
Abstract
The homeodomain transcription factor distal-less homeobox 3 gene (DLX3) is required for hair, tooth and skeletal development. DLX3 mutations have been found to be responsible for Tricho-Dento-Osseous (TDO) syndrome, characterized by kinky hair, thin-pitted enamel and increased bone density. Here we show that the DLX3 mutation (c.533 A>G; Q178R) attenuates osteogenic potential and senescence of bone mesenchymal stem cells (BMSCs) isolated from a TDO patient, providing a molecular explanation for abnormal increased bone density. Both DLX3 mutations (c.533 A>G and c.571_574delGGGG) delayed cellular senescence when they were introduced into pre-osteoblastic cells MC3T3-E1. Furthermore, the attenuated skeletal aging and bone loss in DLX3 (Q178R) transgenic mice not only reconfirmed that DLX3 mutation (Q178R) delayed cellular senescence, but also prevented aging-mediated bone loss. Taken together, these results indicate that DLX3 mutations act as a loss of function in senescence. The delayed senescence of BMSCs leads to increased bone formation by compensating decreased osteogenic potentials with more generations and extended functional lifespan. Our findings in the rare human genetic disease unravel a novel mechanism of DLX3 involving the senescence regulation of bone formation.
Similar content being viewed by others


Introduction
Distal-less homeobox 3 (DLX3), mapped to 17q21.33, is a member of the distal-less vertebrate family (DLX1-6), which has a crucial function in the development and differentiation process1,2. Inactivated Dlx3 in mice leads to transcription factor placental failure, suggesting Dlx3 is indispensable in placental development3. DLX3, regarded as a homeodomain transcription factor in vertebrate, is required for hair follicle differentiation and cycling programs, for tooth morphogenesis, and for skeletal formation and development1,4,5,6. Mutations of DLX3 are closely related to Tricho-Dento-Osseous syndrome (TDO; OMIM 190320), which is a disorder with autosomal dominant inheritance mainly characterized by kinky hair, thin-pitted enamel (with remarkable attrition), dentin hypoplasia, and taurodontism as well as increased thickness and density of cranial and mandibular bones. To date, only six mutations in the DLX3 gene have been identified within minority family groups7,8,9,10 (Supplementary Fig. S1). Among them, the most common mutation in DLX3 is a frameshift mutation (c.571_574delGGGG; G191RfsX66), resulting in recoding and truncating the C-terminal transactivating domain of the DLX3 protein7. However, the de novo missense mutation (c.533 A>G; Q178R), previously reported in our department, was found in the homeodomain of the encoded protein11.
The patients with the TDO syndrome, showing a marked increased bone mineral density (BMD) in endochondral and intramembranous bones, imply that DLX3 exerts an essential role in bone formation. It has been identified that DLX3 is not only detected in osteoprogenitor cells, osteoblasts and osteocytes in vitro, but also is expressed in both developing and postnatal bones in vivo12,13,14. Studies have strongly suggested the importance of Dlx3 in bone by directly regulating bone differentiation determinants, such as osteocalcin (Ocn), Runx2 and osteoactivin15,16,17. Notably, the accrual of bone mass and density is observed in transgenic mice with Dlx3 ablation in osteogenic lineage cells, suggesting that Dlx3 has a negative effect on osteogenesis14.
Aging brings significant changes to skeletal system with gradually bone loss, as well as a shift in tissue microenviroment with cellular senescence in bone marrow, like osteoporosis18,19. The replicative bone marrow-derived mesenchymal stem cells (BMSCs) senescence encompasses a progressive loss of proliferation ability and a declining osteodifferentiation potential20,21. Misexpression of Dlx3 in the basal proliferative layer of epidermis of transgenic mice induces cell cycle arrest and leads to premature terminal differentiation22. In normal human epidermal keratinocytes, exogenous DLX3 promotes cell cycle arrest by activating tumor suppressor gene p53 and cyclin-dependent kinase inhibitor p21, which play critical roles in cell cycle and senescence regulation23. Collectively, these data suggest a possible role for DLX3 in senescence during epidermis proliferation and differentiation. However, whether DLX3 function as a modulator of cellular senescence involving bone formation has not been experimentally investigated.
To address the effect of DLX3 mutations on senescence during bone formation, we firstly isolated naïve BMSCs from a TDO patient with DLX3 mutation (c.533 A>G; Q178R). Interestingly, an attenuated osteoblastic activity accompanying with a delayed cellular senescence was observed in the BMSCs harboring the c.533 A>G mutation when compared with gender- and age-matched healthy BMSCs. This finding was further verified and was mechanistically studied by transfecting plasmids of the wild type DLX3, the mutant DLX3 (c.533 A>G) and the truncated DLX3 (c.571_574delGGGG) into pre-osteoblastic MC3T3-E1 cells. Consistently, the attenuated bone loss and aging in the aged transgenic mice with DLX3 mutation (c.533 A>G) suggest a regulatory role of DLX3 in the context of senescence during bone formation.
Results
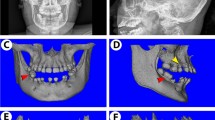
Isolation of BMSCs from the patient with TDO
The 27-yr-old female patient with typical TDO features was enrolled in this study (Supplementary Fig. S2A,B and C)11. Specially, thickened cortical and abnormally increased trabecular bone density of mandibular was clearly observed in the patient’s panoramic radiograph. The cephalometric radiograph of the patient showed a bossed frontal bone, a square jaw, increased bone density and thickness in parietal and calvaria bones, with a lack of pneumatization of the mastoids. Mandibular bone specimens of the patient were collected during implant surgery (Supplementary Fig. S3A). Bone marrow derived stem cells were isolated from the specimen (named as TDO-BMSCs) and reached 80% confluence after 14 days culture (Supplementary Fig. S3B). The missense mutation of DLX3 (c.533 A>G; Q178R) was confirmed by Sanger sequencing in the blood sample as well as in the BMSCs of the patient (Supplementary Fig. S2D). Further, BMSCs isolated from three gender- and age-matched normal donors as controls (named as CON-BMSCs). The flow cytometry assay, which showed that both TDO- and CON-BMSCs were negative for the hematopoietic markers CD34 and CD45, and positive for the mesenchymal stem cell-associated markers CD73, CD90, and CD106, indicated that DLX3 mutation did not affect expression of stem cell markers (Supplementary Fig. S3C).
TDO-BMSCs exhibit weaker osteogenic potential than CON-BMSCs
Due to the increased bone density in TDO patient, we firstly explored the inter-group differences on osteogenic potential. Under osteoinductive condition, the third passage of TDO-BMSCs differentiated into osteoblasts but showed lower levels of alkaline phosphatase (ALP) activity as assessed by ALP staining and ALP activity assays on day 3, 7, and 14 compared with CON-BMSCs (Fig. 1A,C). After osteoinduction for 14 and 21 days, Alizarin red staining (ARS) of calcium deposition with less staining intensity in TDO-BMSCs further confirmed these results (Fig. 1B,D). Western blot analysis indicated that the protein levels of RUNX2 and BSP were lower in TDO-BMSCs at 14 days post-osteoinduction (Fig. 1E). Meanwhile, by real-time PCR, the mRNA levels of RUNX2, ALP, Collagen I, BSP, and OCN were decreased in TDO-BMSCs on day 7 and 14 after osteoinduction, whereas the opposite effects were observed in CON-BMSCs (Fig. 1F). In addition, both real-time PCR and western blot analysis revealed lower levels of RANKL/OPG in TDO- relative to CON-BMSCs (Fig. 1E,F). Of note, DLX3 expression in TDO-BMSCs was half of that in normal controls (Supplementary Fig. S4).

TDO-BMSCs exhibit weaker osteogenic potential than CON-BMSCs.
TDO- and CON-BMSCs at passage 3 were cultured under osteoinduction medium. (A) ALP staining assay on days 3, 7, and 14 after osteoinduction. (B) Alizarin red staining assay at 14 and 21 days post-osteoinduction. (C) ALP activity assay on days 3, 7, and 14 after osteoinduction. (D) Quantification of alizarin red staining on days 14 and 21 post-osteoinduction. (E) Western blots of RUNX2, BSP, RANKL, OPG, and β-actin protein 14 days after osteogenic stimulation. (F) Osteogenesis-related genes (RUNX2, ALP, COLLAGEN I, BSP, OCN, RANKL and OPG) mRNA expression after osteoinduction. GAPDH served as an internal control. Data were presented as the mean ± S.D. of 3 independent experiments. *p < 0.05; **p < 0.01.
TDO-BMSCs exhibit slower senescence rate than CON-BMSCs
Since the panoramic radiograph indicated that the mandibular was heavily mineralized, decreased osteogenic potential apparently could not account for the phenotype of TDO. After literature reviewing, an important consideration is emphasized that bone formation is not only dependent on the osteogenic activity of individual osteoblast but also on the status of cellular senescence18,19,24. Of note, the classic senescence cell morphology, which was enlarged, flattened cellular morphology and a decline replicate proliferation, was observed in gender-, age- and passage-matched CON-BMSCs. Whereas, TDO-BMSCs, cultured for the same condition, showed relative younger state, with regularly long spindle-shaped fibroblast-like morphology (Supplementary Fig. S5). Thus, we examined whether DLX3 mutation intrinsically affected BMSCs senescence with further research.
Senescence-associated β-galactosidase (SA-β-gal), defined as a β-galactosidase activity detectable at pH 6.0, has been used as a common marker for aged cells as it has been demonstrated to increase with aging of various cells both in vitro and in vivo25,26. The number of SA-β-gal+ staining cells 38% in TDO-BMSCs and 55% in CON-BMSCs suggested that BMSCs with DLX3 mutation remained a younger status while the normal BMSCs entered a premature senescence (Fig. 2A,B). Consistently, GLB1 expression, the gene encoding lysosomal β-galactosidase27, was measured by flow cytometry and showed a lower level in TDO-BMSCs (Fig. 2C).

TDO-BMSCs exhibit slower senescence rate than CON-BMSCs.
(A) Representative images of cells assayed for SA-β-gal activity (blue) (scale bar, 100 μm). (B) Percentages of SA-β-gal+ cells/total cells from experiments as in panel (A) β-gal activity normalized to total cells. (C) Flow cytometry of the percentage of GLB1 expression compared with isotype controls. (D) Relative telomere length expressed as relative T/S ratio measured by quantitative real-time PCR analysis showing remarkably longer telomeres of TDO-BMSCs at passage 5 and 9. (E) Absolute telomere length expressed kb/diploid genome determined by qPCR also displaying longer telomeres of TDO-BMSCs at passage 5 and 9. (F) Real-time PCR for p16INK4a, p15INK4b, p53, OCT4, and NANOG in TDO- and CON-BMSCs at the passages 6, 7, and 8. (G) Western blots of p16INK4a, OCT4, and NANOG protein levels; GAPDH served as an internal control. Data were presented as the mean ± S.D. of 3 independent experiments. *p < 0.05; **p < 0.01.
Previous studies have identified that accelerated telomere shortening is observed in senescence cells28. Relative telomere length (expressed as relative ratio of telomere to single-copy gene (36B4) [relative T/S ratio]) and absolute telomere length (expressed kb/diploid genome) at passage 5 and 9 was measured by quantitative real-time PCR. It has been acknowledged that the decrease in the relative T/S ratio positively correlates with telomere shortening29,30. The results showed that the relative T/S ratio was higher in TDO-BMSCs (Fig. 2D). Moreover, absolute telomere length results further confirmed that TDO-BMSCs had longer telomeres (Fig. 2E).
p16INK4a, p15INK4b, and p21, encoded by CDKN2A gene, CDKN2B gene and CDKN1A respectively, together with tumor suppressive p53 transcription factor, were considered core elements of signaling pathways involved in cell senescence for their activation inducing cells into irreversible cell cycle arrest31,32,33. On the other hand, OCT4 and NANOG were stem cell specific transcription factors for maintaining pluripotency of stem cells during development34,35. The real-time PCR results showed that aging-related genes (p16INK4a, p15INK4b, and p53) were down-regulated and stem cell-associated genes (OCT4 and NANOG) were up-regulated in the passages 6, 7, and 8 in TDO-BMSCs (Fig. 2F). Correspondingly, western blot analysis also exhibited that p16INK4a protein expression was significantly reduced, while OCT4 expression was clearly increased in TDO-BMSCs (Fig. 2G). These results not only confirm that DLX3 mutation prevents the premature senescence of BMSCs but also indicates that DLX3 mutation stabilizes the pluripotency of BMSCs. Prolonged expression of stemness-related genes allow TDO-BMSCs to produce more “stem”-like osteogenic cells, to generate more daughter cells, and to extend the functional lifespan of osteoblastic cells to perform bone remodeling.
Overexpression of WT-DLX3, MT-DLX3 and TR-DLX3 in MC3T3-E1 cells
To abrogate background difference generated by different donors’ cells and to further identify whether the novel mutation is a gain- or loss- of function, we constructed plasmids of wild type DLX3, mutant DLX3 (c.533 A>G) and mutant DLX3 (c.571_574delGGGG), and transfected into osteoprogenitor cells MC3T3-E1 to investigate their function on osteogenic differentiation and aging in vitro. Of note, the mutant DLX3 (c.571_574delGGGG) was used to confirm the effect of DLX3 mutation on osteogenic potential and senescence.
The recombinant plasmids in empty vector pEGFP-C1, carrying wild type DLX3 and mutant DLX3 (c.533 A>G) and (c.571_574delGGGG) expression cassette were transfected into pre-osteoblastic cell line MC3T3-E1 and named as WT-DLX3, MT-DLX3 and TR-DLX3, respectively. MC3T3-E1 cells transfected with pEGFP-C1 empty vector (named as EGFP-EV) serves as control. The green fluorescence was detected under fluorescent microscope at 48 h post-transfection, and the results showed that transfection efficiency was over 30% (Supplementary Fig. S7). EGFP expression tagged to WT-DLX3, MT-DLX3 and TR-DLX3 was detected in nucleus, whereas EGFP expression in empty pEGFP-C1 was in cytoplasm, suggesting that whether DLX3 missense mutation (c.533 A>G) or DLX3 truncation mutation (c.571_574delGGGG) could enter into nucleus, like wild type DLX3 (Fig. 3A). In addition, both real-time PCR analysis and western blot analysis showed that the expression of DLX3 was substantially increased in WT-DLX3, MT-DLX3 and TR-DLX3 cells (Fig. 3B,C).

Cellular localization, DNA binding property to RUNX2 and overexpression of WT-DLX3, MT-DLX3 or TR-DLX3 in MC3T3-E1 cells.
(A) Panels showing EGFP and EGFP-DLX3 fusion protein expression in MC3T3-E1 cells 48 h after transfection with pEGFP-C1, pWT-DLX3, pMT-DLX3 and pTR-DLX3. Differential interference contrast (DIC) images were acquired to visualize cell shape (a, d, g and j). Images of EGFP expression (b, e, h and k) were detected by fluorescent microscope. WT-DLX3, MT-DLX3 or TR-DLX3 was located in nuclear while the EGFP was in cytoplasm, which was shown in the merged images (c, f, i and l). (B) DLX3 mRNA expression was highly enhanced in WT-DLX3, MT-DLX3 and TR-DLX3 compared with EGFP-EV after 48 h transfection by real-time PCR. (C) DLX3-GFP protein expression was detected by western blot analysis with anti-DLX3 (a) and anti-EGFP (b) antibodies after 48 h transfection. Only EGFP protein was detected in EGFP-EV samples. EGFP-DLX3 fusion protein was detected in WT-DLX3, MT-DLX3 and TR-DLX3 samples. Data were presented as the mean ± S.D. of 3 independent experiments. **p < 0.01; Scale bars = 100 μm. (D) DLX3 occupies its affinity site in the RUNX2 promoter. EMSA assay was performed by using a biotin-labeled double-stranded probe (containing the DLX3 affinity site). Nuclear extracts (10 μg) from HEK 293 T cells expressing GFP-WT-DLX3, GFP-MT-DLX3 or GFP-TR-DLX3 were used. The wild type probe (GCATTTTGTAATTTATTTCAAAGC) and the mutant probe (GCATTTTGTGGGTTATTTCAAAGC) were used as unlabeled competitors at a 100-fold excess. The presence of specific complexes, including supershifted GFP/DLX3 in the complexes, are indicated.
Previous study have shown that DLX3 acted as a molecular switch that regulated RUNX2 expression throughout bone formation, we here tested the ability of mutant DLX3 to bind RUNX2 promoter to determine whether deceased osteogenic potential associated with TDO-BMSCs were directly due to DLX3 mutation. The electrophoretic mobility shift (EMSA) experiments (Fig. 3D) showed that endogenous DLX3 could bind to the specific oligonucleotide probe. WT-DLX3, MT-DLX3 and TR-DLX3 were also able to bind the probe. The DLX3 EMSA signal was specific, as determined by competition with a 100-fold excess of unlabeled probe (wild-type [WT] probe), but not by an unlabeled mutant probe (MT probe) bearing point mutations in the high-affinity site. In addition, the stronger band in the lanes with the transfected WT-DLX3, MT-DLX3 and TR-DLX3 could be supershifted with anti-GFP, confirming that WT-DLX3, MT-DLX3 and TR-DLX3 do specifically bind the high-affinity site probe.
WT-DLX3 and TR-DLX3 increase while MT-DLX3 decreases cellular osteogenesis in MC3T3-E1 cells
Since the mutations in DLX3 did not change the DNA bind activity to RUNX2 promoter, we examined whether DLX3 mutations could affect osteogenic potential of MC3T3-E1 cells. After osteoinduction for 72 h, osteoblastic markers Runx2, Alp, Bsp, and Ocn mRNA expression detected by real-time PCR at mRNA level (Fig. 4A) and by western blot analysis at protein level (Fig. 4B,C) were slightly increased in WT-DLX3 and significantly increased in TR-DLX3, but remarkably decreased in MT-DLX3 when compared with EGFP-EV. ALP staining assay further confirmed these results by showing higher ALP activity in WT-DLX3 and TR-DLX3, lower in MT-DLX3 (Fig. 4D). These results suggest that different DLX3 mutations have different function on osteogenesis even though both of them could locate into nucleus and bind to RUNX2 promoter. Of note, real-time PCR and western blot analysis showed that BMP2 expression was lower in TDO-BMSCs and was decreased in MT-DLX3 but increased in TR-DLX3 (Supplementary Fig. S8).

WT-DLX3 and TR-DLX3 increase while MT-DLX3 decreases cellular osteogenesis in MC3T3-E1 cells.
MC3T3-E1 cells were transfected with pEGFP-C1, pWT-DLX3, pMT-DLX3 and pTR-DLX3 respectively. After 24 h transfection, these cells were further cultured in osteoinduction medium for 72 h. Then, the cells were harvested and subjected to real-time PCR and western blot. (A) Osteogenesis-related genes (Runx2, Alp, Collagen I, Bsp, and Ocn) mRNA expression were determined by real-time PCR. GAPDH served as an internal control. (B) Protein expression of RUNX2, BSP, OCN, and GAPDH were detected by western blot. GAPDH served as loading control. (C) Quantification of protein expression related to GAPDH. (D) ALP staining assay after osteoinduction for 72 h was performed. Data were presented as the mean ± S.D. of 3 independent experiments. *p < 0.05; **p < 0.01.
WT-DLX3 pushes while MT-DLX3 and TR-DLX3 delays cellular senescence in MC3T3-E1 cells
Apparently, the inhibition effect of DLX3 mutation (Q178R) on osteogenic differentiation had been revalidated, we further examined its effect on senescence. After 72 h osteoinduction, aging-related markers p16INK4a and p15INK4b mRNA expression detected by real-time PCR (Fig. 5A) and p16INK4a and GLB1 protein expression examined by western blot (Fig. 5B,C) were significantly increased in WT-DLX3 but decreased in MT-DLX3 and TR-DLX3 when compared with EGFP-EV. Stemness markers Oct4 and Nanog mRNA expression detected by real-time PCR (Fig. 5A) were significantly decreased in WT-DLX3 but increased in MT-DLX3 and TR-DLX3 when compared with EGFP-EV.

WT-DLX3 pushes while MT-DLX3 and TR-DLX3 delay cellular senescence in MC3T3-E1 cells.
MC3T3-E1 cells were transfected with pEGFP-C1, pWT-DLX3, pMT-DLX3 and pTR-DLX3 respectively, and were further cultured for 72 h under osteodifferentiation induction. Then, the cells were harvested and subjected to do real-time PCR and western blot. (A) Senescence-related genes (p16INK4a and p15INK4b) and stemness-related genes (Oct4 and Nanog) mRNA expression were determined by real-time PCR. GAPDH served as an internal control. (B) p16INK4a, GLB1 and GAPDH protein expression were detected by western blot. GAPDH served as loading control. (C) Quantification of protein expression related to GAPDH. (D) SA-β-gal staining was performed in WT-DLX3, MT-DLX3 or TR-DLX3 overexpression-MC3T3-E1 cells exposed to 100 μM H2O2 for 96 h. Representative images of SA-β-gal-positive cells (blue) (scale bar, 100 μm). (E) Percentages of SA-β-gal+ cells/total cells from experiments as in panel (D) β-gal activity normalized to total cells. (F) Relative telomere length expressed as relative T/S ratio measured by quantitative real-time PCR analysis. Data were presented as the mean ± S.D. of 3 independent experiments. **p < 0.01.
Exogenous H2O2 has been used extensively as an inducer of oxidative stress in vitro aging models. Therefore, SA-β-gal staining and relative telomere length was performed in WT-DLX3, MT-DLX3 or TR-DLX3 overexpression-MC3T3-E1 cells exposed to 100 μM H2O2 for 96 h. The results showed that percentage of SA-β-gal-positive cells was significantly higher in WT-DLX3, but much lower in MT-DLX3 and TR-DLX3 (Fig. 5D,E). Telomere length assay by quantitative real-time PCR (Fig. 5F) demonstrated that T/S ration was aggravated in MT-DLX3 and TR-DLX3, but reduced in WT-DLX3, when compared with EGFP-EV. H2O2 exposure further confirmed the functional discrepancy between the DLX3 mutations and wild type DLX3. These verified that both DLX3 mutations had negative effect on senescence.
DLX3 (Q178R) prevents skeletal aging and bone loss in vivo
Since DLX3 mutation (Q178R) could increase bone formation by preventing cell senescence even though with decreased osteogenic potential in vitro, we further generated transgenic mice expressing DLX3 mutation (Q178R) (named as DLX3 (Q178R)-Tg mice) to examine whether this novel mutation could protect against natural aging-mediated bone loss or osteopenia in vivo. Western blot analysis of DLX3 (Q178R) transgene was highly expressed in femur and tibia bone of founder mice (Supplementary Fig. S9A). Micro-CT analysis revealed that there was a gradual loss of trabecular bone mass with advancing age from 8 to 18 months in wild type mice (named as WT mice) (Fig. 6A). Further quantitative analysis showed that the most dramatic trabecular bone loss occurred between 8 and 16 months of age and average bone mineral density (BMD) dropped 62%, while bone volume/total volume (BV/TV) dropped 79%. However, the reduction was significantly less with DLX3 (Q178R)-Tg mice, with 43% in BMD and 65% in BV/TV respectively (Fig. 6B). Of note, we confirmed that DLX3 transgene mRNA in bone tissues was still expressed in aged DLX3 (Q178R)-Tg mice using RT-PCR although the levels of DLX3 mRNA declined over age (Supplementary Fig. S9B). We also examined that whether natural aging-associated p16INK4a activation was suppressed by DLX3 mutation (Q178R). While immunostaining of p16INK4a expression revealed p16INK4a activities in the vicinity of trabecular bones of WT mice. And p16INK4a activities appeared less prevalent in DLX3 (Q178R)-Tg mice (Fig. 6D). Consistently, the western blot analysis showed that the expression of p16INK4a, p53 and p21 were markedly weaker in DLX3 (Q178R)-Tg mice compared with WT mice (Fig. 6E).

DLX3 (Q178R) attenuates skeletal aging and bone loss in vivo.
(A and B) Micro-CT analysis: 3D trabecular reconstructions (A) Trabecular BMD (Tb. BMD), Trabecular bone volume ratio (Tb. BV/TV) (B) of distal femoral metaphysis regions from 8- and 16-months-old WT and DLX3 (Q178R)-Tg mice. (C) ALP staining and Alizarin red staining (ARS) of femur bone BMSCs from 8-months-old DLX3 (Q178R)-Tg vs WT mice, after osteogenic induction. (D) Immunostaining with p16INK4a surrounding the trabecular bones from 8-months-old WT and DLX3 (Q178R)-Tg mice. Scale bars, 25 μm. (E) Western blot of p16INK4a, p53 and p21 in tibia and femur of 8 months mice.
To examine if DLX3 mutation (Q178R) attenuates osteoblastic activity in a cell-autonomous manner, we isolated BMSCs from femurs of DLX3 (Q178R)-Tg and WT mice. Primary BMSCs from DLX3 (Q178R)-Tg mice demonstrated reduced osteogenic potential, as evidenced by ALP staining and Alizarin Red staining (ARS), when cells were induced by osteogenic medium (Fig. 6C).
Discussion
Herein we demonstrate a distinct role for DLX3 as an essential regulator of two integral processes in bone formation: osteogenesis and senescence. And our studies provide a possible explanation for abnormal bone formation of TDO patients and transgenic mice with DLX3 mutation (Q178R). Owing to the inhibition of osteogenic potential found in BMSCs isolated from the TDO patient or transgenic mice, the phenotype of increased bone mineral density is attributed to delayed cellular senescence caused by DLX3 mutation (Q178R). Additionally, the delayed skeletal aging and attenuated bone loss in DLX3 mutation (Q178R)-Tg suggest that DLX3 mutation is able to prevent aging-related bone loss. Mechanistically, the function studies of WT-DLX3, MT-DLX3 and TR-DLX3 in MC3T3-E1 extend our knowledge of DLX3 function in osteogenesis and senescence, as well as function alteration caused by DLX3 mutations.
Previous studies have proved that DLX3 is a key regulator of osteoblastogenesis. Yet, what’s the function of DLX3 in osteoblast differentiation? Most in vitro function studies have shown that DLX3 have the capability to directly regulate osteogenic markers such as OCN, RUNX2, and osteoactivin15,16,17. Overexpression of DLX3 in osteoprogenitors promotes osteogenic differentiation, while knockdown of DLX3 suppresses13,15. However, these results are challenged by the experiments in vivo. Both of transgenic mice Dlx3Prx1-cKO and Dlx3OCN-cKO models, conditional loss-of-function mutations of Dlx3 in mesenchymal cells and in osteogenic lineage cells respectively, present an increased bone mass accrual throughout the lifespan due to direct enhancement of osteoblast activity and up-regulation expression of osteogenic essential markers, including Runx2 and its downstream target Sp714. Undoubtedly, these results show that DLX3 is a negative regulator of bone mass accrual. Taken together, the studies above provide contrasting results and emphasize that DLX3 plays an implicated and sophisticated role in bone formation.
Given the fact that the bone homeostasis is an intricate process orchestrated by hundreds of factors, an interesting concept that emerges is that DLX3 performs its role in osteogenesis by coordinating with other osteogenic differentiation essential factors and constituting a regulatory network to keep the maintenance of bone homeostasis36. Firstly, DLX3 gene belongs to a Dlx homeobox transcription factors family, which consists of three inverted and convergent clusters (Dlx1/2, Dlx3/4 and Dlx5/6), and have essential function in skeleton development2,37,38. Dlx5 and Dlx6 deficient mice exhibits bone and ossification defects2,38. Increased levels of Dlx5 and Dlx6 are induced in transgenic mice with Dlx3 deletion in osteogenic cells and are considered as a contributor to increased bone mineral density in the mice14. Hassan et al. show that the selection association of Dlx5, Msx2 and Dlx3 binding to the Runx2 and Ocn promoter occurs in different stages of osteoblast maturation process15,16. Secondly, transcriptome studies exhibit that in vivo Dlx3 inactivation in osteogenic cells affects a wide range of developmental signals including Wnt, TGF-β/BMP and Notch pathways14. Previous studies have already shown that BMP2 induces the osteogenic differentiation, and Dlx3 are rapidly induced in response to BMP2-mediated osteoblast differentiation. Hassan et al. prove a regulatory pathway from BMP2 to commitment to osteogenesis via Dlx3 regulation16. Furthermore, the mechanism of Dlx3 and BMP2 regulation each other with a feedback control is inspected in the osteogenic differentiation of dental follicle cells39. Of note, our study showed that BMP2 expression was lower in TDO-BMSCs and was decreased in MT-DLX3 but increased in TR-DLX3 (Supplementary Fig. S8), suggesting that these two different DLX3 mutations act as different proteins in triggering osteogenic differentiation regulatory network. Meanwhile, the different expression levels of BMP2 also explain the reason of inhibited osteogenic potential in MT-DLX3 and promoted in TR-DLX3. Thirdly, as the fatal member in bone homeostasis, osteoclast activity is a force to be reckoned with. The ration of receptor activator of NF-κB ligand (RANKL) and osteoprotegerin (OPG) is considered a marker of bone metabolism balance40. Isaac et al. demonstrate that Dlx3 deletion in osteoblasts gives rise to high levels of OPG, consequently resulting a decreased RANKL/OPG ratio and down-regulated bone resorption, and finally contributing to bone formation augment14. The phenotypes of increased bone volume and trabecular BMD in transgenic TDO mice (a mutant DLX3 (571_574delGGGG) driven by 2.3 Col1A1 promoter) are owing to decreased osteoclast formation and activity caused by increased IFN-γ, because the mice lack of enhanced dynamic bone formation in vivo41. Our study showed that DLX3 mutation (Q178R) in BMSCs leaded to a decreased RANKL/OPG ratio, which indicates a regulation balance against bone resorption. These results are further supported by our previous studies which reveal that DLX3 mutation (Q178R) inhibits osteoclast differentiation in Raw 264.7 cells by up-regulating osteoclastogenesis inhibitor microRNA-124 expression42. From this point, the two different DLX3 mutations share the same negative effect on osteoclasts activity.
Our functional studies in MC3T3-E1 showed that wild type DLX3 overexpression in vitro promoted osteogenic differentiation, which is consistent with the studies in vitro but opposite to the studies in vivo. Based on the concept of regulatory network to maintain bone homeostasis, it is possible that the in vitro studies exhibit DLX3 direct function due to the lack of regulatory network in vitro, whereas the in vivo studies demonstrate that other regulators concerning bone homeostasis in the network compensate or overcompensate the function alteration caused by DLX3 inactivation or mutations. This hypothesis is supported by the studies in the TDO transgenic mouse which demonstrated that the BMSCs isolated from these mice exhibit enhanced osteogenic potentials, but the dynamic bone formation is lacked in the mouse41. Of note, DLX3 expression in TDO-BMSCs was almost half of that in normal controls (Supplementary Fig. S4) and the TDO-BMSCs exhibited lower osteogenic potentials in vitro. Yet, increased bone density was observed in the TDO patient in vivo. These studies not only further testify the existence of regulatory network to keep bone homeostasis but also indicate that DLX3 mutation (Q178R) act as a loss of function in vitro. Furthermore, our DLX3 mutation (Q178R) transgenic mice studies reconfirm this concept by showing that BMSCs isolated from these mice exhibited decreased osteogenic potentials in vitro, but these mice demonstrated increased BMD and trabecular bone volume in vivo. Collectively, we are inclined to take DLX3 as a direct positive regulator in osteogenesis in vitro. As for the reason responsible for the conflicts, our studies offer the notion of senescence as the answer.
Our findings that delayed senescence in TDO-BMSCs remedied and enhanced the bone formation in TDO patient in spite of declined osteogenic potentials cast a novel insight of DLX3 function in bone formation. In Isaac’s study, compared with the same age of wild type mice, loss of function of Dlx3 in Dlx3OCN-cKO mice results in femur BMD decreasing at 2 weeks to 5 weeks, but increasing from 4 months to the whole lifespan when compared with age matched wild-type mice14. These results indicate that DLX3 plays an important role in bone formation when age is increased. Our immunohistochemical analysis demonstrated that osteoblasts senescence was delayed by DLX3 (Q178R) mutation, and our Micro-CT analysis revealed that this delayed senescence attenuated aging-mediated bone loss in vivo. Overall, these experiments determine the role of DLX3 in senescence. Mechanistically, the functional studies in MC3T3-E1 showed that wild type DLX3 resulted in cell premature senescence, whereas both DLX3 mutations prevented cellular senescence. These results imply that DLX3 positively regulates cellular senescence and both DLX3 mutations show a loss of function. In Palazzo’s study, the luciferase and EMSA assays demonstrates that Dlx3 regulated p21 expression by directly binding to p21 promoter or by co-regulating with p5323. Of note, DLX3 expression was lower in TDO-BMSCs (Supplementary Fig. S4). Taken together, DLX3 is therefore a positive regulator in senescence and it might regulate senescence through p53/p21-regulted network.
In general, evidence has accumulated that DLX3 plays a pleiotropic role in several biological process to maintain bone homeostasis in normal bone, whereas, in TDO patients, this homeostasis is jeopardized by DLX3 mutation through triggering different pathways in the regulatory network to rectify this mistake. In this TDO patient, DLX3 (Q178R) mutation results in lower levels of expression of RUNX2, OCN, ALP, BMP2, p16INK4a, p53, p21, and so on, decreases osteogenesis and senescence. Mice lacking p16INK4a demonstrate increased proliferation and reduced frequency of apoptotic events, as well as enhanced repopulation ability by raising expression of stem cell self-renewal genes31,43. p21 deletion rescues proliferation and improves the repopulation capacity and self-renewal of stem cells from mice with dysfunctional telomeres44. The repressive expression of senescence genes rejuvenate BMSCs to keep the “young” state for their ability to self-renewal, proliferate, resistance to apoptosis and differentiate24,45,46, thereby more generations and longer lifespan of BMSCs compensate the decreased osteogenic differentiation propensity and finally lead to increased bone density.
Nevertheless, herein we focus on the role of DLX3 mutation in senescence, even though it might not be the only reason for the increased bone formation. Its role highlights a concept that DLX3 regulates bone formation by preventing senescence, gives us a novel sight to view DLX3 function in bone formation. Furthermore, increasing bone-and age-related disorder, especially in the aging population, is becoming a major socioeconomic burden to society. The osteoporosis exhibits net bone loss on account of skeletal aging47. The application of BMSCs in bone repair and regeneration is limited because of its senescence causing a high rate of failure48. To tackle the challenge, novel insight into transcriptional programs bound up with bone formation and aging process are urgent to excavate novel therapeutic targets in future clinical trials. Our research of DLX3 mutation protecting aging-related bone loss opens the possibility of its therapeutic potential in bone regeneration and bone loss disease.
Methods
Isolation and identification of BMSCs from the TDO patient and normal donors
A 26-year-old female with typical TDO features11 and gender- and age-matched normal donors (n = 3) participated in this study with informed consent. The study was approved by the Ethics Committee of Peking University School and Hospital of Stomatology. And the whole methods were carried out in accordance with the relevant guidelines, including any relevant details. BMSCs were isolated from alveolar bone of the involved patient (TDO-BMSCs) and healthy donors (CON-BMSCs) during implantation surgery as described below. Briefly, the bone tissue was rinsed in sterile phosphate-buffered saline (PBS), cut into 1 mm3 cubes, and digested in collagenase I (Invitrogen Life Technologies, Grand Island, NY, USA) and dispase II (Roche, Indianapolis, IN) for 1 h at 37 °C. Cells were collected by centrifugation at 200 × g and seeded in a 100-mm culture dish in alpha minimum essential medium (Alpha-MEM, Gibco, Paisley, Scotland, UK) supplemented with 10% fetal bovine serum (FBS) (Gibco) and penicillin-streptomycin and L-glutamine. Following overnight culture at 37 °C in a humidified atmosphere of 5% CO2 and 95% air, unattached cells were discarded when the medium was changed. Cells at passage 3 were analyzed for stem cell-associated markers by flow cytometry. Details were described in supplementary.
Generation of transgenic mice and experimental animals
All animal experiments were approved by the Peking University Animal Care and Use Committee. All methods were performed in accordance with the relevant guidelines and regulations. Human DLX3 mutation (Q178R) gene flanked by the mouse cytokeratin 14 promoter was subcloned into plasmid pGL647. The fragments of the DLX3 mutation (Q178R) transgene were purified and microinjected into C57BL/6 mouse oocytes, and the oocytes were surgically transferred to pseudopregnant C57BL/6 mouse (Cyagen Biosciences Inc., Guangzhou, China). Initial genotyping of founder transgenic mice was performed by PCR using tail biopsies from pups. Positive founders for transgene were reconfirmed by western blot analysis to test the overexpression of DLX3. Transgenic mice were bred with wild type mice to generate (to obtain a defined genetic background) hemizygotes for the analysis of bone phenotype changes. In all experiments, female transgenic mice and female wild type littermates as controls were used. We established a sample of at least 5 mice per group.
Screening DLX3 status in BMSCs at DNA level
To screen DLX3 status in TDO- and CON-BMSCs, we collected cells from the patient and control donors at passage 1 and extracted DNA with the General AllGen Kit (Cwbiotech, Beijing, China) according to the manufacturer’s instruction. Exon 3 of DLX3 was amplified using GoTaq Green Master Mix (Promega, Madison, WI) and exon 3 primer pair. The forward exon 3 primer was 5′-AGCATTCTGAGAGGCTAACTAGCTAC-3′, and the reverse primer was 5′-AGGTTCTGTGCGTGATACCA-3′. The PCR products were then sequenced on ABI 3730 sequencer.
Plasmids generation and cellular localization examination
To investigate the function of WT-DLX3 and MT-DLX3 protein, GFP-tagged DLX3 fusion gene expression plasmids were constructed. Briefly, Human full-length DLX3 cDNA was synthesized and subcloned into pEGFP-C1 vector. The novel mutant cDLX3 (c.533 A>G) and cDLX3 (c.571_574delGGGG) were generated from pEGFP-WT-DLX3 using single-site mutation kit (Promega, Madison, WI). The sequences of WT-DLX3, MT-DLX3 and TR-DLX3 were confirmed by DNA sequencing. To investigate the cellular localization, different plasmids were respectively transfected into MC3T3-E1 cells using Mirus TransIT-X2® Dynamic Delivery System following the manufacturer’s instruction. At 48 h post-transfection, the cellular localization of WT-DLX3, MT-DLX3 and TR-DLX3 was examined using an inverted fluorescence microscope.
Alkaline phosphatase (ALP) histochemistry and quantification of ALP activity
BMSCs were cultured in osteogenic medium for 3, 7, and 14 days. At the indicated times, the ALP histochemistry was stained with BCIP/NBT solution (Cwbiotech) and ALP activity was measured with an ALP assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer’s instruction. The details were described in supplementary.
Alizarin red staining and quantification of matrix mineralization
BMSCs were cultured in osteogenic medium for 14 and 21 days. At the indicated times, Alizarin red staining and quantification was performed. Details were described in supplementary.
Measurement of β-galactosidase activity and expression in BMSCs
Senescence-associated β-galactosidase (SA-β-gal) activity was assessed using two methods. Firstly, the same number (5 × 105) of BMSCs from both groups were seed into 60-mm culture dishes, and the medium was changed every 3 days. At 7 days post-inoculation, senescence was assessed using the senescence β-galactosidase staining kit (Beyotime Institute of Biotechnology, Jiangsu, China) according to the manufacturer’s instruction. To quantify SA-β-gal activity, we counted at least 300 cells in randomly choose fields and calculated the ratio of SA-β-gal+ cells to total cells. Secondly, to confirm the above result, BMSCs at passage 8 th were collected and analyzed by flow cytometry after incubation with anti-GLB1 antibody (Abcam, Cambridge, MA) and anti-human IgG isotype (GeneTex, Irvine, CA) for 1 h at 4 °C. The recommended PE-conjugated non-specific rabbit IgG [F(ab1)2 donkey anti-rabbit IgG PE secondary antibody (eBioscience, San Diego, CA)] was used as secondary antibody to measure GLB1 expression. Flow cytometry was performed on a Coulter Epics XL (Beckman Coulter, Inc.).
Electrophoretic mobility shift assay (EMSA)
The nuclear extract (10 μg of protein) was mixed with biotin-labeled DNA probe or cold competitor in binding buffer (LightShift® Chemiluminescent EMSA Kit). The reaction mixture was incubated for 30 min at room temperature. GFP antibody was included for supershift. Then, the reaction mixture was incubated for additional 30 min at room temperature. Samples were run on an acrylamide gel at 120 V for about 2 h. The gel was transferred to a nylon membrane. The biotin-labeled DNA was detected using the streptavidin-horseradish peroxidase conjugate and LightShift chemiluminescent substrate.
Telomere measurement by quantitative real-time PCR
Genome DNA was prepared using DNeasy Blood & Tissue Kit (Cwbiotech). Average telomere length was measured from total genomic DNA using a real-time PCR as previously described29,30. PCR reactions were performed on the ABI 7500 real-time PCR system (Life Technologies Corporation), using telomeric primers, primers for the reference control gene (human/mouse 36B4 single copy gene). For each PCR reaction, a standard curve was made by serial dilutions of known amounts of DNA. The telomere signal was normalized to the signal from the single copy gene to generate a T/S ratio indicative of relative telomere length. For human absolute telomere length calculation, the telomere kb per reaction value was divided genome copy number (calculated from 36B4 Ct and standard curve) to generate a total telomeric length in kb per diploid genome49. Equal amounts of DNA (20 ng) were used for each reaction, with at least three replicates for each specimen. The telomere standard curve and 36B4 standard curve was supplied in Supplementary Fig. S6.
Real-time PCR analysis
Total RNA was isolated with TRIzol® reagent (Invitrogen Life Technologies) and 2 μg of RNA was reverse-transcribed into cDNA using the Superscript first-strand synthesis system (Invitrogen Life Technologies) according to protocol. Real-time PCR reactions were conducted in a 20-μl reaction mixture (containing cDNA and SYBR green master mix) using an ABI 7500 real-time PCR system (Life Technologies Corporation). The mRNA expression relative to osteoclastogenesis was normalized to murine glyceraldehydephosphate dehydrogenase (GAPDH) and calculated using the 2−∆∆ Ct method. The sequences of each primer are listed in Supplementary Tables S1 and S2.
Western blot assay
Western blot assay was used to measure the protein expression, and the detailed procedures were described as below. Cells were harvested and lysed in RIPA buffer containing protease inhibitors. After determining the concentration, protein samples were subjected to 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to a polyvinylidenedifluoride membrane. The membranes were blocked in 5% skim milk for 1 h and incubated with antibodies against RUNX2, BSP, RANKL, OPG (Santa Cruz Biotechnology, Santa Cruz, CA) human p16INK4a, OCN, OCT4, EGFP (Cell Signaling Technology, Beverly, MA), p16INK4a, GLB1, DLX3 (Abcam), β-actin and GAPDH (Proteintech, Chicago, IL) separately at 4 °C overnight. After incubation with peroxidase-linked secondary antibodies, immunoreactive proteins were visualized on an Odyssey infrared imaging system (Odyssey LI-COR Biosciences, Lincoln, NE).
Micro-CT analysis
Micro-CT analysis of specimens was performed using a high-resolution Inveon Micro-CT (Siemens, Munich, Germany). Briefly, the x-ray source was set at 60 kV, with a node current of 220 μA and an exposure time of 400 ms for each of the 360 rotational steps. Image slices were then reconstructed using micro-CT image analysis software (Inveon Research Workplace). The regions of interest were defined as the areas between 0.5 mm and 1.0 mm proximal to the growth plate in the distal femurs, in order to include the secondary trabecular spongiosa. A threshold of 500 was used for evaluation of all scans. For visualization, we imported the segmented data and reconstructed them as a 3D-image displayed in micro-CT image analysis software (Inveon Research Workplace).
Immunohistochemical analysis
The specimens were decalcified in 10% ethylene diamine tetraacetic acid (EDTA) for 1 month, and sectioned (5 μm) for staining as previously described50. The specimens were incubated with primary antibody against p16INK4a (Abcam) at 4 °C overnight. Then, samples were processed using the DAB detection kit (Zhangshanjinqiao, Beijing, China) and visualized under an Olympus BX51 light microscope equipped with an Olympus DP70 camera (Olympus, Co., Tokyo, Japan).
Statistical analysis
All data was representative of each assay repeated independently at least three times with similar results. Statistical significance was determined using the two-tailed Student’s t-test, assuming equal variances. The χ2 test was used to compare rates. Significance is indicated as follows: *p < 0.05; **p < 0.01.
Additional Information
How to cite this article: Zhao, N. et al. Senescence: novel insight into DLX3 mutations leading to enhanced bone formation in Tricho-Dento-Osseous syndrome. Sci. Rep. 6, 38680; doi: 10.1038/srep38680 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Beanan, M. J. & Sargent, T. D. Regulation and Function of Dlx3 in Vertebrate Development. Dev Dyn. 218, 545–553 (2000).
Merlo, G. R. et al. Multiple Functions of Dlx Genes. Int J Dev Biol. 44, 619–626 (2000).
Morasso, M. I., Grinberg, A., Robinson, G., Sargent, T. D. & Mahon, K. A. Placental Failure in Mice Lacking the Homeobox Gene Dlx3. Proc Natl Acad Sci USA 96, 162–167 (1999).
Feledy, J. A., Morasso, M. I., Jang, S. I. & Sargent, T. D. Transcriptional Activation by the Homeodomain Protein Distal-Less 3. Nucleic Acids Res. 27, 764–770 (1999).
Hwang, J., Mehrani, T., Millar, S. E. & Morasso, M. I. Dlx3 is a Crucial Regulator of Hair Follicle Differentiation and Cycling. Development. 135, 3149–3159 (2008).
Choi, S. J. et al. Mutant Dlx3 Disrupts Odontoblast Polarization and Dentin Formation. Dev Biol. 344, 682–692 (2010).
Price, J. A., Bowden, D. W., Wright, J. T., Pettenati, M. J. & Hart, T. C. Identification of a Mutation in Dlx3 Associated with Tricho-Dento-Osseous (Tdo) Syndrome. Hum Mol Genet. 7, 563–569 (1998).
Lee, S. K. et al. Dlx3 Mutation in a New Family and its Phenotypic Variations. J Dent Res. 87, 354–357 (2008).
Mayer, D. E., Baal, C., Litschauer-Poursadrollah, M., Hemmer, W. & Jarisch, R. Uncombable Hair and Atopic Dermatitis in a Case of Trichodento-Osseous Syndrome. J Dtsch Dermatol Ges. 8, 102–104 (2010).
Nieminen, P. et al. Dlx3 Homeodomain Mutations Cause Tricho-Dento-Osseous Syndrome with Novel Phenotypes. Cells Tissues Organs. 194, 49–59 (2011).
Li, Y. et al. Morphological Analyses and a Novel De Novo Dlx3 Mutation Associated with Tricho-Dento-Osseous Syndrome in a Chinese Family. Eur J Oral Sci. 123, 228–234 (2015).
Ghoul-Mazgar, S. et al. Expression Pattern of Dlx3 During Cell Differentiation in Mineralized Tissues. Bone. 37, 799–809 (2005).
Li, H. et al. Expression and Function of Dlx Genes in the Osteoblast Lineage. Dev Biol. 316, 458–470 (2008).
Isaac, J. et al. Dlx3 Regulates Bone Mass by Targeting Genes Supporting Osteoblast Differentiation and Mineral Homeostasis in Vivo. Cell Death Differ. 21, 1365–1376 (2014).
Hassan, M. Q. et al. Dlx3 Transcriptional Regulation of Osteoblast Differentiation: Temporal Recruitment of Msx2, Dlx3, and Dlx5 Homeodomain Proteins to Chromatin of the Osteocalcin Gene. Mol Cell Biol. 24, 9248–9261 (2004).
Hassan, M. Q. et al. Bmp2 Commitment to the Osteogenic Lineage Involves Activation of Runx2 by Dlx3 and a Homeodomain Transcriptional Network. J Biol Chem. 281, 40515–40526 (2006).
Singh, M., Del, C. F., Monroy, M. A., Popoff, S. N. & Safadi, F. F. Homeodomain Transcription Factors Regulate Bmp-2-Induced Osteoactivin Transcription in Osteoblasts. J Cell Physiol. 227, 390–399 (2012).
Manolagas, S. C. & Parfitt, A. M. What Old Means to Bone. Trends Endocrinol Metab. 21, 369–374 (2010).
Marie, P. J. Bone Cell Senescence: Mechanisms and Perspectives. J Bone Miner Res. 29, 1311–1321 (2014).
Izadpanah, R. et al. Long-Term in Vitro Expansion Alters the Biology of Adult Mesenchymal Stem Cells. Cancer Res. 68, 4229–4238 (2008).
Wilson, A., Shehadeh, L. A., Yu, H. & Webster, K. A. Age-Related Molecular Genetic Changes of Murine Bone Marrow Mesenchymal Stem Cells. BMC Genomics. 11, 229 (2010).
Morasso, M. I., Markova, N. G. & Sargent, T. D. Regulation of Epidermal Differentiation by a Distal-Less Homeodomain Gene. J Cell Biol. 135, 1879–1887 (1996).
Palazzo, E. et al. The Homeoprotein Dlx3 and Tumor Suppressor P53 Co-Regulate Cell Cycle Progression and Squamous Tumor Growth. Oncogene. 35, 3114–3124 (2016).
Baker, N., Boyette, L. B. & Tuan, R. S. Characterization of Bone Marrow-Derived Mesenchymal Stem Cells in Aging. Bone. 70, 37–47 (2015).
Dimri, G. P. et al. A Biomarker that Identifies Senescent Human Cells in Culture and in Aging Skin in Vivo. Proc Natl Acad Sci USA. 92, 9363–9367 (1995).
Lee, B. Y. et al. Senescence-Associated Beta-Galactosidase is Lysosomal Beta-Galactosidase. Aging Cell. 5, 187–195 (2006).
Wagner, J. et al. Overexpression of the Novel Senescence Marker Beta-Galactosidase (Glb1) in Prostate Cancer Predicts Reduced Psa Recurrence. PLoS One. 10, e124366 (2015).
von Zglinicki, T., Pilger, R. & Sitte, N. Accumulation of Single-Strand Breaks is the Major Cause of Telomere Shortening in Human Fibroblasts. Free Radic Biol Med. 28, 64–74 (2000).
Gil, M. E. & Coetzer, T. L. Real-Time Quantitative Pcr of Telomere Length. Mol Biotechnol. 27, 169–172 (2004).
Callicott, R. J. & Womack, J. E. Real-Time Pcr Assay for Measurement of Mouse Telomeres. Comp Med. 56, 17–22 (2006).
Janzen, V. et al. Stem-Cell Ageing Modified by the Cyclin-Dependent Kinase Inhibitor P16Ink4a. Nature. 443, 421–426 (2006).
Liu, D. & Xu, Y. P53, Oxidative Stress, and Aging. Antioxid Redox Signal. 15, 1669–1678 (2011).
Sharpless, N. E. & Sherr, C. J. Forging a Signature of in Vivo Senescence. Nat Rev Cancer. 15, 397–408 (2015).
Chambers, I. et al. Nanog Safeguards Pluripotency and Mediates Germline Development. Nature. 450, 1230–1234 (2007).
van den Berg, D. L. et al. An Oct4-Centered Protein Interaction Network in Embryonic Stem Cells. Cell Stem Cell. 6, 369–381 (2010).
Levi, G. & Gitton, Y. Dlx Genes and the Maintenance of Bone Homeostasis and Skeletal Integrity. Cell Death Differ. 21, 1345–1346 (2014).
Cohen, S. M., Bronner, G., Kuttner, F., Jurgens, G. & Jackle, H. Distal-Less Encodes a Homoeodomain Protein Required for Limb Development in Drosophila. Nature. 338, 432–434 (1989).
Depew, M. J., Lufkin, T. & Rubenstein, J. L. Specification of Jaw Subdivisions by Dlx Genes. Science. 298, 381–385 (2002).
Li, X., Yang, G. & Fan, M. Effects of Homeobox Gene Distal-Less 3 On Proliferation and Odontoblastic Differentiation of Human Dental Pulp Cells. J Endod. 38, 1504–1510 (2012).
Walsh, M. C. & Choi, Y. Biology of the Rankl-Rank-Opg System in Immunity, Bone, and Beyond. Front Immunol. 5, 511 (2014).
Choi, S. J. et al. In Vivo Impact of a 4 Bp Deletion Mutation in the Dlx3 Gene On Bone Development. Dev Biol. 325, 129–137 (2009).
Zhao, N. et al. Dlx3 Negatively Regulates Osteoclastic Differentiation through Microrna-124. Exp Cell Res. 341, 166–176 (2016).
Krishnamurthy, J. et al. P16Ink4a Induces an Age-Dependent Decline in Islet Regenerative Potential. Nature. 443, 453–457 (2006).
Choudhury, A. R. et al. Cdkn1a Deletion Improves Stem Cell Function and Lifespan of Mice with Dysfunctional Telomeres without Accelerating Cancer Formation. Nat Genet. 39, 99–105 (2007).
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The Hallmarks of Aging. Cell. 153, 1194–1217 (2013).
Boyette, L. B. & Tuan, R. S. Adult Stem Cells and Diseases of Aging. J Clin Med. 3, 88–134 (2014).
Raisz, L. G. Pathogenesis of Osteoporosis: Concepts, Conflicts, and Prospects. J Clin Invest. 115, 3318–3325 (2005).
Xin, Y. et al. Aging Adversely Impacts Biological Properties of Human Bone Marrow-Derived Mesenchymal Stem Cells: Implications for Tissue Engineering Heart Valve Construction. Artif Organs. 34, 215–222 (2010).
O’Callaghan, N. J. & Fenech, M. A Quantitative Pcr Method for Measuring Absolute Telomere Length. Biol Proced Online. 13, 3 (2011).
Chang, J. et al. Inhibition of Osteoblastic Bone Formation by Nuclear Factor-Kappab. Nat Med. 15, 682–689 (2009).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81570961), the Natural Science Foundation of Beijing Municipality (7092113), the National Key Health Research Project Foundation of China during the 11th Five-Year Plan Period (2006BAI05A07), the Capital Medical Developing Foundation (2007-1005), the Beijing Natural Science Foundation Grant (5162013) and National Institute of Dental and Craniofacial Research (R90DE022527). We are grateful to the TDO patient and the normal healthy donors and for participating in this study.
Author information
Authors and Affiliations
Contributions
Na Zhao performed the measurement of all biomarkers, analyzed the data, created figures and drafted the manuscript. Dong Han and Yue Li carried out sample preparation and were involved in the analysis and interpretation of the data. Haochen Liu and Singwai Wong carried out plasmid construction. Zhengyi Cao carried out the EMSA experiments. Jian Xu assisted in figures creating. Xiaowei Zhang and Tao Cai provided a lot of technical guidance and assisted in manuscript revising. Yixiang Wang and Hailan Feng designed the study, supervised the experiments, supervised all data acquisition and supervised writing of article.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhao, N., Han, D., Liu, H. et al. Senescence: novel insight into DLX3 mutations leading to enhanced bone formation in Tricho-Dento-Osseous syndrome. Sci Rep 6, 38680 (2016). https://doi.org/10.1038/srep38680
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep38680
This article is cited by
-
Temporal Modulation of DNA Methylation and Gene Expression in Monolayer and 3D Spheroids of Dental Pulp Stem Cells during Osteogenic Differentiation: A Comparative Study
Tissue Engineering and Regenerative Medicine (2022)
-
Human Genetics of Sclerosing Bone Disorders
Current Osteoporosis Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
