
Abstract
While fecal microbiota transplantation (FMT) presents an attractive therapeutic strategy, it remains unclear how to choose the microbiota repertoire that most effectively transfers benefit to recipients. We identified a beneficial taxonomic repertoire in a transgenic mouse model (RIP140mϕKD) which resists the development of high fat diet (HFD)-induced metabolic diseases due to enhanced anti-inflammation engineered by lowering receptor interacting protein (RIP140) expression in macrophage. We confirmed using FMT from HFD-fed RIP140mϕKD to wild type (WT) mice that recipient mice acquired the microbiota repertoire of donor mice. Importantly, FMT from RIP140mϕKD to WT not only effectively transferred the beneficial taxonomic repertoire to WT recipients, but also enabled recipient animals acquiring the anti-inflammatory status of RIP140mϕKD donor animals and avoid HFD-induced insulin resistance, which is associated with significantly improved intestinal integrity. We conclude that FMT can transfer not only microbiota but also the donors’ intestinal innate immune status and improved intestinal integrity.
Similar content being viewed by others


Introduction
Metabolic syndrome, including obesity, high blood pressure, hyperglycemia, high levels of serum triglyceride level, and low level of high-density lipoprotein (HDL), presents a serious health condition associated with metabolic diseases such as type 2 diabetes and cardiovascular diseases1. Recent studies have demonstrated that intestinal homeostasis, harmonious interactions between the microbiota and the host immune system, is critical to the maintenance of normal physiology, metabolism and immune regulations2,3, and that the gut microbiota are involved in the development of numerous diseases4. The diversity and composition of intestinal microbiota could be altered by the host’s diet and/or genome, and is associated with the host’s systemic and local inflammatory responses that are critically important in the development of metabolic diseases4,5,6,7. Manipulation of the gut microbiota by fecal microbiota transplantation (FMT) has been applied in various disease models and clinical trials, such as inflammatory bowel diseases (IBD), Clostridium difficile infection (CDI) and neurodevelopmental disorders8,9,10,11; however, the specific taxonomic repertoire for protection, particularly under a disease-prone condition such as high fat diet (HFD) feeding, has never been identified. Further, the underlying mechanism remains elusive.
Receptor interacting protein 140 (RIP140, also known as NRIP1) is a wide-spectrum nuclear coregulator involved in various biological processes leading to diseases12,13. Specifically related to metabolic regulation where inflammation plays a key role, RIP140 is a critical regulator of innate immunity, mediated by its functions in macrophage polarization. RIP140 is a M1 activator by co-activating NF-κB14, and a M2 repressor through suppressing STAT615. We previously generated mice with macrophage-specific knockdown of RIP140 (RIP140mϕKD)14 where the M2 macrophage population is dramatically expanded and the M1 macrophage population is reduced. These mice are protected against endotoxin shock14 and HFD-induced metabolic diseases16,17, and exhibit improved wound healing15. Further, their local innate immunity, such as anti-inflammatory potential in adipose tissues, is dramatically improved17,18. We therefore speculated possible beneficial shifts in their gut microbiota and intestinal innate immune status, and tested the potential to transfer, via FMT, their protective phenotype to recipient wild type (WT) animals also under HFD feeding.
We first determined the repertoire of intestinal microbiota affected by genotype, i.e., RIP140mϕKD mice vs. WT mice, and then carried out FMT, reciprocally, to validate efficacy of the transfer of gut microbiota. We found that FMT, from RIP140mϕKD to WT, transferred not only the specific taxonomic repertoire of RIP140mϕKD to WT recipient but also their intestinal innate immune feature (elevated M2 anti-inflammatory population) and tissue integrity. Both of these transferred features are associated with protection against HFD-induced metabolic abnormality in recipient animals. This study defines a specific taxonomic repertoire that confers protection against diet-induced metabolic diseases, and validates the efficacy of FMT in treating metabolic syndrome. The study also suggests a protective mechanism via transferring microbiome-associated intestinal innate immunity and tissue integrity.
Results
Genotype alters the composition and functional repertoire of intestinal microbiota
We first confirmed that HFD affected the composition of the gut microbiota (Fig. S1A). Interestingly, when comparing the effect of genotype, we found that the effect was most profound under HFD feeding, based on 16 s rRNA sequences of unweighted UniFrac distance metric (Fig. 1A, S1B and S1C) (PERMANOVA, p = 0.04). Moreover, supervised learning using Random Forests, a machine learning method using OTUs as predictive features, accurately assigned samples to their source population based on taxonomic profiles at the OTU level (83.3% accuracy, 3 times better than the baseline error rate for random guessing).

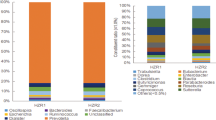
Macrophage RIP140 level alters the composition and functional repertoire of intestinal microbiota.
(A) Beta diversity comparisons of the gut microbiomes of the fecal samples collected from WT and RIP140mϕKD mice receiving High Fat Diet. Analyses were performed on 16 S rRNA V4 regions data with a rarefaction depth of 66677 reads per sample. Principal coordinates analysis of Unweighted UniFrac distances. Proportion of variance explained by each principal coordinate axis is denoted in the corresponding axis label. The plot shows a separation between samples from WT and RIP140mϕKD mice receiving High Fat Diet (PERMANOVA, p = 0.04). (B) Summary of the taxa that differentiate WT from RIP140mϕKD mice receiving HFD using Linear discriminant analysis Effect Size analysis (LEfSe). (C) Left: KD microbiome index corresponding to the sum of number of genera among the differentiating taxa. Data were presented with Mann–Whitney U test: p-value = 0.007. Right: KD microbiome index corresponding to the total relative abundance of the differentiating taxa. Data were presented with Mann–Whitney U test: p-value = 0.002. (D) ROC curve analysis for KD microbiome index. (E) Beta-diversity plots from Bray-Curtis distance matrices for genome analysis. (F) CAZY GH assignments for glycoside hydrolase families analysis.
Using LEfSe19, we found that 6 genera were significantly different between WT and RIP140mϕKD mice fed a HFD (LDA log10 score >2). Specifically, RIP140mϕKD profile was associated with a significant gain in Odoribacter, Coprococcus, Lautropia, Luteimonas, Candidatus Arthromitus and Pseudomonadaceae when compared to WT mice. (Fig. 1B, S1D). We then constructed a KD microbiome index from this panel of taxa that highly differentiated between WT and RIP140mϕKD mice under HFD. This KD index corresponded to the sum of relative abundances of the 6 differentiating taxa. We found that the median KD index was 0.30 (IQR = 0.05) in RIP140mϕKD mice and 0.17 (IQR = 0.01) in WT mice (Mann-Whitney U test, p = 0.002) (Fig. 1C, right panel). Moreover, ROC curve analysis showed that our KD index was a strong predictor of the genotype, with an area under the curve of 0.91 (Fig. 1D). In order to determine the KD index threshold that best predicts genotype, we performed leave-one-out cross-validation on our KD indices. Each held-out KD index was treated as a new sample, independently from the initial cohort, on whom we tested and subsequently refined the optimal index cutoff to separate WT and RIP140mϕKD. This Leave-one-out (LOO) cross-validation procedure demonstrated that the taxon panel was able to predict the genotype in a new sample, with an accuracy of 83% at a specificity of 80%. Thus, our LOO analysis predicted genotype with reasonable accuracy and identified taxa associated with resistance to diet induced metabolic diseases that can serve as future biomarkers. These data demonstrated that genotype, that is WT vs. RIP140mϕKD profile, is strongly associated with a specific taxonomic repertoire in mice fed a HFD and that only 6 taxa can be used to predict the genotype of a mouse
Using PICRUSt20, we found that the gut microbiota functional repertoire is significantly different between WT and RIP140mϕKD mice. This algorithm estimates the functional potential of microbial communities given the current 16 S rRNA gene survey and a set of currently sequenced reference genomes. PICRUSt predictions in human and mouse gut microbiomes are expected to have 80–90% accuracy. First, beta-diversity plots generated from Bray–Curtis distance matrices showed a separation between WT and RIP140mϕKD mice fed a HFD (PERMANOVA, p = 0.041) (Fig. 1E). Moreover, supervised learning using Random Forests, with the predicted metagenome table collapsed at level 3 KEGG Orthology groups as predictive features, accurately assigned samples to their source population based on predicted metagenomic profiles (75% accuracy, 2 times better than the baseline error rate for random guessing). We also identified significant differences in microbial functional pathways in the fecal samples of WT and RIP140mϕKD mice fed a HFD (level 3 KEGG Orthology groups, Mann-Whitney U test, False Discovery Risk corrected p-value <0.20). The fecal microbiome of RIP140mϕKD mice is enriched in functional categories associated with fatty acid metabolism, lysine, valine, leucine and isoleucine degradation, caprolactam degradation, styrene and atrazine degradation, and depleted in categories associated with galactose metabolism, purine metabolism, cysteine and methionine metabolism, D-Glutamine and D-glutamate metabolism, nicotinate and nicotinamide metabolism, amino sugar and nucleotide sugar, thiamine metabolism and primary bile acid biosynthesis (Table S1).
Using CAZY GH assignments, we found that 5 glycoside hydrolase families were increased in RIP140mϕKD mice as compared to WT mice, including GH17, PL5, GT4, GT9 and AA1, whereas 10 glycoside hydrolase families showed a loss of abundance in KD mice as compared to WT mice, including GT14, GH36, GH3, GH18, GH115, GH78, GH65, GH130, GT32 and GH127 (LDA score (log10) >2) (Fig. 1F)
Together, these findings demonstrate that, in addition to a change in taxonomy, the genotype is associated with marked change in functional profile and glycoside hydrolase repertoire in animals fed a HFD.
Fecal microbiota transplantation transfers gut microbiota from donor to recipient mice
Our previous studies showed that RIP140mϕKD mice are resistant to diet-induced metabolic diseases16,21. Since gut microbiota are strongly associated with hosts’ health, we proposed that gut microbiota in RIP140mϕKD mice could be associated with their metabolic protection features particularly under HFD. We thus performed reciprocal FMTs, from WT or RIP140mϕKD fed a HFD into WT or RIP140mϕKD mice in four groups: WT → WT (WT receiving WT), KD → WT (WT receiving KD), WT → KD (KD receiving WT) and KD → KD (KD receiving KD). The experimental design is depicted in Fig. 2A.

FMT transfers gut microbiota from donor to recipient.
(A) A scheme showing FMT experiment. (B) Left: Unweighted UniFrac based PCoA from RIP140mϕKD mice receiving FMT from WT (WT → KD) mice. Right: Unweighted UniFrac based PCoA from WT mice receiving FMT from RIP140mϕKD (KD → WT) mice. (C) Left panel: KD microbiome index in RIP140mϕKD mice receiving FMT from WT (WT → KD) mice. Right panel: KD microbiome index in WT mice receiving FMT from RIP140mϕKD (KD → WT) mice. (D) Representative pie chart of Bayesian source-tracking analysis of taxonomy, predicted metagenome and predicted GH of WT → KD mice post FMT 4 weeks (left panel) and KD → WT mice post FMT 4 weeks (right panel). Source contributions were averaged across samples within the population. WT → WT: WT receiving WT, KD → WT: WT receiving KD, WT → KD: KD receiving WT and KD → KD: KD receiving KD.
To first validate FMT efficiency, we examined changes in the diversity of recipient KD mice receiving FMT from donor WT mice (WT → KD). Unweighted UniFrac based PCoA showed differences between fecal samples collected from recipient RIP140mϕKD mice before FMT (i.e. native RIP140mϕKD mice) and four weeks after FMT (WT → KD) (PERMANOVA, p = 0.04), but did not show significant differences between fecal samples of WT → KD mice four weeks after FMT and native WT mice (PERMANOVA, p = 0.19). Moreover, fecal samples of recipient RIP140mϕKD mice after FMT (WT → KD) differed in terms of overall diversity when comparing to fecal samples of native RIP140mϕKD mice (PERMANOVA, p = p 0.048), but did not differ when comparing to fecal samples of native WT mice (PERMANOVA, p = 0.192) (Fig. 2B, left panel). We also examined changes in diversity of recipient WT mice receiving FMT from donor RIP140mϕKD mice (KD → WT). Unweighted UniFrac PCoA showed differences between fecal samples of recipient WT mice before FMT (i.e. native WT mice) and four weeks after FMT (KD → WT) (PERMANOVA, p = 0.02), but did not show significant differences between fecal samples of KD → WT mice and native RIP140mϕKD mice (PERMANOVA, p = 0.06). Moreover, fecal samples of recipient WT mice after FMT (KD → WT) differed in terms of diversity when comparing to fecal samples of native WT mice (PERMANOVA, p = p 0.043), but did not differ when comparing to fecal samples of native RIP140mϕKD mice (PERMANOVA, p = 0.071) (Fig. 2B, right panel).
Using the KD microbiome index described above, we found that WT → KD mice acquired a WT fecal microbiota signature; KD → WT mice acquired a KD fecal microbiota signature. The KD index differed between native RIP140ϕKD mice (i.e. fecal sample collected before FMT) and WT → KD mice post FMT (MWU test, p = 0.012) but the KD index did not differ between native WT mice and WT → KD mice post FMT (MWU test, p = 0.28) (Fig. 2C, left panel). Following the same trend, the KD index differed between native WT mice (i.e. fecal sample collected before FMT) and KD → WT mice post FMT (MWU test, p = 0.0018) but the KD index did not differ between native KD mice and KD → WT mice post FMT (MWU test, p = 0.28) (Fig. 2C, right panel).
To further explore this trend, we performed Bayesian source tracking on each of the post FMT fecal samples. This allows us to estimate the contribution of bacteria from the native WT mice, native RIP140mϕKD mice or from ‘unknown’ sources (one or more sources absent from the training data) in the post FMT samples. We found that the fecal samples of WT → KD mice were dominated by bacteria, predicted genes or CAZY GH of native WT mice (Fig. 2D, left panel); on the other hand, fecal samples of KD → WT mice were dominated by bacteria, predicted genes or CAZY GH of native RIP140mϕKD mice (Fig. 2D, right panel).
Based on these findings, we conclude that WT → KD mice post FMT acquired the microbiota signature of the WT genotype and KD → WT mice acquired that of the KD genotype.
Healthy gut microbiota ameliorate diet-induced metabolic syndrome
To determine if gut microbiota from RIP140mϕKD mice could benefit recipient mice under HFD, we performed a series of metabolic tests on post-FMT mice. RIP140mϕKD typically have elevated anti-inflammatory activities in their adipose tissues17,22. We therefore first examined if adipose innate immunity was affected by FMT. It appeared that mice receiving FMT from RIP140mϕKD mice (KD → WT) indeed have reduced M1 inflammatory marker (Tnfα) and elevated level of M2 anti-inflammatory marker (Arg1) in white adipose tissue (Fig. 3A). Furthermore, these mice express stronger beige (Cd137) and brown (Ucp1) fat markers (Fig. 3B). Mice receiving FMT from KD mice (KD → WT or KD → KD) were resistant to HFD-induced weight gain and insulin resistance compared to mice receiving FMT from WT mice (WT → WT or WT → KD) (Fig. 3C, Fig. S2). KD → WT mice show a higher rate of energy expenditure (O2 consumption) in both the light and dark phases (Fig. 3D). Taken together, our data show that FMT could transfer not only the “good” gut microbiota, but also the HFD-resistant, protective phenotype of the donor, RIP140mϕKD.

Receiving FMT from RIP140mϕKD mice ameliorates diet-induced diabetic traits.
(A) qPCR of Tnfa (M1) and Arg1 (M2) in visceral white adipose tissues. Student test (n = 3) was used and presented as mean ± SD, *P < 0.05. (B) qPCR of Ucp1 (brown fat) and Cd137 (beige fat) in visceral white adipose tissues. Student test (n = 3) was used and presented as mean ± SD, *P < 0.05; **P < 0.01. (C) GTT (left) and ITT (right) determined after 14 weeks of HFD feeding Student test (n = 3) was used and presented as mean ± SD, *P < 0.05, **P < 0.01 (WT → WT vs. KD → WT); †P < 0.05 (WT → KD vs. KD → KD). (D) Energy expenditure of FMT mice. O2 consumption was measured in the dark and light phases and presented as vO2 (ml/kg/hr) (n = 3 in each group). Student test was used and presented as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001 (WT → WT vs. KD → WT), †P < 0.05; ††P < 0.01; †††P < 0.001 (WT → KD vs. KD → KD).
To gain insights into the possible mechanism, we examined the innate immune potential and tissue integrity of the gastrointestinal (GI) tract, because gut microbiota make direct contact with GI tract, affecting the microenvironment. We first monitored intestinal permeability (Fig. 4A) and found that both KD → WT and KD → KD mice have decreased intestinal permeability, indicating that they are less susceptible to low-grade inflammation that could contribute to the development of metabolic syndrome23. These mice also exhibit fewer pathological features in the colon, with apparently decreased hyperplasia (Fig. 4B). In terms of local (intestinal) innate immunity as monitored by M1 vs. M2 ratio (Fig. 4C), these mice have elevated M2 and lowered M1 markers and a reduced M1/M2 ratio indicative of improved anti-inflammation in the GI tract.

Receiving FMT from RIP140mϕKD mice improves colon health.
(A) Serum levels of FITC-dextran in FMT mice. Student test was used and presented as mean ± SD. *P < 0.05; **P < 0.01 for comparison of WT → WT mice with KD → WT mice. †P < 0.05 for comparison of WT → WT mice with KD → WT mice. (n = 3 in each group). (B) Representative H&E staining of colon sections. (C) FACS analysis of gut macrophage population in colon. Student test was used and presented as mean ± SD. *P < 0.05; **P < 0.01. (n = 3 in each group).
Discussion
Our study is the first to identify microbiota-associated biomarkers that correlate with protection against diet-induced metabolic syndrome. The study also uncovers a specific taxonomic repertoire in mice with an elevated anti-inflammatory potential under HFD feeding, and validates the efficacy of FMT to transfer protection against HFD-induced metabolic diseases. The study also elucidates the mechanism involving augmentation in GI innate immune status and its tissue integrity.
Several taxa associated with RIP140mϕKD mice were previously found protective against insulin resistance or dyslipidemia. Thus our findings of specific intestinal microbiota enriched in RIP140mϕKD mice are consistent with a proposed role in glucose homeostasis for these commensal bacteria. In the present study, we found the specific taxonomic repertoire consists of 6 genera of bacteria enriched in RIP140mϕKD mice, which are correlated with resistance to diet-induced metabolic syndrome: Odoribacter, Coprococcus, Lautropia, Luteimonas, Candidatus Arthromitus and Pseudomonadaceae. Odoribacter splanchnicus was previously reported to be associated with a healthy fasting serum lipid profile, defined as a positive correlation with HDL cholesterol and a negative correlation with total- or LDL cholesterol. Importantly, Odoribacter splanchnicus was also positively correlated with HbA1c24. Another study reported that eradicating Candidatus arthromitus in the mouse gut microbiota resulted in metabolic changes that led to obesity25. A study also showed that non-caloric artificial sweeteners drove the development of glucose intolerance through induction of compositional and functional alterations to the intestinal microbiota including a decrease in Candidatus arthromitus26. Moreover, a study in humans reported a substantial reduction in abundance of Coprococcus in obese and nonalcoholic steatohepatitis (NASH) subjects as compared to healthy subjects27. Furthermore, Pseudomonadaceae was found increased in zebrafish larvae following probiotic supplementation that resulted in a down-regulation of the expression of genes that decrease glucose levels in the blood28. Thus the presence of some taxa could be helpful in assessing risk of developing diet-induced metabolic syndrome.
In addition, we found suggestive evidence that microbiota-associated functional modules are significantly different between WT and RIP140mϕKD mice using PICRUSt. Importantly, several of the KEGG modules differentiating WT and RIP140mϕKD mice are implicated in lipid and glucose metabolism. Nicotinamide, found depleted in RIP140mϕKD, was previously reported to be involved in the development of type 2 diabetes, through an increase in reactive oxygen species, subsequent oxidative stress and insulin resistance29. Purine, found depleted in RIP140mϕKD mice, could modulate insulin sensitivity and was associated with the development of diabetic microvascular complication30. D-Glutamine and D-glutamate metabolism, found depleted in RIP140mϕKD mice, was found strongly associated with insulin resistance in a previous study in humans31. Other investigations have highlighted associations of branched-chain amino acids (i.e., isoleucine, leucine, and valine) with obesity, impaired glucose tolerance, and insulin resistance, which is consistent with our findings that isoleucine, leucine, and valine degradation pathways are enriched in RIP140mϕKD mice32,33,34. Valine, of which degradation module was found increased in RIP140mϕKD mice, was associated with insulin resistance35. Cysteine and methionine metabolism, found decreased in RIP140mϕKD mice, were also previously associated with obesity and insulin resistance. In mice, cysteine intake decreases glucose tolerance, with up-regulation of lipogenic and diabetogenic enzymes that induce fat gain36. In humans, increased plasma total cysteine was associated with obesity and insulin resistance37,38. A study in rats showed that long-term exposure to atrazine caused obesity in those fed a HFD, leading to diabetes. This is also consistent with our findings that styrene and atrazine degradation are enriched in RIP140mϕKD mice39. Furthermore, amino sugar and nucleotide sugar metabolism, found decreased in RIP140mϕKD mice, was reported to have a direct linkage with diabetes and metabolic syndrome40.
We also found that several glycoside hydrolase families are correlated with RIP140mϕKD mice and protection against diet-induced metabolic syndrome. GH18, found decreased in RIP140mϕKD mice was reported to be a biomarker for endothelial dysfunction, atherosclerosis, insulin resistance, and Type 2 Diabetes Mellitus41,42. Thus the presence of some microbiota-associated glycoside hydrolase families could also assess risk in developing diet-induced metabolic syndrome.
We also demonstrated here that FMT can be performed reciprocally and effectively. WT mice receiving FMT from RIP140mϕKD mice acquired the RIP140mϕKD phenotype, and RIP140mϕKD mice receiving FMT from WT mice acquired the WT phenotype. Using Bayesian source tracking43, we showed that the taxonomy and functional modules associated with RIP140mϕKD were transferred to WT mice, suggesting that commensal bacteria and microbiota-associated modules correlated with resistance to diet-induced metabolic syndrome are substantially transferred to the diseased animals and augment disease outcome. Mechanistically, we found that FMT effectively transferred the protective innate immune phenotype (enhanced M2, anti-inflammatory potential) of the donor animals to the recipients under HFD, which also is associated with the improved GI tissue integrity in the recipients (Fig. 4C). These results provide a proof-of-concept that innate immunity affects gut microbiota, and vise versa, and that the protection can be effectively transferred by FMT. How microbiota affects host innate immunity, either systemically or locally, remains to be examined.
A small, randomized, double-blind controlled study showed that FMT, using stool from lean donors, significantly improves insulin sensitivity in obese male individuals, with butyrate-producing intestinal bacteria increasing in intestinal samples8. Our results validate the therapeutic efficacy of FMT from animals resistant to diet-induced metabolic syndrome, particularly those with improved innate immunity.
Methods
Animals and FMT procedure
Male C57Bl/6 mice were purchased from the Jackson Laboratory and maintained in the animal facility of University of Minnesota on a 12-h light/dark photocycle. All animal studies were approved and conducted according to guidelines of the University of Minnesota Institutional Animal Care and Use Committee. RIP140 mϕKD transgenic mice were generated as described before14. For experiments in Fig. S1A, 18 WT and 18 RIP140 mϕKD mice were used. For FMT experiments, we adopted a commonly used FMT experimental design where three individually housed animals had been used per group44,45,46,47,48,49. The procedure is descried as following. Briefly, eight-week-old male mice (6 WT and 6 KD) were housed individually and fed a high fat diet containing 60% calories from fat and 345 mg cholesterol/kg (F3282; Bio-Serv, West Chester, PA). After 7-weeks HFD feeding, feces was collected and stored for microbiome analyses and FMT later (pre-FMT samples). Fresh ampicillin solution (0.5 g/L) was added in drinking water every 3 days for a week50. FMT was then performed using pre-FMT samples, daily for three days, starting from 8-weeks of HFD feeding (one week post antibiotics treatment). Fecal samples were then collected, one week after FMT, for four more weeks. Metabolic measurement (see later) were performed. Animals were then sacrificed for collecting blood, colons and visceral white adipose tissues. Detailed sequence analyses and statistical analyses were provided in supplemental information.
16 S rRNA gene amplification and sequencing
Fecal samples were kept frozen at −80 °C until they were processed. After fecal DNA isolation (MoBio, Carlsbad, CA fecal DNA kit), amplicons spanning the variable region 4 of bacterial 16 S rRNA were generated and sequenced using Illumina Mi-seq platform at the University of Minnesota Genomic Center, Twin Cities, MN. The 16 S rRNA sequencing data from the Illumina runs were trimmed and chimera filtered using Quantitative Insights Into Microbial Ecology (QIIME) 1.9.151 with default parameters which include several quality filtering steps for sequence lengths, end-trimming, and minimum quality score. We performed operational taxonomic units (OTUs) assignment using ‘NINJA-OPS’ against the Greengenes 13.8 database as a ref. 52.
Hematoxylin and Eosin (H&E) Staining
After mice were sacrificed, colon tissues were fixed, embedded in paraffin, sectioned and stained as described previously17. Images were taken and analyzed by Zeiss Axioplan 2 Upright Microscope in University Imaging Centers in U. of Minnesota. Hemotoxylin and eosin staining kit was purchased from Fisher Scientific.
Metabolic Measurement
Indirect calorimetry was performed as described previously17 to measure volumetric oxygen consumption (vO2) on mice. Briefly, animals were housed in individual chambers with free access to food and water in a 12-h light/dark cycle. vO2 was recorded and normalized to body weight using Oxymax (Columbus Instrument, Columbus OH).
Glucose tolerance test (GTT) and insulin tolerance test (ITT)
GTT and ITT were performed after overnight fasting. Animal baseline blood samples were collected. D–glucose (2 g/kg) or insulin (0.75 units/kg) was i.p. injected into animals. Blood glucose levels were measured at indicated time points with a glucometer (OneTouch Ultra).
Gut permeability assay
In vivo gut permeability assay was performed as described previously53. Briefly, mice were fed 0.6 mg/g FITC-labeled dextran (Sigma) after fasting for 5 hrs. Serum samples were collected at indicated time points and analyzed (485 nm/535 nm) using a microplate reader (Tecan Infinite M100 pro).
RNA isolation and quantitative real-time PCR
RNA was isolated using Trizol. Reverse transcription was performed using a High-Capacity cDNA Reverse Transcription Kit containing RNase Inhibitor (Applied Biosystems). Quantitative real-time PCR (qPCR) was carried out with Maxima SYBR Green qPCR Master Mixes (Thermo Scientific) as described previously16. Primers for Tnfα (QT00104006), Arg1 (QT00134288), Ucp1 (QT00097300) and Cd137 (QT00147266) were purchased from Qiagen. Each analysis was performed triplicate and normalized to β-actin.
Flow Cytometry
Fc Block (20 m g/mL; BD Biosciences) was used to block cell-surface antigens. After blocking cells were stained with fluorophore conjugated antibodies or isotype control antibodies. Fluorophore-conjugated primary antibodies were purchased from BioLegend: F4/80-Alexa Fluor 488, CD11b-PerCP/Cy5.5, CD11c-phycoerythrin, and CD206-Alexa Fluor 647. After incubation with antibodies, cells were washed and centrifuged, re-suspended in a washing buffer, and analyzed on a FACSCalibur using FlowJo 10.0.6.
Additional Information
How to cite this article: Lin, Y.-W. et al. Gut microbiota from metabolic disease-resistant, macrophage-specific RIP140 knockdown mice improves metabolic phenotype and gastrointestinal integrity. Sci. Rep. 6, 38599; doi: 10.1038/srep38599 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Kaur, J. A comprehensive review on metabolic syndrome. Cardiology research and practice 2014, 943162, doi: 10.1155/2014/943162 (2014).
Claesson, M. J. et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 488, 178–184, doi: 10.1038/nature11319 (2012).
Nicholson, J. K. et al. Host-gut microbiota metabolic interactions. Science 336, 1262–1267, doi: 10.1126/science.1223813 (2012).
Round, J. L. & Mazmanian, S. K. The gut microbiota shapes intestinal immune responses during health and disease. Nature reviews. Immunology 9, 313–323, doi: 10.1038/nri2515 (2009).
Maslowski, K. M. & Mackay, C. R. Diet, gut microbiota and immune responses. Nature immunology 12, 5–9, doi: 10.1038/ni0111-5 (2011).
Kamada, N., Seo, S. U., Chen, G. Y. & Nunez, G. Role of the gut microbiota in immunity and inflammatory disease. Nature reviews. Immunology 13, 321–335, doi: 10.1038/nri3430 (2013).
Knights, D. et al. Complex host genetics influence the microbiome in inflammatory bowel disease. Genome medicine 6, 107, doi: 10.1186/s13073-014-0107-1 (2014).
Vrieze, A. et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 143, 913–916 e917, doi: 10.1053/j.gastro.2012.06.031 (2012).
Singh, V., Yeoh, B. S. & Vijay-Kumar, M. Gut microbiome as a novel cardiovascular therapeutic target. Current opinion in pharmacology 27, 8–12, doi: 10.1016/j.coph.2016.01.002 (2016).
Bakken, J. S. et al. Treating Clostridium difficile infection with fecal microbiota transplantation. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association 9, 1044–1049, doi: 10.1016/j.cgh.2011.08.014 (2011).
Borody, T. J. & Khoruts, A. Fecal microbiota transplantation and emerging applications. Nature reviews. Gastroenterology & hepatology 9, 88–96, doi: 10.1038/nrgastro.2011.244 (2012).
Mostaqul Huq, M. D., Gupta, P. & Wei, L. N. Post-translational modifications of nuclear co-repressor RIP140: a therapeutic target for metabolic diseases. Current medicinal chemistry 15, 386–392 (2008).
Nautiyal, J., Christian, M. & Parker, M. G. Distinct functions for RIP140 in development, inflammation, and metabolism. Trends in endocrinology and metabolism: TEM 24, 451–459, doi: 10.1016/j.tem.2013.05.001 (2013).
Ho, P. C., Tsui, Y. C., Feng, X., Greaves, D. R. & Wei, L. N. NF-kappaB-mediated degradation of the coactivator RIP140 regulates inflammatory responses and contributes to endotoxin tolerance. Nature immunology 13, 379–386, doi: 10.1038/ni.2238 (2012).
Lin, Y. W., Lee, B., Liu, P. S. & Wei, L. N. Receptor-Interacting Protein 140 Orchestrates the Dynamics of Macrophage M1/M2 Polarization. Journal of innate immunity 8, 97–107, doi: 10.1159/000433539 (2016).
Lin, Y. W., Liu, P. S., Adhikari, N., Hall, J. L. & Wei, L. N. RIP140 contributes to foam cell formation and atherosclerosis by regulating cholesterol homeostasis in macrophages. Journal of molecular and cellular cardiology 79, 287–294, doi: 10.1016/j.yjmcc.2014.12.009 (2015).
Liu, P. S. et al. Reducing RIP140 expression in macrophage alters ATM infiltration, facilitates white adipose tissue browning, and prevents high-fat diet-induced insulin resistance. Diabetes 63, 4021–4031, doi: 10.2337/db14-0619 (2014).
Liu, P. S., Lin, Y. W., Burton, F. H. & Wei, L. N. Injecting engineered anti-inflammatory macrophages therapeutically induces white adipose tissue browning and improves diet-induced insulin resistance. Adipocyte 4, 123–128, doi: 10.4161/21623945.2014.981438 (2015).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome biology 12, R60, doi: 10.1186/gb-2011-12-6-r60 (2011).
Langille, M. G. et al. Predictive functional profiling of microbial communities using 16 S rRNA marker gene sequences. Nature biotechnology 31, 814–821, doi: 10.1038/nbt.2676 (2013).
Liu, P. S. et al. Reducing RIP140 expression in macrophage alters ATM infiltration, facilitates white adipose tissue browning and prevents high fat diet-induced insulin resistance. Diabetes, doi: 10.2337/db14-0619 (2014).
Liu, P. S., Lin, Y. W., Burton, F. H. & Wei, L. N. M1-M2 balancing act in white adipose tissue browning - a new role for RIP140. Adipocyte 4, 146–148, doi: 10.4161/21623945.2014.981428 (2015).
Tilg, H. & Kaser, A. Gut microbiome, obesity, and metabolic dysfunction. The Journal of clinical investigation 121, 2126–2132, doi: 10.1172/JCI58109 (2011).
Brahe, L. K. et al. Specific gut microbiota features and metabolic markers in postmenopausal women with obesity. Nutrition & diabetes 5, e159, doi: 10.1038/nutd.2015.9 (2015).
Cox, L. M. et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 158, 705–721, doi: 10.1016/j.cell.2014.05.052 (2014).
Suez, J. et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature 514, 181–186, doi: 10.1038/nature13793 (2014).
Zhu, L. et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology 57, 601–609, doi: 10.1002/hep.26093 (2013).
Falcinelli, S. et al. Probiotic treatment reduces appetite and glucose level in the zebrafish model. Scientific reports 6, 18061, doi: 10.1038/srep18061 (2016).
Zhou, S. S. et al. Nicotinamide overload may play a role in the development of type 2 diabetes. World journal of gastroenterology 15, 5674–5684 (2009).
Xia, J., Wang, Z. & Zhang, F. Association between Related Purine Metabolites and Diabetic Retinopathy in Type 2 Diabetic Patients. International journal of endocrinology 2014, 651050, doi: 10.1155/2014/651050 (2014).
Cheng, S. et al. Metabolite profiling identifies pathways associated with metabolic risk in humans. Circulation 125, 2222–2231, doi: 10.1161/CIRCULATIONAHA.111.067827 (2012).
Newgard, C. B. et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell metabolism 9, 311–326, doi: 10.1016/j.cmet.2009.02.002 (2009).
Shaham, O. et al. Metabolic profiling of the human response to a glucose challenge reveals distinct axes of insulin sensitivity. Molecular systems biology 4, 214, doi: 10.1038/msb.2008.50 (2008).
Wopereis, S. et al. Metabolic profiling of the response to an oral glucose tolerance test detects subtle metabolic changes. PloS one 4, e4525, doi: 10.1371/journal.pone.0004525 (2009).
Park, S., Park, J. Y., Lee, J. H. & Kim, S. H. Plasma levels of lysine, tyrosine, and valine during pregnancy are independent risk factors of insulin resistance and gestational diabetes. Metabolic syndrome and related disorders 13, 64–70, doi: 10.1089/met.2014.0113 (2015).
Elshorbagy, A. K. et al. Dietary cystine level affects metabolic rate and glycaemic control in adult mice. The Journal of nutritional biochemistry 23, 332–340, doi: 10.1016/j.jnutbio.2010.12.009 (2012).
Elshorbagy, A. K., Refsum, H., Smith, A. D. & Graham, I. M. The association of plasma cysteine and gamma-glutamyltransferase with BMI and obesity. Obesity 17, 1435–1440, doi: 10.1038/oby.2008.671 (2009).
Elshorbagy, A. K., Valdivia-Garcia, M., Refsum, H. & Butte, N. The association of cysteine with obesity, inflammatory cytokines and insulin resistance in Hispanic children and adolescents. PloS one 7, e44166, doi: 10.1371/journal.pone.0044166 (2012).
Hayes, T. B. et al. Hermaphroditic, demasculinized frogs after exposure to the herbicide atrazine at low ecologically relevant doses. Proceedings of the National Academy of Sciences of the United States of America 99, 5476–5480, doi: 10.1073/pnas.082121499 (2002).
Zhao, C. et al. Integrated lipidomics and transcriptomic analysis of peripheral blood reveals significantly enriched pathways in type 2 diabetes mellitus. BMC medical genomics 6 Suppl 1, S12, doi: 10.1186/1755-8794-6-S1-S12 (2013).
Rathcke, C. N., Johansen, J. S. & Vestergaard, H. YKL-40, a biomarker of inflammation, is elevated in patients with type 2 diabetes and is related to insulin resistance. Inflammation research: official journal of the European Histamine Research Society … [et al.] 55, 53–59, doi: 10.1007/s00011-005-0010-8 (2006).
Kyrgios, I. et al. Elevated circulating levels of the serum acute-phase protein YKL-40 (chitinase 3-like protein 1) are a marker of obesity and insulin resistance in prepubertal children. Metabolism: clinical and experimental 61, 562–568, doi: 10.1016/j.metabol.2011.09.004 (2012).
Knights, D. et al. Bayesian community-wide culture-independent microbial source tracking. Nature methods 8, 761–763, doi: 10.1038/nmeth.1650 (2011).
Buffie, C. G. et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517, 205–208, doi: 10.1038/nature13828 (2015).
Salzman, N. H. et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nature immunology 11, 76–83, doi: 10.1038/ni.1825 (2010).
Turnbaugh, P. J. et al. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Science translational medicine 1, 6ra14, doi: 10.1126/scitranslmed.3000322 (2009).
Nagalingam, N. A. et al. The effects of intestinal microbial community structure on disease manifestation in IL-10−/− mice infected with Helicobacter hepaticus. Microbiome 1, 15, doi: 10.1186/2049-2618-1-15 (2013).
Deriu, E. et al. Influenza Virus Affects Intestinal Microbiota and Secondary Salmonella Infection in the Gut through Type I Interferons. PLoS pathogens 12, e1005572, doi: 10.1371/journal.ppat.1005572 (2016).
Fransen, F. et al. BALB/c and C57BL/6 Mice Differ in Polyreactive IgA Abundance, which Impacts the Generation of Antigen-Specific IgA and Microbiota Diversity. Immunity 43, 527–540, doi: 10.1016/j.immuni.2015.08.011 (2015).
Ubeda, C. et al. Intestinal microbiota containing Barnesiella species cures vancomycin-resistant Enterococcus faecium colonization. Infection and immunity 81, 965–973, doi: 10.1128/IAI.01197-12 (2013).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods 7, 335–336, doi: 10.1038/nmeth.f.303 (2010).
DeSantis, T. Z. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and environmental microbiology 72, 5069–5072, doi: 10.1128/AEM.03006-05 (2006).
Wernstedt Asterholm, I. et al. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell metabolism 20, 103–118, doi: 10.1016/j.cmet.2014.05.005 (2014).
Acknowledgements
This work was supported by DK54733, DK60521, DK54733-11S, DK60521-12S1, and the Dean’s Commitment and the Distinguished McKnight Professorship of University of Minnesota to LNW. EM was supported by the Robert Tournut award from the French National Society of Gastroenterology.
Author information
Authors and Affiliations
Contributions
Y.W.L. and E.M. designed the experiments, analyzed the data, and performed the experiments. D.K. and L.N.W. designed the experiments, analyzed the data, L.N.W. provided financial support and is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Lin, YW., Montassier, E., Knights, D. et al. Gut microbiota from metabolic disease-resistant, macrophage-specific RIP140 knockdown mice improves metabolic phenotype and gastrointestinal integrity. Sci Rep 6, 38599 (2016). https://doi.org/10.1038/srep38599
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep38599
This article is cited by
-
Downregulating carnitine palmitoyl transferase 1 affects disease progression in the SOD1 G93A mouse model of ALS
Communications Biology (2021)
-
Glyburide and retinoic acid synergize to promote wound healing by anti-inflammation and RIP140 degradation
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
