
Abstract
Lanthipeptides are a large class of bacteria-produced, ribosomally-synthesized and post-translationally modified peptides. They are recognized as peptide antibiotics because most of them exhibit potent antimicrobial activities against Gram-positive bacteria especially those that are phylogenetically related to producers. Maturation of class II lanthipeptide like bovicin HJ50 undergoes precursor modification by LanM and a subsequent leader peptide cleavage by LanT. Herein, via co-expression of precursor gene bovA, modification gene bovM and transporter gene bovT in Escherichia coli C43 (DE3), bioactive bovicin HJ50 was successfully produced and secreted. To further achieve in vitro one-pot synthesis of bovicin HJ50, an engineered bovicin HJ50 synthetase BovT150M was obtained by fusing the peptidase domain of BovT (BovT150) to the N-terminus of BovM. BovT150M exhibited dual functions of precursor modification and leader peptide cleavage to release mature bovicin HJ50. Under the guidance of BovA leader peptide, BovT150M exhibited substrate tolerance to modify non-native substrates including suicin and lacticin 481. This work exemplifies the feasibility of enzyme chimera of peptidase domain (LanT150) and modification enzyme (LanM) as a one-pot lanthipeptide synthetase.
Similar content being viewed by others

Introduction
Drug-resistant bacteria have posed increasing threats to human health and have raised global concern for lack of effective resorts1,2,3. Lanthipeptides are polycyclic peptides featured by the presence of unusual lanthionine/methyl lanthionine and belong to a growing family of natural products known as ribosomally synthesized and post-translationally modified peptides (RiPPs)4. Lanthipeptides that exert antimicrobial activities are referred to lantibiotics, which are regarded as ideal alternatives to antibiotics because of their extraordinary efficacy, remarkable stability and low possibility to raise bacterial resistance5. Most lantibiotics act either by inhibition of cell wall biosynthesis via binding and sequestration of essential peptidoglycan precursor lipid II and/or disruption of the membrane integrity via pore formation4,6. Nisin, the prototype lanthipeptide, has been long used as food preservative worldwide for over 50 years without occurrence of bacteria resistance and has been in clinical trials to treat diseases like bovine mastitis7. Recent genome-based mining over increasing microbial genomes envisioned the unanticipated wide distribution of lanthipeptide gene clusters, which greatly outnumbered known lanthipeptides and are thus a fascinating arsenal for peptide antibiotics8.
Lanthipeptides are produced exclusively by Gram-positive bacteria and their biosynthesis-related genes are assembled in gene clusters encoding precursor peptides (LanA), modification enzymes (LanBC/LanM/LanKC/LanL), transporters (LanT), processing proteases (LanP), immunity proteins (LanFEG/LanH/LanI) and regulation machineries. Posttranslational modification of precursor peptide and subsequent leader peptide cleavage are the most pivotal steps for lanthipeptide maturation. Initially, precursor LanA is ribosomally produced as a linear peptide composed of an N-terminal leader peptide and a C-terminal core peptide. Under the guidance of leader peptide, the modification enzyme executes a sequential dehydration and cyclization process, during which certain Ser or Thr residues in the core peptide are firstly dehydrated and then cyclized with Cys residues to form intramolecular thioether bridges9. Removal of the N-terminal leader peptide by protease is required to release bioactive product referred as lanthipeptide. Lanthipeptides are mainly divided into four classes based on the diversity of modification enzymes10. Class II lanthipeptides, one of the most extensively investigated family members of lanthipeptides, are modified by a bifunctional LanM which contains an N-terminal dehydratase domain and a C-terminal LanC-like cyclase domain8. After precursor modification, the leader peptides of class II lanthipeptides are in most cases processed by LanT, which is an ABC transporter with an N-terminal cysteine peptidase domain and a C-terminal ATP-binding domain11. LanT is responsible for cleavage of the leader peptide at a highly conserved double glycine motif (GlyGly/GlyAla/GlySer), concomitant with exporting the mature lanthipeptides outside the cell12. While in a few cases, a second trimming of an additional hexa-peptide is required by a serine protease LanP like lichenicidin and cerecidin13,14.
Traditional methods to discover novel lanthipeptides are mainly via bioactivity-based screening of producer strains or heterologous expression of lanthipeptide gene clusters in related model hosts, which are somewhat time-consuming and cost-extensive15,16. Thus, efficient and potent synthetic strategy would no doubt facilitate expedient generation and bioengineering of lanthipeptides. Reconstitution of LctM, the synthetase of a typical class II lanthipeptide lacticin 481, unlocked the gate to in vitro bioengineering of lanthipeptides17. Until recently, this in vitro modification strategy combined with leader peptide cleavage by commercial protease was fully exploited in lanthipeptide biosynthesis18,19. Bovicin HJ50 (PDB ID: 2M8V), produced by Streptococcus bovis HJ50, is reminiscent of lacticin 481 but differed by an unusual disulfide bridge20,21,22. Bovicin HJ50 contains a conserved putative lipid II binding motif that indicates its mode of action by inhibiting cell wall biosynthesis as well as pore formation on membrane21. The gene cluster for bovicin HJ50 biosynthesis includes 9 genes namely bovAMTFEGKRI23,24. Bovicin HJ50 has been produced by way of a two-step semi-in vitro biosynthesis system (SIVB) consisting of (1) in vivo modification of precursor peptide by co-expression of precursor gene bovA and modification gene bovM in E. coli and (2) in vitro digestion of leader peptide by N-terminal peptidase domain of BovT (BovT150) (Fig. 1a)20. Thus, the reconstituted BovM and BovT150 could function as the minimal machinery for bovicin HJ50 biosynthesis.

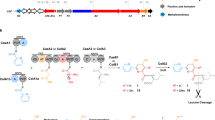
Biosynthesis of bovicin HJ50 and crucial residues in BovT150, BovM and BovT150M.
(a) Maturation of bovicin HJ50 requires modification by BovM and leader peptide cleavage at the double glycine motif by BovT150. (b) BovT150 is a cysteine peptidase containing a crucial Cys15 and BovM is the modification enzyme of bovicin HJ50 containing a crucial Asp264. The engineered bovicin HJ50 synthetase BovT150M contains an N-terminal BovT150 and a C-terminal BovM, in which Cys15 corresponds to Cys15 in BvoT and Asp416 corresponds to Asp264 in BovM.
In the present study, we established a one-pot synthesis system for bovicin HJ50. First, we achieved bovicin HJ50 production in E. coli via co-expression of bovA and bovM with either bovT150 or intact bovT. Prompted by that, an engineered lanthipeptide synthetase BovT150M (BovT150-BovM fusion enzyme) was successfully reconstituted that exerted dual functions of precursor modification and leader peptide cleavage. Bovicin HJ50 as well as bovicin HJ50-like lanthipeptide suicin, were successfully produced via BovT150M. This work provided a new approach to release bioactive lanthipeptides via a modification and processing enzyme chimera, which could be potentially applied for expedient biosynthesis and bioengineering of novel lanthipeptides.
Results
One-pot synthesis of bovicin HJ50 in vivo
BovA could be successfully modified by BovM when they are co-expressed in E. coli20. To exploit E. coli as microbial biofactory for bovicin HJ50 production, bovA and bovM were co-expressed with N-terminal peptidase bovT150or intact bovT. Expression of single bovA or co-expression of bovA and bovM in E. coli BL21 (DE3) could not produce any antimicrobial activities when assayed against sensitive indicator strain Micrococcus flavus NCIB8166 (Fig. 2a). However, when bovA, bovM and bovT150 were co-expressed, the cell lysates that were induced for more than 10 h exhibited antimicrobial activity, whereas the supernatants exhibited no activity (Fig. 2a). MALDI-TOF MS analysis of the cell lysates detected the presence of bovicin HJ50 with m/z as 3428.6 Da (Fig. 2b). Co-expression of bovA, bovM and bovT in E. coli BL21 (DE3) did not produce any bioactive agents in neither supernatants nor cell lysates (Fig. 2a). This indicated that the peptidase domain BovT150 was capable to cleave the leader peptide of BovM modified precursor in vivo, whereas the full length BovT might not be functionally expressed in E. coli BL21 (DE3).

Production of bovicin HJ50 in E. coli.
(a) Antimicrobial assay of supernatants and cell lysates of E. coli BL21 (DE3) after induced by IPTG for 5, 10, 20 h. E. coli cells are transformed with (1) pET28a-bovA; (2) pET28a-bovAM; (3) pET28a-bovAM + pACYC-Duet-bovT150; (4) pET28a-bovAM + pACYC-Duet-bovT; (5) pET28a-bovA + pACYC-Duet-bovT150M. M. flavus NCIB8166 was used as the indicator strain for antimicrobial assay. (b) MS analysis of cell lysates of E. coli BL21 (DE3) co-transformed with pET28a-bovA and pACYC-Duet-bovT150M. (c) MS analysis and antimicrobial assay of supernatants of E. coli C43 (DE3) co-transformed with pET28a-bovAM and pACYC-Duet-bovT after induced by IPTG. A, culture supernatants; B, cell lysates.
To reconstitute the full length BovT in vivo, we adopted a host strain E. coli C43 (DE3), a BL21 (DE3) derived strain suitable for membrane protein expression25. Interestingly, co-expression of bovA, bovM and bovT in E. coli C43 (DE3) produced bioactive bovicin HJ50 in supernatants but not in cell lysates, which was further confirmed by MS analysis (Fig. 2c). This indicated that mature bovicin HJ50 was produced and exported outside from E. coli cells. Thus, bioactive lanthipeptide bovicin HJ50 was successfully produced in E. coli via manipulating the minimal biosynthetic machinery.
One-pot synthesis of bovicin HJ50 via engineered BovT150M
Prompted by the concomitant functionality of BovM and BovT150 in E. coli, we created a recombinant bovicin HJ50 synthetase BovT150M (BovT150-BovM) by fusing the BovT150 to the N-terminus of BovM. To reconstitute the in vivo function of the engineered BovT150M, bovA was co-expressed with bovT150M in E. coli BL21 (DE3) via transformation with pET28a-bovA and pACYC-Duet-bovT150M. When induced by IPTG, bovicin HJ50 was produced in vivo but not exported outside the cell membrane as only cell lysates showed antimicrobial activity (Fig. 2a). This indicated that BovT150M might exert precursor modification and leader peptide digestion functions simultaneously. Quantification of bovicin HJ50 in the cell lysates using agar diffusion bioassay showed that 0.20 μg/ml to 0.91 μg/ml bovicin HJ50 were produced (Supplementary Information, Figure S1). Cell growth curve indicated that in vivo production of bovicin HJ50 led to slight growth retardation (Supplementary Information, Figure S1).
To reconstitute BovT150M in vitro, BovT150M was expressed and purified by immobilized ion metal affinity chromatography (IMAC) (Supplementary Information, Figure S2a). Precursor hexahistidine-tagged BovA (His6-BovA) was expressed and purified from inclusion bodies by IMAC and C18 reversed phase high performance liquid chromatography (RP-HPLC) as described previously26. When incubating His6-BovA (20 μM) and BovT150M (2 μM) in assay buffer containing 10 mM Mg2+, 1 mM DL-dithiothreitol (DTT) and 2.5 mM adenosine triphosphate (ATP) for 1 h, bioactive bovicin HJ50 was produced (Fig. 3a). MS analysis of the product exhibited an [M+H]+ of 3430.6 Da, which was in good accordance with authentic bovicin HJ50 with an unfolded disulfide bridge (Fig. 3a).

Enzymatic activity assay of the engineered bovicin HJ50 synthetase BovT150M.
MS analysis and antimicrobial assay of reaction products after incubation of His6-BovA and BovT150M in the presence of DTT and ATP (a), in the absence of DTT (b), and in the absence of ATP (c).
Reductive agents like DTT are supposed to be required for the catalytic function of BovT150M in that BovT150 is a cysteine protease and BovM also needs reductive conditions to modify substrate BovA24. When DTT was absent, incubation of His6-BovA and BovT150M for more than 4 h could not produce any bioactive agents (Fig. 3b). MS analysis showed the mass peak of 8275.7 Da, which was about 4 Da decrease compared with calculated mass of 8279.3 Da of His6-BovA (Fig. 3b). This indicated that 2 disulfide bridges were spontaneously formed in His6-BovA and BovT150M could hardly modify or digest the disulfide cross-linked His6-BovA as indicated in previous research17,24. Although reductive conditions are required for the fusion enzyme to correctly modify and release bovicin HJ50, the unfolded disulfide bridge could be spontaneously re-formed when DTT was removed or in other oxidative conditions24. ATP was also demonstrated to be indispensable for the catalytic function of BovT150M as absence of ATP eliminated the dehydration activity of BovT150M (Fig. 3c). ATP might serve as phosphate donors during the phosphorylation of substrate BovA by BovM domain17.
Mutational analysis of BovT150M to elucidate its functionality
The leader peptide has been indicated to be nonessential for modification function of LanM because LctM can still produce partially processed LctA in the absence of leader peptide27. However, without the leader peptide, the modification activity of LanM was greatly impaired28,29. Recently, an engineered leader-LctM fusion enzyme LctCE was generated that was constitutively active to modify LctA core peptide to produce authentic lacticin 481 but with limited efficacy29. Herein, BovT150M could efficiently produce fully modified bovicin HJ50 when incubated with His6-BovA, suggesting that the leader peptide directed precursor modification by BovM domain might commence before leader peptide cleavage by BovT150 domain.
To demonstrate this hypothesis, we eliminated either peptidase or modification function via mutation of active sites in BovT150M. Cys15 is the enzymatic center of peptidase BovT150 while Asp264 of BovM are crucial and conserved in LanM proteins (Fig. 1b)30,31,32. As expected, BovT150 C15A was unable to cleave leader peptide of modified BovA (His6-mBovA) while BovM D264N was unable to modify His6-BovA even in 4 h (Fig. 4a,b and c). However, incubation of His6-BovA with BovT150M C15A produced fully modified His6-mBovA (m/z 8243.3 Da) but not active bovicin HJ50, indicating that BovT150M C15A maintained modification function but abolished peptidase activity (Fig. 4d). BovT150M D416N, corresponding to D264N mutation in BovM domain, could not modify His6-BovA while only trace amounts of unmodified core peptide was produced after 4 h (Fig. 4e). BovT150M D416N actually retained efficient peptidase activity towards modified precursor His6-mBovA as bioactive bovicin HJ50 (m/z 3430.6 Da) was produced in 1 h (Fig. 4f). The mutagenesis analyses indicated that BovT150 domain and BovM domain could function independently whereas loss-of-function of BovM domain will significantly impair the proteolytic activity of BovT150 domain towards unmodified precursor His6-BovA. Thus we proposed a successive working mode for BovT150M that leader peptide guided precursor modification via BovM domain precedes leader peptide proteolysis via BovT150 domain (Fig. 5).

Mutagenesis of BovT150M.
MS analysis and antimicrobial assay of reaction products after incubation of His6-mBovA and BovT150 C15A (a,b), His6-BovA and BovM D264N (c), His6-BovA and BovT150M C15A (d), His6-BovA and BovT150M D416N (e), His6-mBovA and BovT150M D416N (f). In panel d, dotted line indicated the MS spectrum of unmodified His6-BovA and red line indicated the MS spectrum of His6-BovA treated by BovT150M C15A.

Proposed model for the catalytic mechanism of engineered BovT150M.
Pink and light blue represent BovT150 domain and BovM domain, respectively. Red and black lines represent leader peptide and core peptide of BovA, respectively. BovA core peptide is firstly modified by BovM domain to introduce thioether rings under the guidance of BovA leader peptide. Then mBovA is digested by BovT150 domain at the double glycine motif to release bioactive bovicin HJ50.
One-pot synthesis of other lanthipeptides via BovT150M
To test the generality of BovT150M on substrates other than bovicin HJ50, two short peptides BovsuiA and BovlctA were designed and expressed as chimeras with BovA leader peptide at the N-terminus (Supplementary Information, Figure S2b). Suicin was a bovicin HJ50-like lanthipeptide restored from a remnant lan locus of S. suis serotype 2 and lacticin 481 was a typical class II lanthipeptide with no disulfide bridge26. The chimeric peptide His6-BovsuiA consisting of BovA leader peptide and suicin core peptide was purified and incubated with BovT150M. Antimicrobial assay indicated that the reaction product was inhibitory against M. flavus NCIB8166 and MS analysis showed the mass peak of 3343.7 Da (Fig. 6a), which was in good accordance with authentic suicin with unfolded disulfide bridge. Chimeric peptide His6-BovlctA (lacticin 481) consisting of BovA leader peptide and lacticin 481 core peptide was expressed, purified and subjected to BovT150M. MS analysis of the reaction product showed a mass peak of 2954.1 Da, which was 18.2 Da decrease compared with calculated mass of lacticin 481 core peptide (2972.3 Da) (Fig. 6b). This indicated that lacticin 481 was one-fold dehydrated by BovT150M. Antimicrobial assay indicated that the modified product was inactive (Fig. 6b). Thus, BovT150M was capable of fully modifying and generating bioactive bovicin HJ50-like lanthipeptides while partially modifying other class II lanthipeptides like lacticin 481. However, dehydration of non-native lanthipeptides by BovT150M implied the substrate tolerance of BovM domain and the capability of BovT150 domain to release the core peptides that were appended to BovA leader peptide.

Production of suicin and lacticin 481 using BovT150M.
MS analysis and antimicrobial assay after incubation of His6-BovsuiA with BovT150M (a) and incubation of His6-BovlctA with BovT150M (b).
Discussion
Lanthipeptides are a fast-growing class of gene-encoded and ribosomally-synthesized peptides with multi-functions, most of which are conferred with potent antimicrobial activities against Gram-positive bacteria. Because of their high efficacy and stability, lanthipeptides are regarded as promising candidates for novel antimicrobial applications in many areas like food preservatives and antibiotics6. With accumulating elucidation of lanthipeptide biosynthetic pathways and modification machineries, genome-based mining enabled revealing of lanthipeptide repertoire in a wider variety of species than anticipated8,33,34. However, isolation of lanthipeptides from natural resources is a tremendous work, let alone certain lanthipeptide clusters are even cryptic or conditionally expressed13,26,35.
To facilitate bioengineering of lanthipeptides, we first achieved lanthipeptide production in a model biofactory like E. coli. Bovicin HJ50, a typical class II lanthipeptide, was produced in E. coli C43 (DE3) via introduction of minimal biosynthetic genes bovA, bovM and bovT. Recently, production of the two-component lanthipeptide lichenicidin (Bliα and Bliβ) has also been achieved in E. coli via co-expression of licA, licM, licT and/or licP36. Thus, it is feasible to manipulate minimal genetic prerequisites in E. coli to obtain bioactive lanthipeptides. However, the growth retardation of bovicin HJ50 producing cells indicated that bovicin HJ50 might interfere with the cell wall synthesis by sequestration of intracellular lipid II.
The successful production of bovicin HJ50 via co-expression of bovA, bovM and bovT150 in E. coli cells was encouraging, as BovA could be processed by BovM and BovT150 simultaneously to release bioactive bovicin HJ50. Prompted by that, we reconstituted an engineered lanthipeptide synthetase BovT150M by fusing the peptidase domain BovT150 to the N-terminus of the modification enzyme BovM. A similar approach fused the leader peptide of lacticin 481 or Halβ to their corresponding modification enzyme LctM or HalM2, generating constitutively active lanthipeptide synthetases LctCE or HalCE2 but with limited efficiency29,37. The leader peptide was important for precursor modifications due to its role in LanM recognition and binding28,37. With intact leader peptide, the modification activity of BovM domain towards precursor was fully maintained. Additionally, fusing protease domain BovT150 to BovM could facilitate the stability of BovT150, which was unstable in in vitro conditions as also observed in LctT150 30. Thus, BovT150M facilitated both precursor modification and leader peptide cleavage. Our results further demonstrated that the separate domains of BovT150M exerted functions successively; first, the leader peptide guided precursor modification via BovM domain and then the leader peptide was cleaved by BovT150 domain. One interesting finding was that a novel LanT and LanM fusion protein (SBI_06987) was recently identified in Streptomyces bingchenggensis BCW-1 genome, suggesting the possible co-functionality of LanT and LanM proteins in native microbes38.
The fusion lanthipeptide synthetase could be applied for efficient and rapid one-pot synthesis of lanthipeptides. We focused on using BovT150M to generate class II lanthipeptides that are similar to bovicin HJ50. Under the guidance of BovA leader peptide, authentic suicin is obtained while lacticin 481 is one-fold dehydrated but not bioactive. Suicin is a bovicin HJ50-like lanthipeptide with two thioether bridges (ring A and B) and a disulfide bridge, while lacticin 481 has three thioether bridges instead. All of them share a conserved ring A structure, which is the proposed lipid II binding motif with two dehydratable Thr/Ser. This also demonstrated that the modification machinery of bovicin HJ50 was different from that of lacticin 481, though bovicin HJ50 is structurally resembled with lacticin 481 with an N-terminal linear and C-terminal globular structure. Furthermore, the potential application of this lanthipeptide synthetase approach may be extended to introducing non-native thioether rings or nonproteinogenic amino acids into short artificial peptides39,40.
In conclusion, we achieved bovicin HJ50 production in E. coli via co-expression of minimal biosynthetic genes. Specifically, an engineered lanthipeptide synthetase BovT150M was reconstituted both in vivo and in vitro to produce mature bovicin HJ50. This one-pot synthesis system provides new options for production and in vitro bioengineering of novel lanthipeptides. Moreover, BovT150M implies potential application in introducing dehydro amino acids or thioether bridges into non-native substrate peptide drugs, which might enhance thermostability or maintain structural conformation.
Materials and Methods
Materials
Escherichia coli DH5α was used for plasmid construction and E. coli BL21 (DE3) and C43 (DE3) for protein expression. Plasmid pET28a and pACYC-Duet-1 were used as expression vectors. Kanamycin of 50 μg/ml and chloramphenicol of 10 μg/ml were used when needed. E. coli strains were incubated in Luria-Bertani (LB) medium at 37 °C and Micrococcus flavus NCIB8166 was inoculated in S1 medium at 30 °C13.
Cloning, Mutagenesis and Protein Expression
Molecular biology methods were performed according to standard protocols41. Plasmid pET28a-bovA, pET28a-bovAM and pET28a-bovT150 were constructed previously20,24. bovT150 and bovT were amplified from genomic DNA of S. bovis HJ50 with primers containing NdeI and KpnI and were then respectively constructed into pACYC-Duet-1 to obtain pACYC-Duet-bovT150 and pACYC-Duet-bovT. bovT150 with non-stop codon was constructed into pACYC-Duet-1 between NdeI and KpnI and bovM was ligated between KpnI and XhoI subsequently. This plasmid was named pACYC-Duet-bovT150M. pET28a-bovT150M was obtained by constructing bovT150M into pET28a. Chimeric genes bovsuiA and bovlctA were synthesized by Sango Biotech (Shanghai, China) and constructed into pET28a, respectively. Site-directed ligase-independent mutagenesis (SLIM) was performed to introduce mutations by a PCR method as described by Chiu42. Protein expression and purification were conducted as described previously26. Purified proteins were identified by 16% acrylamide SDS-PAGE and protein concentrations were determined by BCA assay kit (Thermo Scientific, USA) according to instructions.
Enzyme Activity Assay
The reaction buffer (50 mM Tris-HCl, 150 mM NaCl, 10 mM MgCl2, pH 7.4) was used for activity assay of BovT150 and 1 mM DTT and 2.5 mM ATP were needed for BovM and BovT150M. His6-BovA, BovM and BovT150M were respectively used with a final concentration of 20 μM, 2 μM and 2 μM. The reactions were proceeded at 25 °C for 1 h to 4 h and quenched by 0.5% trifluoroacetic Acid (TFA). The reaction products were analyzed by mass spectrometry (MS) analysis.
Bioactivity Detection
Antimicrobial activity was determined by well-diffusion method against indicator strain M. flavus NCIB8166. 25 μl samples of culture supernatants, cell lysates or in vitro biosynthesized products were applied to wells with diameter of 5 mm on agar plates containing M. flavus NCIB8166 and the agar plates were incubated in 30 °C for 24 h. Concentration of bovicin HJ50 from cell lysates were determined by measuring the diameter of inhibition zone with agar diffusion bioassay as described by Pongtharangkul43. E. coli BL21 (DE3) containing pET28a-bovA and pACYC-Duet-bovT150M were induced by 0.5 mM IPTG. Cell growth curve were recorded by measuring OD600 and induced E. coli cells of 5 ml were pelleted and resuspended in 1 ml PBS solution, and further lysed via sonication. The lysates were centrifuged and 25 μl aliquot was loaded to the agar plate containing sensitive indicator strain M. flavus NCIB8166. Purified bovicin HJ50 via SIVB was diluted into a gradient concentration of 20, 10, 5, 2.5, 1.25 and 0.625 μg/ml, which were used as for construction of the standard curve.
MS Analysis
MALDI-TOF (matrix-assisted laser desorption/ionization-time of flight) MS analysis was performed on 4700 Proteomics Analyzer mass spectrometer (Applied Biosystems, USA). Samples were prepared by acidification with adding 0.1% TFA and subsequent processing via C18 ZipTip column. CHCA (α-cyano-4-hydroxycinnamic acid) matrix was prepared by dissolving 5 mg in 1 ml of 50:50 acetonitrile/water containing 0.1% TFA. Mass spectra for 1–5 kDa were obtained in positive reflectron mode and 5–10 kDa in linear mode.
Additional Information
How to cite this article: Wang, J. et al. One-pot synthesis of class II lanthipeptide bovicin HJ50 via an engineered lanthipeptide synthetase. Sci. Rep. 6, 38630; doi: 10.1038/srep38630 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Paulsen, I. et al. Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 299, 2071–2074 (2003).
Rice, L. B. Tn916 family conjugative transposons and dissemination of antimicrobial resistance determinants. Antimicrob. Agents Chemother. 42, 1871–1877 (1998).
Arias, C. A. & Murray, B. E. Antibiotic-resistant bugs in the 21st century-a clinical super-challenge. N. Engl. J. Med. 360, 439–443 (2009).
Arnison, P. G. et al. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 30, 108–160 (2013).
van Heel, A. J., Montalban-Lopez, M. & Kuipers, O. P. Evaluating the feasibility of lantibiotics as an alternative therapy against bacterial infections in humans. Exp. Opin. Drug Metab. Toxicol. 7, 675–680 (2011).
Dischinger, J., Chipalu, S. B. & Bierbaum, G. Lantibiotics: promising candidates for future applications in health care. Int. J. Med. Microbiol. 304, 51–62 (2014).
Hancock, R. E. & Sahl, H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24, 1551–1557 (2006).
Zhang, Q., Yu, Y., Vélasquez, J. E. & van der Donk, W. A. Evolution of lanthipeptide synthetases. Proc. Natl. Acad. Sci. USA 109, 18361–18366 (2012).
van der Donk, W. A. & Nair, S. K. Structure and mechanism of lanthipeptide biosynthetic enzymes. Curr. Opin. Struct. Biol. 29, 58–66 (2014).
Yang, X. & van der Donk, W. A. Ribosomally synthesized and post-translationally modified peptide natural products: new insights into the role of leader and core peptides during biosynthesis. Chemistry 19, 7662–7677 (2013).
Nishie, M., Shioya, K., Nagao, J.-i., Jikuya, H. & Sonomoto, K. ATP-dependent leader peptide cleavage by NukT, a bifunctional ABC transporter, during lantibiotic biosynthesis. J. Biosci. Bioeng. 108, 460–464 (2009).
Nishie, M. et al. Lantibiotic transporter requires cooperative functioning of the peptidase domain and the ATP binding domain. J. Biol. Chem. 286, 11163–11169 (2011).
Wang, J. et al. Cerecidins, novel lantibiotics from Bacillus cereus with potent antimicrobial activity. Appl. Environ. Microbiol. 80, 2633–2643 (2014).
Caetano, T., Krawczyk, J. M., Mosker, E., Sussmuth, R. D. & Mendo, S. Heterologous expression, biosynthesis, and mutagenesis of type II lantibiotics from Bacillus licheniformis in Escherichia coli. Chem. Biol. 18, 90–100 (2011).
Jabes, D. & Donadio, S. Strategies for the isolation and characterization of antibacterial lantibiotics. Methods Mol. Biol. 618, 31–45 (2010).
Gomez-Escribano, J. P. & Bibb, M. J. Heterologous expression of natural product biosynthetic gene clusters in Streptomyces coelicolor: from genome mining to manipulation of biosynthetic pathways. J. Ind. Microbiol. Biotechnol. 41, 425–431 (2014).
Xie, L. et al. Lacticin 481: in vitro reconstitution of lantibiotic synthetase activity. Science 303, 679–681 (2004).
McClerren, A. L. et al. Discovery and in vitro biosynthesis of haloduracin, a two-component lantibiotic. Proc. Natl. Acad. Sci. USA 103, 17243–17248 (2006).
Garg, N., Tang, W., Goto, Y., Nair, S. K. & van der Donk, W. A. Lantibiotics from Geobacillus thermodenitrificans. Proc. Natl. Acad. Sci. USA 109, 5241–5246 (2012).
Lin, Y., Teng, K., Huan, L. & Zhong, J. Dissection of the bridging pattern of bovicin HJ50, a lantibiotic containing a characteristic disulfide bridge. Microbiol. Res. 166, 146–154 (2011).
Jie, Z. et al. Type AII lantibiotic bovicin HJ50 with a rare disulfide bond: structure, structure-activity relationships and mode of action. Biochem. J. 461(3), 497–508 (2014).
Xiao, H. et al. Bovicin HJ50, a novel lantibiotic produced by Streptococcus bovis HJ50. Microbiology 150, 103–108 (2004).
Liu, G. et al. Characteristics of the bovicin HJ50 gene cluster in Streptococcus bovis HJ50. Microbiology 155, 584–593 (2009).
Wang, J. et al. Bovicin HJ50-like lantibiotics, a novel subgroup of lantibiotics featured by an indispensable disulfide bridge. PLoS One 9, e97121 (2014).
Miroux, B. & Walker, J. E. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260, 289–298 (1996).
Wang, J. et al. Restoration of bioactive lantibiotic suicin from a remnant lan locus of pathogenic Streptococcus suis serotype 2. Appl. Environ. Microbiol. 80, 1062–1071 (2014).
Levengood, M. R., Patton, G. C. & van der Donk, W. A. The leader peptide is not required for post-translational modification by lacticin 481 synthetase. J. Am. Chem. Soc. 129, 10314–10315 (2007).
Oman, T. J. & van der Donk, W. A. Follow the leader: the use of leader peptides to guide natural product biosynthesis. Nat. Chem. Biol. 6, 9–18 (2010).
Oman, T. J., Knerr, P. J., Bindman, N. A., Velásquez, J. E. & van der Donk, W. A. An engineered lantibiotic synthetase that does not require a leader peptide on its substrate. J. Am. Chem. Soc. 134, 6952–6955 (2012).
Furgerson Ihnken, L., Chatterjee, C. & van der Donk, W. A. In vitro reconstitution and substrate specificity of a lantibiotic protease. Biochemistry 47, 7352–7363 (2008).
You, Y. O. & van der Donk, W. A. Mechanistic investigations of the dehydration reaction of lacticin 481 synthetase using site-directed mutagenesis. Biochemistry 46, 5991–6000 (2007).
Ma, H. et al. Dissecting the catalytic and substrate binding activity of a class II lanthipeptide synthetase BovM. Biochem. Biophys. Res. Commun. 450, 1126–1132 (2014).
Blin, K., Kazempour, D., Wohlleben, W. & Weber, T. Improved lanthipeptide detection and prediction for antiSMASH. PLoS One 9, e89420 (2014).
van Heel, A. J., de Jong, A., Montalbán-López, M., Kok, J. & Kuipers, O. P. BAGEL3: automated identification of genes encoding bacteriocins and (non-) bactericidal posttranslationally modified peptides. Nucleic Acids Res. 41, 448–453 (2013).
Haas, W., Shepard, B. D. & Gilmore, M. S. Two-component regulator of Enterococcus faecalis cytolysin responds to quorum-sensing autoinduction. Nature 415, 84–87 (2002).
Kuthning, A., Mösker, E. & Süssmuth, R. D. Engineering the heterologous expression of lanthipeptides in Escherichia coli by multigene assembly. Appl. Microbiol. Biotechnol. 99, 6351–6361 (2015).
Thibodeaux, G. N., McClerren, A. L., Ma, Y., Gancayco, M. R. & van der Donk, W. A. Synergistic binding of the leader and core peptides by the lantibiotic synthetase HalM2. ACS Chem. Biol. 10, 970–977 (2015).
Singh, M. & Sareen, D. Novel LanT associated lantibiotic clusters tdentified by genome database mining. PLoS One 9, e91352 (2014).
Chatterjee, C., Patton, G. C., Cooper, L., Paul, M. & van der Donk, W. A. Engineering dehydro amino acids and thioethers into peptides using lacticin 481 synthetase. Chem. Biol. 13, 1109–1117 (2006).
Levengood, M. R., Knerr, P. J., Oman, T. J. & van der Donk, W. A. In vitro mutasynthesis of lantibiotic analogues containing nonproteinogenic amino acids. J. Am. Chem. Soc. 131, 12024–12025 (2009).
Sambrook, J., Fritsch, E. F. & Maniatis, T. Molecular cloning. Vol. 2 (Cold spring harbor laboratory press, New York, 1989).
Chiu, J., March, P. E., Lee, R. & Tillett, D. Site-directed, ligase-independent mutagenesis (SLIM): a single-tube methodology approaching 100% efficiency in 4 h. Nucleic Acids Res. 32, e174 (2004).
Pongtharangkul, T. & Demirci, A. Evaluation of agar diffusion bioassay for nisin quantification. Appl. Microbiol. Biotechnol. 65, 268–272 (2004).
Acknowledgements
The authors thank Shutao Sun at Core Facility of Institute of Microbiology Chinese Academy of Sciences for providing MALDI-TOF MS analysis. This research was supported by the National Natural Science Foundation of China (31570114), Special Fund for Agro-scientific Research in the Public Interest (201503134) and Key Deployment Project of the Chinese Academy of Sciences (KSZD-EW-Z-012-2, KFZD-SW-101-4).
Author information
Authors and Affiliations
Contributions
J.W., K.T. and J.Z. conceived and designed the experiments. J.W., X.G. and L.Z. performed the experiments and analyzed the data. J.W. and J.Z. contributed to the writing of the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wang, J., Ge, X., Zhang, L. et al. One-pot synthesis of class II lanthipeptide bovicin HJ50 via an engineered lanthipeptide synthetase. Sci Rep 6, 38630 (2016). https://doi.org/10.1038/srep38630
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep38630
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
