neonates. Ventricular myocardial tissues from C57BL⁄6 mice born within 24 h were minced in a nominally Ca2+- and Mg2+-free Hanks’ balanced solution. Cardiomyocytes were dispersed using 0.625 mg/mL collagenase (type II) at 37 °C for 40 min. The isolated cells were preplated for 90 min to remove non-cardiomyocytes. The cardiomyocytes were plated in M199 medium containing 10% fetal calf serum in 35 mm Petri dishes precoated with 1% gelatin. The cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2. After the cardiomyocytes were cultured for 48 h, they were plated in dishes for the following studies. Cardiomyocytes were attached to the cell culture dishes (with approximately 70–80% confluency) and started to contract spontaneously. H2O2 (0 to 100 μM) was incubated with the cells according the experiment designs. 2-AP (Sigma) (4 mM) was dissolved in medium and incubated with cells 30 min before the addition of H2O2 (50 μM) to the cultures. RNase (Sigma) (100 μg/ml) was dissolved in medium and incubated with cells 30 min before the addition of H2O2 (50 μM) to the cultures.
Measurement of intracellular ROS
Equal numbers of cells (10000/well in 96-well plates in Hanks Balanced Salt Solution) were treated with 10 μmol/L 2,7-dichlorofluoroscein diacetate (DCF-DA) for 3 h. Cells were washed with phosphate-buffered saline and treated with 50 μM HO during different time intervals. DCF-DA penetrates into viable cells, and inside the cells, it is converted to DCF, which later reacts with ROS and fluorescence. At the indicated time intervals, the intensity of the fluorescence was measured at an excitation wavelength of 485 nm and an wavelength of 527 nm and is expressed as percent of control fluorescence.
Small interfering RNA (siRNA) transfection
For transient silencing of PKR or STAT-2, NCMs were transfected with mouse PKR siRNA or STAT-2 siRNA (Santa Cruz) using the siRNA Transfection Regent 24 or 48 h before following experiments. Scrambled siRNA (Santa Cruz) was also transfected as a negative control.
Western blot
Proteins from cell-free supernatants were extracted by methanol/chloroform precipitation as described previously42. Cell extracts were also prepared. Samples were separated on SDS-polyacrylamide gels and electrotransferred onto nitrocellulose membranes. The membranes were blocked with 3% bovine serum albumin for 2 h and then incubated overnight at 4 °C with antibodies specific for phosphor-Thr466 on PKR (Abcam), total PKR (Abcam), caspase-3 (Cell Signaling), BAX (Abcam), IL-1β (Santa Cruz), caspase-1 (Santa Cruz), NLRP3 (Adipogen), STAT-2 (Santa Cruz), ADAR1 (Santa Cruz) and GAPDH (Sigma) followed by incubation with HRP-conjugated secondary antibody. The quantitative relative expression of proteins were calculated by normalizing the densitometric analysis to GAPDH.
RNA preparation
Cultured cells were mock treated or 50 μM H2O2-treated for 12 h. RNA was extracted with TRIzol, followed by DNase treatment to eliminate any traces of genomic DNA contamination. RNA precipitations were collected after ethanol treatment. RNA was quantified by UV spectrometry and electrophorsed on a 1% agarose gel to verify purity and integrity prior to use.
PKR binding assay
Cultured cells were washed with cold PBS and resuspended in RIPA lysis buffer. Then cellular lysates were precleared with protein G-Sepharose beads (Santa Cruz) and incubated with anti-PKR polyclonal antibody. The beads were washed with RIPA, collected. Immunoprecipitated PKR was incubated in kinase buffer containing 50 mM Tris-HCl, PH 7.5, 2 mM MgCl2, 2 mM MnCl2, 50 mM KCl, 0.1 mM EDTA, 1 μg/mL aprotinin, 1 mM DTT, 20% glycerol, 10 μCi (γ-32P) and 1 μg/mL RNA (or polyI:C). The beads were washed with salt buffer and the reaction was terminated by addition of SDS sample buffer and loaded on a 10% SDS-PAGE gel.
Real-time PCR
Total RNA was isolated from cells using the RNeasy Mini Kit (Qiagen). Total RNA (2.5 μg) was reverse transcribed to cDNA with the SuperScript II RT kit (GIBCO BRL). mRNA expressions were determined by real time PCR using SYBR Premix Ex TaqTM (Takara, Tokoyo, Japan). The primers used for real-time PCR were as follows: IL-6, sense 5′- tgg cta agg acc aag ac cat cca a-3′, antisense 5′- aac gca cta ggt ttg ccg agt aga -3′; TNF-α, sense 5′- ctg tga agg gaa tgg gtg tt -3′, antisense 5′- ccc agc atc ttg gtt tctg -3′; ADAR1 p150, sense 5′- ggc act atg tct caa ggg ttc -3′, antisense 5′- gct gaa gct gga aac tcc tag -3′; ADAR1 p110, sense 5′- ctg aag gtg gaa gac tag gc -3′, antisense 5′- gtc caa gta cga ctg tgt ctg -3′; GAPDH 5′- cac ttg aag ggt gga gc -3′, antisense 5′- ggg cta agc agt tgg tg-3′. Data were collected and quantitatively analyzed on an ABI PRISM 7900HT sequence detection system (Applied Biosystems, Warrington, Cheshire, UK). All data were normalized automatically using GAPDH as the loading control.
LDH measurement
The LDH content was detected using a chromatometry assay kit using a commercially available detection kit (Nanjing Jiancheng Biochemical Reagent Co., Nanjing, China) according to the manufacturer’s instructions. The absorbance of supernatant was measured at 532 nm.
Cell viability determination
Cell viability was evaluated by the ability to reduce MTT, which is an indicator of metabolic activity. This viability assay was conducted in 96-well plates; MTT reduction was determined by spectrophotometry using a microplate reader (Bio-Tek Instruments, USA).
Cell apoptosis analysis
The cells were harvested, washed and incubated with the solution of Annexin V-FITC for 20 min and then PI (50 μg/mL) for 10 min. All staining operations must be carried out on ice and in dark. The cells were analyzed by flow cytometer.
Caspase3/7 activity analysis
Caspase3/7 intracellular activity was detected by Caspase-3/7 Assay Kit (Promega), and cell fluorescence intensity at 499 nm was measured by ELISA Tablet counter for quantitative assessment.
Preparation of ADAR1 adenoviruses and virus transfection
Mouse ADAR1 cDNA (NM_001146296) and ADAR1 shRNA (sequence: gcc aag aac tac ttc aag aaa) were cloned into Pme1/Xho1 sites of a pENTCMV vector and recombined with the Ad5 backbone for virus preparation through a commercial company (Welgen, Inc. Worcester, MA). Control virus was purchased from same company. Equal amounts of adenovirus were added to cells and incubated for 24 h. The following experiments were conducted as designed.
Electrophoretic mobility shift assay (EMSA)
The nuclear extracts (20 μg) prepared as described previously43 were pre-incubated for 10 min in binding buffer (1 μg poly dI-dC, 10 mmol/L Tris-HCl (pH 7.5), 50 mmol/L NaCl, 1 mmol/L EDTA, 5% glycerol, 1 mmol/L DTT, and 1 μg/μL BSA) on ice, followed by 30 min of incubation at room temperature with 1 × 105 dpm (approximately 0.5 ng) of a γ-32P-labeled probe (Amersham) containing the NFκB binding site 5′-agt tga ggg gac ttt ccc agg c-3′ (Santa Cruz). DNA-protein complexes were run on a 6% polyacrylamide gel.
TransAM assay
Whole cell extracts (5 μg) was used to detect activation of the p65 subunit of NFκB by the commercially available TransAM p65 NFκB assay kit (Active Motif) following the instructions of the manufacturer.
ELISA
TNF-α, IL-6 and IL-1β levels were detected in culture medium using ELISA (R&D) according to the manufacturer’s instructions.
Statistical analyses
All statistical analyses were performed using SPSS (version 17.0, SPSS Inc.). Values were reported as the mean ± SD. Samples were analyzed by two-tailed unpaired Student’s t-test and one-way analysis of variance (ANOVA) when appropriate. Statistical significance was defined as P < 0.05.
Additional Information
How to cite this article: Wang, Y. et al. Inhibition of PKR protects against H2O2-induced injury on neonatal cardiac myocytes by attenuating apoptosis and inflammation. Sci. Rep. 6, 38753; doi: 10.1038/srep38753 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Lefer, D. J. & Granger, D. N. Oxidative stress and cardiac disease. Am J Med 109, 315–23 (2000).
Yellon, D. M. & Hausenloy, D. J. Myocardial reperfusion injury. N Engl J Med 357, 1121–1135 (2007).
Clerk, A., Kemp, T. J., Zoumpoulidou, G. & Sugden, P. H. Cardiac myocyte gene expression profiling during H2O2-induced apoptosis. Physiol Genomics 29, 118–27 (2007).
Minutoli, L. et al. ROS-Mediated NLRP3 Inflammasome Activation in Brain, Heart, Kidney, and Testis Ischemia/Reperfusion Injury. Oxid Med Cell Longev 2016, 2183026, doi: 10.1155/2016/2183026 (2016).
Neri, M. et al. Cardiac oxidative stress and inflammatory cytokines response after myocardial infarction. Curr Vasc Pharmacol 13, 26–36 (2015).
Toldo, S. et al. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia-reperfusion in the mouse. Int J Cardiol 209, 215–20, doi: 10.1016/j.ijcard.2016.02.043 (2016).
Shi, Z., Lian, A. & Zhang, F. Nuclear factor-κB activation inhibitor attenuates ischemia reperfusion injury and inhibits Hmgb1 expression. Inflamm Res 63, 919–25, doi: 10.1007/s00011-014-0765-x (2014).
Quan, W. et al. Magnesium lithospermate B reduces myocardial ischemia/reperfusion injury in rats via regulating the inflammation response. Pharm Biol 51, 1355–62, doi: 10.3109/13880209.2013.791324 (2013).
Park, B. M., Cha, S. A., Lee, S. H. & Kim, S. H. Angiotensin IV protects cardiac reperfusion injury by inhibiting apoptosis and inflammation via AT4R in rats. Peptides 79, 66–74, doi: 10.1016/j.peptides.2016.03.017 (2016).
Yamaguchi, S. et al. Dental pulp-derived stem cell conditioned medium reduces cardiac injury following ischemia-reperfusion. Sci Rep 5, 16295, doi: 10.1038/srep16295 (2015).
Lin, S. S. et al. A role for protein kinase PKR in the mediation of Epstein-Barr virus latent membrane protein-1-induced IL-6 and IL-10 expression. Cytokine 50, 210–9, doi: 10.1016/j.cyto.2010.01.008 (2010).
Lu, B. et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488, 670–4, doi: 10.1038/nature11290 (2012).
Boriushkin, E., Wang, J. J., Li, J., Bhatta, M. & Zhang, S. X. p58(IPK) suppresses NLRP3 inflammasome activation and IL-1β production via inhibition of PKR in macrophages. Sci Rep 6, 25013, doi: 10.1038/srep25013 (2016).
Kumar, A., Haque, J., Lacoste, J., Hiscott, J. & Williams, B. R. Double-stranded RNA-dependent protein kinase activates transcription factor NF-kappa B by phosphorylating I kappa B. Proc Natl Acad Sci USA 91, 6288–92 (1994).
von Roretz, C. & Gallouzi, I. E. Protein kinase RNA/FADD/caspase-8 pathway mediates the proapoptotic activity of the RNA-binding protein human antigen R (HuR). J Biol Chem 285, 16806–13, doi: 10.1074/jbc.M109.087320 (2010).
Balachandran, S. et al. Activation of the dsRNA-dependent protein kinase, PKR, induces apoptosis through FADD-mediated death signaling. EMBO J 17, 6888–902 (1998).
Wu, Y. et al. Adenosine deaminase that acts on RNA 1 p150 in alveolar macrophage is involved in LPS-induced lung injury. Shock 31, 410–15, doi: 10.1097/SHK.0b013e31817c1068 (2009).
Meltzer, M. et al. The RNA editor gene ADAR1 is induced in myoblasts by inflammatory ligands and buffers stress response. Clin Transl Sci 3, 73–80, doi: 10.1111/j.1752-8062.2010.00199.x (2010).
Wang, H. et al. ADAR1 Suppresses the Activation of Cytosolic RNA-Sensing Signaling Pathways to Protect the Liver from Ischemia/Reperfusion Injury. Sci Rep 6, 20248, doi: 10.1038/srep20248 (2016).
Bleiblo, F. et al. Bacterial RNA induces myocyte cellular dysfunction through the activation of PKR. J Thorac Dis 4, 114–25, doi: 10.3978/j.issn.2072-1439.2012.01.07 (2012).
Wang, H. et al. Double-stranded RNA-dependent protein kinase deficiency protects the heart from systolic overload-induced congestive heart failure. Circulation 129, 1397–406, doi: 10.1161/CIRCULATIONAHA.113.002209 (2014).
Li, Z., Wolff, K. C. & Samuel, C. E. RNA adenosine deaminase ADAR1 deficiency leads to increased activation of protein kinase PKR and reduced vesicular stomatitis virus growth following interferon treatment. Virology 396, 316–22, doi: 10.1016/j.virol.2009.10.026 (2010).
Nakamura, T. et al. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell 140, 338–48, doi: 10.1016/j.cell.2010.01.001 (2010).
Cabanski, M. et al. PKR regulates TLR2/TLR4-dependent signaling in murine alveolar macrophages. Am J Respir Cell Mol Biol 38, 26–31 (2008).
Liu, X. et al. PKR regulates proliferation, differentiation, and survival of murine hematopoietic stem/progenitor cells. Blood 121, 3364–74, doi: 10.1182/blood-2012-09-456400 (2013).
Shinohara, H. et al. Double Stranded RNA-Dependent Protein Kinase is Necessary for TNF-α-Induced Osteoclast Formation In Vitro and In Vivo. J Cell Biochem 116, 1957–67, doi: 10.1002/jcb.25151 (2015).
Heil, F. et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303, 1526–9 (2004).
Diebold, S. S. et al. Nucleic acid agonists for Toll-like receptor 7 are defined by the presence of uridine ribonucleotides. Eur J Immunol 36, 3256–67 (2006).
Brentano, F., Schorr, O., Gay, R. E., Gay, S. & Kyburz, D. RNA released from necrotic synovial fluid cells activates rheumatoid arthritis synovial fibroblasts via Toll-like receptor 3. Arthritis Rheum 52, 2656–65 (2005).
Youssef, O. A. et al. Potential role for snoRNAs in PKR activation during metabolic stress. Proc Natl Acad Sci USA 112, 5023–8, doi: 10.1073/pnas.1424044112 (2015).
Christman, J. W., Lancaster, L. H. & Blackwell, T. S. Nuclear factor kappa B: a pivotal role in the systemic inflammatory response syndrome and new target for therapy. Intensive Care Med 24(11), 1131–8 (1998).
Gao, C. et al. TNF-α antagonism ameliorates myocardial ischemia-reperfusion injury in mice by upregulating adiponectin. Am J Physiol Heart Circ Physiol 308, H1583–91, doi: 10.1152/ajpheart.00346.2014 (2015).
Narula, J., Hajjar, R. J. & Dec, G. W. Apoptosis in the failing heart. Cardiol Clin 16, 691–710, ix (1998).
Scheuner, D. et al. Double-stranded RNA-dependent protein kinase phosphorylation of the alpha-subunit of eukaryotic translation initiation factor 2 mediates apoptosis. J Biol Chem. 281, 21458–68 (2006).
Haneji, T., Hirashima, K., Teramachi, J. & Morimoto, H. Okadaic acid activates the PKR pathway and induces apoptosis through PKR stimulation in MG63 osteoblast-like cells. Int J Oncol. 42, 1904–10, doi: 10.3892/ijo.2013.1911 (2013).
Takada, Y., Ichikawa, H., Pataer, A., Swisher, S. & Aggarwal, B. B. Genetic deletion of PKR abrogates TNF-induced activation of IkappaBalpha kinase, JNK, Akt and cell proliferation but potentiates p44/p42 MAPK and p38 MAPK activation. Oncogene. 26, 1201–12 (2007).
Yang, J. H. et al. Widespread inosine-containing mRNA in lymphocytes regulated by ADAR1 in response to inflammation. Immunology. 109, 15–23 (2003).
George, C. X. & Samuel, C. E. STAT2-dependent induction of RNA adenosine deaminase ADAR1 by type I interferon differs between mouse and human cells in the requirement for STAT1. Virology. 485, 363–70, doi: 10.1016/j.virol.2015.08.001 (2015).
George, C. X., Wagner, M. V. & Samuel, C. E. Expression of interferon-inducible RNA adenosine deaminase ADAR1 during pathogen infection and mouse embryo development involves tissue-selective promoter utilization and alternative splicing. J Biol Chem. 280, 15020–8 (2005).
Randall, R. E. & Goodbourn, S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol. 89 (Pt 1), 1–47 (2008).
Liu, X. et al. Identification and characterization of a constitutively expressed Ctenopharyngodon idella ADAR1 splicing isoform (CiADAR1a). Dev Comp Immunol. 63, 10–7, doi: 10.1016/j.dci.2016.05.008 (2016).
Martinon, F., Pétrilli, V., Mayor, A., Tardivel, A. & Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–41 (2006).
Wang, Y., Men, M., Yang, W., Zheng, H. & Xue, S. MiR-31 downregulation protects against cardiac ischemia/reperfusion injury by targeting protein kinase C epsilon (PKCε) directly. Cell Physiol Biochem 36, 179–90, doi: 10.1159/000374062 (2015).
Acknowledgements
The study was supported by the Ren Ji Seed Foundation (Grant No. RJZZ13-010).
Author information
Authors and Affiliations
Contributions
Yongyi Wang, Changfa Guo and Song Xue designed the research; Yongyi Wang, Min Men, Chengxi Wang, Bo Xie, Jidong Liu, Jianggui Shan, Hui Zheng and Wengang Yang performed the research; Jidong Liu, Hui Zheng and Yongyi Wang analyzed the data; Yongyi Wang and Changfa Guo wrote the manuscript; all authors discussed the results, reviewed and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wang, Y., Men, M., Xie, B. et al. Inhibition of PKR protects against H2O2-induced injury on neonatal cardiac myocytes by attenuating apoptosis and inflammation. Sci Rep 6, 38753 (2016). https://doi.org/10.1038/srep38753
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep38753
This article is cited by
-
Integration of epigenetic regulatory mechanisms in heart failure
Basic Research in Cardiology (2023)
-
Protein kinase RNA-activated controls mitotic progression and determines paclitaxel chemosensitivity through B-cell lymphoma 2 in ovarian cancer
Oncogene (2021)
-
2-aminopurine suppresses the TGF-β1-induced epithelial–mesenchymal transition and attenuates bleomycin-induced pulmonary fibrosis
Cell Death Discovery (2018)
-
Combinatory RNA-Sequencing Analyses Reveal a Dual Mode of Gene Regulation by ADAR1 in Gastric Cancer
Digestive Diseases and Sciences (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
