
Abstract
CRISPR/Cas9 systems are nowadays applied extensively to effect genome editing in various organisms including plants. CRISPR from Prevotella and Francisella 1 (Cpf1) is a newly characterized RNA-guided endonuclease that has two distinct features as compared to Cas9. First, Cpf1 utilizes a thymidine-rich protospacer adjacent motif (PAM) while Cas9 prefers a guanidine-rich PAM. Cpf1 could be used as a sequence-specific nuclease to target AT-rich regions of a genome that Cas9 had difficulty accessing. Second, Cpf1 generates DNA ends with a 5′ overhang, whereas Cas9 creates blunt DNA ends after cleavage. “Sticky” DNA ends should increase the efficiency of insertion of a desired DNA fragment into the Cpf1-cleaved site using complementary DNA ends. Therefore, Cpf1 could be a potent tool for precise genome engineering. To evaluate whether Cpf1 can be applied to plant genome editing, we selected Cpf1 from Francisella novicida (FnCpf1), which recognizes a shorter PAM (TTN) within known Cpf1 proteins, and applied it to targeted mutagenesis in tobacco and rice. Our results show that targeted mutagenesis had occurred in transgenic plants expressing FnCpf1 with crRNA. Deletions of the targeted region were the most frequently observed mutations. Our results demonstrate that FnCpf1 can be applied successfully to genome engineering in plants.
Similar content being viewed by others

Introduction
Targeted mutagenesis and gene targeting using sequence specific nucleases (SSNs) are powerful strategies used to accelerate molecular breeding of crops. Several types of SSN, such as ZFNs (zinc-finger nucleases), TALENs (transcription-activator-like effector nucleases) and CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9) have been intensively adapted for use in genome editing in plants1,2,3. The number of papers reporting plant genome engineering with CRISPR/Cas9 has increased markedly during the last few years4, highlighting the fact that CRISPR/Cas9 is a versatile tool with which to perform targeted mutagenesis in plants5.
Several types of CRISPR/Cas9 systems are known to function as adaptive immune systems in archaea and bacteria6. The well-characterized CRISPR/Cas9 system is categorized as a class 2/type II immune system comprised of single-component effector proteins, and has been engineered for genome editing7,8. Cas9 protein is an endonuclease functioning with CRISPR RNA (crRNA) and transactivating crRNA (tracr RNA)9. The Cas9 RNA complex scans double-stranded DNA to find a DNA sequence complementary to the 20-nucleotide (nt) spacer region (target sequence) within the crRNA, as well as a protospacer adjacent motif (PAM), and then cleaves the target sequence on the invader DNA9. The recognition sequence of the PAM, which is located immediately downstream of the target sequence, varies in each Cas9 protein6,7,10. Widely used Cas9 proteins from Streptococcus pyogenes (SpCas9) and Staphylococcus aureus (SaCas9) prefer a guanidine-rich PAM, with the PAM sequences of SpCas9 and SaCas9 being NGG and NGRRT, respectively6,11,12.
Recently, Cpf1–a new type of RNA-directed endonuclease—was classified as class 2/type V in the CRISPR/Cas system8,13. Some features of Cpf1 differ from those of Cas9 although their functions are similar. While the Cas9 RNA complex contains two RNA molecules in nature, Cpf1 functions with a single crRNA to search and cleave target sequences in infiltrator DNA. A PAM sequence, located immediately upstream of the spacer sequence (target sequence), is also necessary for recognition of the 24 nt target sequence of Cpf113. PAM recognition sequences of Cpf1 are different among bacterial species, and known Cpf1 proteins tend to utilize a thymidine-rich PAM13. The PAM sequence of Cpf1 from Francisella novicida is TTN. TTTN is recognized as PAM by Cpf1 isolated from Acidaminococcus sp. BV3L6 (AsCpf1) and Lachnospiraceae bacterium MA2020 (LbCpf1)13. In addition, Cpf1 and Cas9 generate different types of DNA ends after cleavage of the target sequence. Cpf1 creates DNA ends with a 5′ overhang, while Cas9 generates blunt ends13. Cleavage by FnCpf1 occurs at the 18th base from PAM on the non-targeted (+) strand, and at the 23rd base from PAM on the targeted (–) strand within 24 nt spacer sequence13. Since DSBs with compatible overhangs can be repaired via precise end joining14, the sticky DNA ends generated by Cpf1 are thought to be ideally suited to precise genome editing such as knock-in or replacement of a desired DNA fragment using compatible DNA ends. These specific features of Cpf1 can broaden the spectrum of genome editing that is possible using SSNs.
To apply Cpf1 to plant genome engineering, we selected FnCpf1 for the following reasons. Since FnCpf1 recognizes TTN as PAM sequence, the frequency of target sequences for FnCpf1 in plant genomes is thought to be higher than that of AsCpf1 and LbCpf1, which utilize TTTN as a PAM sequence13. The shorter PAM of FnCpf1 is thus a practical and favorable feature for targeted mutagenesis, although the genome editing activity of FnCpf1 in human cells is reported to be lower than that of AsCpf1 and LbCpf113. Targeted mutagenesis of plants has been performed mostly via stable transformation, introducing T-DNA harboring SSNs into plant genomes15,16. We hypothesized that the lower genome editing activity of FnCpf1 might be compensated by the constitutive expression of FnCpf1 in plants. We first engineered a binary vector to optimize the expression of FnCpf1 in plants, then designed a targeted mutagenesis experiment in tobacco and rice.
Results
FnCpf1 expression vectors for targeted mutagenesis in tobacco and rice
To perform targeted mutagenesis using FnCpf1 in tobacco and rice, we first constructed binary vectors harboring FnCpf1 and antibiotic resistance genes in the T-DNA region (Fig. 1a,b) (cf. our previous construction of binary vectors to express SpCas912,17). The codon usage of FnCpf1 ORF was optimized for effective translation in A. thaliana and rice, respectively. Codon-optimized FnCpf1 (At) and FnCpf1 (Os) were cloned into the binary vectors, pRI201-AN and pPZP200, respectively. FnCpf1 (At) was driven by the ubiquitin 4–2 promoter from Petroselinum crispum (Fig. 1a)18,19. On the other hand, FnCpf1 (Os) was placed under the control of the ubiqcruitin promoter from Zea mays (Fig. 1b)20. To express crRNA of FnCpf1, Arabidopsis U6-26 and rice U6-2 small nuclear RNA gene promoters were used in tobacco and rice, respectively (Fig. 1a,b)17,19.

T-DNA constructions for FnCpf1 expression in tobacco and rice.
(a) Construct for targeted mutagenesis in tobacco. FnCpf1 (At) was inserted downstream of the PcUbi promoter. The Athsp terminator was placed at the end of FnCpf1 ORF. The AtADH 5′-UTR was introduced between the PcUbi promoter and FnCpf1 (At) to enhance translation. the nuclear localization signal (NLS) from the SV40 large T-antigen was fused translationally to the C-terminus of FnCpf1. The crRNA is under the control of Arabidopsis U6-26 promoter. To isolate transformants with kanamycin resistance, an NPT II cassette was included in the construct. AtADH 5′-UTR: 5′ untranslated region of Arabidopsis thaliana ALCOHOLDEHYDROGENASE gene. Athsp ter: the terminator region of Arabidopsis thaliana HEAT SHOCK PROTEIN 18.2 gene. (b) Construct used for targeted mutagenesis in rice. The ZmUbi-1 promoter drives expression of FnCpf1 (Os). The OsADH 5′-UTR was introduced between the ZmUbi promoter and FnCpf1 (At) to enhance translation. An NLS was fused translationally to the C-terminus of FnCpf1 (Os). Pea3A and OsAct1 terminators were inserted tandemly downstream of FnCpf1 to terminate transcription. Expression of crRNA is driven by the rice U6-2 promoter. To screen transformants with hygromycin resistance, HPT cassettes were included in the construct. OsADH 5′-UTR: 5′ untranslated region of Oryza sativa ALCOHOLDEHYDROGENASE gene. Pea3A ter: the terminator region of Pisum sativum rbcS 3A gene. OsAct ter: the terminator region of Oryza sativa Actin gene.
Targeted mutagenesis in tobacco
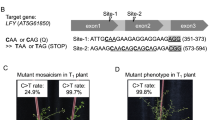
To examine whether FnCpf1 (At) can induce targeted mutation in tobacco, 24 nt target sequences were designed to induce mutations in two genes, i.e., phytoene desaturase (NtPDS) and STENOFOLIA ortholog in Nicotiana tabacum (NtSTF1). Mutation in NtPDS will cause an albino phenotype since a defect in carotenoid biosynthesis leads to loss of pigments such as chlorophyll21. NtSTF1 is thought to be involved in leaf blade expansion since a lam mutant having a defect in LAM (NsSTF1) shows a narrow leaf phenotype in Nicotiana sylvestris22,23. Nicotiana tabacum is an amphidiploid species derived from ancestors that are closely related to the diploid species N. sylvestris and N. tomentosiformis. Therefore, mutant phenotypes can be observed when mutations occur in functionally identical genes located in the N. sylvestris and N. tomentosiformis genomes (S and T genomes), respectively. To select target sequences in these genes on both S and T genomes, TTN, a PAM sequence of FnCpf1, is first searched for within exons of these two genes, and then, 24 nt sequences immediately downstream of the PAM were selected as target sequence. Two and four target sequences, respectively, were designed against NtPDS and NtSTF1 genes (Table 1). These crRNAs were named as follows, crNtPDS-1 and crNtPDS-2, and crNtSTF1-1–crNtSTF1-4. The mutation ratio was estimated by scoring the number of regenerated plants with mutation around the target sequence of FnCpf1 relative to the total number of regenerated plants. The mutation frequency represented the ratio of mutated clones per total randomly sequenced clones.
Transgenic T0 plants showing kanamycin resistance (between 14 and 20 lines) were isolated for each of the target loci. Genomic DNA was isolated from each T0 transgenic plant, and the target loci were then amplified by PCR. To detect mutations in NtPDS genes, PCR products were resolved by performing a heteroduplex mobility assay (HMA). As shown in Fig. 2a, DNA bands with higher molecular weights were observed in several transgenic lines but not in wild-type (WT) (Fig. 2a). Mutation ratios at crNtPDS-1 and crNtPDS-2 loci were around 45%. Mutation patterns of these transgenic lines were analyzed by DNA sequencing, and mutation frequencies were estimated. Deletion mutations were observed around the cleavage site of FnCpf1 (Fig. 2b). Mutation frequencies at crNtPDS-1 and crNtPDS-2 loci were 12.5–65.2% and 4.3–50%, respectively (Fig. 2b, top and middle). Regardless of crRNA, mutation frequencies on the S and T genomes had no consistency in any of the transgenic plants tested (Fig. 2b, top and middle). FnCpf1-induced mutations on the S or T genome seem to occur stochastically at NtPDS loci.

Analyses of FnCpf1-induced mutations in tobacco.
(a) Heteroduplex mobility assay to detect mutation on crNtPDS-1 (upper two panels) and crNtPDS-2 (lower panel) loci. (S) and (T) indicate PCR products amplified from the loci including each target sequence on N. sylvestris and N. tomentosiformis genomes, respectively. (b) Patterns of mutations detected in crNtPDS-1 (top), crNtPDS-2 (middle) and crNtSTF-4 (bottom) loci. The target DNA sequences of each crRNA are shown as wild-type (WT) at the top with underlined. (S) and (T) indicate the target sequence on both S and T genomes. The PAM regions are shown by green. Mismatched nucleotide is indicated in red. Line numbers of transgenic plants were indicated as # at left side of each sequence. DNA deletions are presented as dashes. The length of indel and the number of clones are represented at the right side of each sequence (+, insertion; −, deletion; ×, number of clones). Mut. Freq. (%): Mutation frequency. (c) CAPS analysis of crNtSTF-4 locus in T0 generation. (d) CAPS analysis of crNtSTF-4 locus in T1 generation of line #7. −: Non-digested PCR products, +: EcoRV-digested PCR products. Arrow head indicated the position of undigested PCR products. An undigested band indicates mutation at the crNtSTF-4 locus.
Among the four crRNAs designed to target NtSTF1, crNtSTF1-1 to crNtSTF1-3 did not induce mutation in any of the transgenic lines (Supplemental Fig. 1), while mutations were seen with crNtSTF1-4. This latter crRNA was able to target the NtSTF1 gene in the N. tomentosiformis genome but not in the N. sylvestris genome since crNtSTF1-4 had a one-base mismatched sequence against NtSTF1 (S) (Fig. 2b, lower). To find mutations in NtSTF1, PCR products were subjected to CAPS assay. The mutation ratios of crNtSTF1-4 at the S and T loci were 7.1% and 71.4%, respectively (Fig. 2c). Mutation frequencies of crNtSTF1-4 on the T locus were 28.6–68.2% (Fig. 2b, bottom). These results clearly showed that FnCpf1 could induce mutation at target sites in tobacco. However, we could not recover transgenic plants harboring biallelic mutations at the target sites in the T0 generation.
We next confirmed whether FnCpf1-induced mutations on crNtSTF1-4 locus are genetically transmitted to the next generation. PCR was performed using DNA extracted from progenies of crNtSTF1-t4 line #7 and used for CAPS analysis. As a result, homoallelic mutation was observed in some of the progenies from transgenic tobacco line #7 (Fig. 2d).
Targeted mutagenesis in rice
Next, we applied FnCpf1 (Os) to induce mutation in rice. Agrobacterium-mediated transformation was performed to introduce the T-DNA harboring FnCpf1 (Os) into scutellum-derived calli. The genes OsDrooping leaf (OsDL) and OsAcetolactone synthase (OsALS) were selected as the target genes. dl mutants show a loss of midrib in the leaf blade, resulting in a drooping leaf phenotype24,25. Acetolactone synthase is involved in the synthesis of branched-chain amino acids26,27. Loss of ALS activity leads to lethality. As in tobacco, two target sequences were designed for targeted mutagenesis of each gene. The corresponding crRNAs were named crOsDL-1~2, and crOsALS-1~2 (Table 1). The digestion sites of both FnCpf1 and restriction enzymes were designed to overlap within the target sequences, so that the restriction site would be disrupted if mutations occur at the target loci following cleavage by FnCpf1. The CAPS assay was used to assess the presence of mutations in the target sequence.
To examine FnCpf1-induced mutations in rice calli, genomic DNA was extracted from each callus showing hygromycin resistance. Target loci were amplified by PCR, and the products were subjected to CAPS assay (Fig. 3a). Undigested PCR products were observed in transgenic lines, indicating that the introduction of FnCpf1 with each of the crRNAs was able to induce mutations in rice calli. At the crOsDL-2 and crOsALS-2 target loci, the mutation frequency in rice calli was over 60% (Fig. 3b). Mutation frequencies at the crOsDL-1 and crOsALS-1 target loci were 8.3–25% and 15%, respectively (Fig. 3b). As shown in Fig. 3b, deletions occurred mostly at target sites of all crRNA. In the case of regenerated plants obtained from transgenic calli harboring FnCpf1 with crOsDL-2 and crOsALS-2, the mutation ratio of the regenerated plants at the crOsDL-2 target locus was 85.7% (6/7), and the three plants with bi-allelic mutations showed a drooping leaf phenotype in the T0 generation (Supplemental Fig. 2). In addition, the ratio of regenerated plants with mutation at the crOsALS-2 target locus was 90% (9/10), and five bi-allelic mutant plants were obtained. Because all the bi-allelic mutant plants had deletions with mutations that did not generate frameshifts on the OsALS gene, these als mutants were assumed to be viable (data not shown). In addition, heteroallelic mutation on crOsDL-1 target locus was inherited as homoallelic mutation in the progeny of line #18 (Supplemental Fig. 3). These results clearly indicate that FnCpf1 is able to cleave target sites in rice.

Analyses of FnCpf1-induced mutations in rice.
(a) CAPS analysis of crOsDL-1~2 and crOsALS-1~2 loci. −: Non-digested PCR products, +: PstI or AseI-digested PCR products. Arrow head indicated the position of undigested PCR products. An undigested band indicates mutation in the target loci. (b) Patterns of mutations detected in crOsDL-1~2 and crOsALS-1~2 loci. The target DNA sequences of each crRNA are shown as wild-type (WT) at the top with underlined. (S) and (T) indicate the target sequence on both S and T genomes. The PAM regions are shown by green. Mismatched nucleotide is indicated in red. Line numbers of transgenic plants were indicated as # at left side of each sequence. DNA deletions are presented as dashes. The length of indel and the number of clones are represented at the right side of each sequence (+, insertion; −, deletion; ×, number of clones). Mut. Freq. (%): Mutation frequency.
Off target mutation analysis of FnCpf1 in rice
Zetsche et al. reported that the seed region of the FnCpf1 crRNA is within the first 5 nt of the 5′-end of the spacer sequence in vitro13. We tried to examine the possibility of FnCpf1-induced mutations at off-target genes in rice. To explore this possibility, we selected the 9-cis-epoxycarotenoid dioxygenase (NCED) gene family (OsNCED1–3), and the aldehyde oxidase (AO) gene family (OsAO1–5)28,29. When the crOsNCED1-1 sequence of the OsNCED1 gene was defined as the target sequence, the corresponding sequence of the OsNCED2 has one, and that of OsNCED3 has two, mismatched bases (Table 2). The mutation frequency using FnCpf1 with crOsNCED1-1 in rice calli was 2.14–23.3% in the target gene (OsNCED1), while those in the off-target genes (OsNCED2 and OsNCED3) were 0–6.25% and 0%, respectively (Table 2). Next, crOsAO1-1 was designed to target both the OsAO1 and OsAO2 genes. The crOsAO1-1 has one mismatched base on the corresponding region of the OsAO3 and OsAO4 genes, and two mismatched bases in the OsAO5 gene. When crOsAO-1 was used with FnCpf1, mutation frequencies in the OsAO1, OsAO2, and OsAO4 genes were 38.8–50%, 24.1–36.6% and 0–5%, respectively (Table 2). No mutations were observed in OsAO3 and OsAO5.
Discussion
In this study, we evaluated the use of FnCpf1 in targeted mutagenesis of rice and tobacco genomes; our data showed clearly that FnCpf1 can be applied to targeted mutagenesis in these crops.
Zetsche et al. evaluated the genome editing activity of various Cpf1 proteins via transient assay in human cells13. Their results revealed that AsCpf1 and LbCpf1 exhibited a higher activity to induce mutation than other Cpf1 enzymes, including FnCpf113. In our study, FnCpf1 was able to effectively induce mutations in various target genes upon constitutive expression of FnCpf1 in tobacco and rice. We previously reported that the engineering of binary vectors expressing SpCas9 significantly affected the mutation ratio in rice17. To enhance the expression of FnCpf1 in tobacco and rice, previous studies have introduced a variety of devices such as codon-optimization of FnCpf1, addition of a nuclear localization signal sequence to FnCpf1, translational enhancer, transcriptional terminator and constitutive promoters, to the binary vectors used17,19. It is highly likely that a combination of a stable expression system and these additional tweaks contributed to improving the genome editing activity of FnCpf1 in plant cells.
In our targeted mutagenesis experiments, the average mutation frequencies on targeted loci in tobacco and rice were 28.2% and 47.2%, respectively. The average mutation frequency in tobacco was lower than that in rice. In addition, we successfully isolated bialellic mutants in the T0 generation in rice but not in tobacco. This may be due to differences in the transformation processes between rice and tobacco. In the case of rice transformation, dedifferentiated callus showing relatively higher cell division activity was utilized for Agrobacterium infection, and the callus state was maintained until the regeneration step30. On the other hand, the tobacco transformation process started from leaf discs. Transformed leaf discs were subjected to selection on antibiotics. During this process, screening proceeded in parallel with the other processes, including callus induction and regeneration. When we applied SpCas9 to targeted mutagenesis in rice, mutation ratio and frequency increased in accordance with the duration of the callus state31. Therefore, it may be necessary to prolong the duration of the callus state in tobacco transformation in order to isolate biallelic mutants in the T0 generation.
FnCpf1 induced mostly chimeric mutations in tobacco, with various mutations being observed in each regenerated plant (Fig. 2b). On the other hand, in our previous study using SpCas917,32, each regenerated rice plant possessed monoallelic or biallelic mutation. This difference may due to the difference in the transformation process as described above. Rice plants could be regenerated mostly from genetically homogeneous callus, and SSN-induced mutation rarely occurs in regenerated rice plants33, whereas tobacco might accumulate mutation events in going from regeneration to the reproductive stage. As a result, constitutively occurring mutations could create genetically chimeric plants. When targeted mutagenesis was performed with SpCas9 driven by ubiquitously active promoters, such as 35 S or the ubiquitin promoter, chimeric mutations occurred similarly in tobacco or Arabidopsis12,34. To address this, several groups have already reported the use of tissue-specific promoters to express SpCas9, e.g., in flower meristem or germ line cells, and have succeeded in reducing chimeric mutation in Arabidopsis34,35. Therefore, application of promoters functioning in an inducible or tissue-specific manner to control the expression of FnCpf1 spatiotemporally could contribute to reducing chimeric mutation.
FnCpf1-induced mutations were mostly deletions. The mutation patterns of FnCpf1 were similar to those of TALENs, ZFNs and paired nickases (Cas9)33,36,37,38. FnCpf1 generates DNA ends with 5′ overhangs; TALENs, ZFNs and paired nickases also generate sticky DNA ends after cleavage of target sequences. It is highly possible that similar DNA repair mechanisms operate after cleavage with these nucleases. DSBs activate DNA repair machinery such as homology dependent repair (HDR) or non-homologous end joining (NHEJ)39. DSB repair is affected greatly by the structure of the DNA ends39. Cohesive DSBs with compatibility tend to be repaired by precise end joining, while non-compatible DSB ends with various deletions are repaired via NHEJ40. In our experiments, we tried to introduce mutations in a single gene with a single crRNA. Single digestion of a target locus generates a DSB with compatibility, and most such DSBs could be repaired precisely.
Four crRNAs were designed for the NtSTF1 gene, Three of which did not induce mutations in the target loci. Two possible explanations can be considered: (1) there could be epigenomic modification or chromatin structure around the target regions, which could decrease the accessibility of FnCpf1 to the target site41,42. (2) The secondary structure of FnCpf1 crRNA; FnCpf1 crRNA is 43 nt in length, while the single chimeric guide RNA of Cas9 is around 100 nt9,13. The crRNA of FnCpf1 consists of two parts, which are responsible for scaffold (19 nt) and target recognition (24 nt), respectively13. The interaction between FnCpf1 protein and crRNA could be affected by the secondary structure of the crRNA, which depends strongly on that part of the target sequence. Chemical modification of the crRNA, or an artificially designed crRNA, may improve the interaction between FnCpf1 and crRNA. Chemical modification of the ribonucleotide in the guide RNA of SpCas9 improved mutation frequency by altering RNA stability, and the secondary structure of the guide RNA43,44. Since FnCpf1 has RNase III activity and trims its crRNA by itself45, expression of FnCpf1 crRNA with extra oligonucleotides contributing to preventing unfavorable secondary structure of the crRNA may improve the mutation efficiency of FnCpf 1 as long as a stable transformation system is used to express FnCpf1 with crRNA in plant cells.
Although off-target mutation of FnCpf1 was found in two of five off-target genes in rice (Table 2), mutation frequencies at off-target genes were lower than those at on-target genes (Table 2). Each crRNA had one base mismatch in these two off-target genes, OsNCED2 and OsAAO4 (Table 2). A mismatch was found at 11th nucleotide from PAM. We also found an off-target mutation in the NtSTF1 gene in the S genome of tobacco. The mismatched nucleotide was found just next to the PAM sequence (Fig. 2b,c). Similar to our results, AsCpf1 exhibited consistent tolerance to a single mismatch at positions 1, 8, 9 and 19–23 within the 24 nt spacer sequence in human cells46. The problem of off-target mutation by FnCpf1 should be improved by increasing the fidelity of FnCpf1. In the case of SpCas9, the fidelity of target recognition depends greatly on precise recognition of PAM47,48,49; as the PAM sequence becomes longer, fidelity increases. The length of PAM restricts the number of target sites; as PAM becomes shorter, the frequency of occurrence of target sequences in the genome increases. Crystal-structure-based rational engineering of the positively charged groove between the HNH-, RuvC- and PAM-interacting domains in SpCas9 improved the fidelity of target recognition50, thus proving it is possible to improve the fidelity of FnCpf1 by structure-based engineering without increasing PAM length.
It was shown recently that AsCpf1 only rarely induces off-target mutation during genome editing in human cells and mice46,51,52. AsCpf1 may have higher fidelity than FnCpf1 since the PAM sequences of FnCpf1 and AsCpf1 are TTN and TTTN, respectively. Application of AsCpf1 and LbCpf1 will be the next challenge in expanding the utility of Cpf1 in plant genome editing.
Methods
Vector construction
Two types of FnCpf1 coding sequence were synthesized to optimize codon usage for Arabidopsis thaliana and Oryza sativa, respectively. The coding sequence of each codon-optimized nuclease, FnCpf1 (At) and FnCpf1 (Os), was cloned into the binary vectors, pRI201-AN (TaKaRa, Japan) and pPZP20053, respectively. The crRNA of FnCpf1 was placed under the control of the U6-26 promoter from Arabidopsis, or the U6-2 promoter from rice17,19; 24 nt target sequences were inserted into the Bbs I site next to the crRNA. The expression cassette of FnCpf1 crRNA with target sequences was cloned into the site generated by digestion of the binary vector using two restriction enzymes, Asc I and Pac I.
Transformation of tobacco or rice
For tobacco transformation, the binary vector harboring FnCpf1 (At) was introduced into Agrobacterium strain LBA4404. Leaf discs (8 mm diameter) collected from fully expanded leaves of tobacco (Nicotiana tabacum L. cv. Petit Havana SR-1) were used for Agrobacterium-mediated transformation as described in Kaya et al.12. For rice transformation, the Agrobacterium strain EHA105 transformed with the binary vector containing FnCpf1 (Os), was used to infect scutellum-derived rice callus (Oryza sativa L. ssp. japonica cv. Nipponbare). Details of the rice transformation procedure have been described previously32.
CAPS analysis and heteroduplex mobility assay
Genomic DNA was extracted from regenerated shoots of tobacco, hygromycin-resistant rice calli, or regenerated rice plants, using Agencourt Chloro Pure (BECKMAN COULTER, USA), and target loci were amplified by PCR using the primer sets listed in supplemental table 1. For cleaved amplified polymorphic sequences (CAPS) analysis, PCR products were digested by the appropriate restriction enzymes, and then analyzed by agarose gel electrophoresis. A heteroduplex mobility assay (HMA) was performed using MultiNA (SHIMADZU, Japan) according to our previous report12.
Sequencing analysis
PCR products used in CAPS analysis or HMA were cloned into pCR-BluntII-TOPO (Thermo Fisher Scientific, USA). DNA sequence was determined using a 3500xL genetic analyzer (Applied Biosystems, USA).
Additional Information
How to cite this article: Endo, A. et al. Efficient targeted mutagenesis of rice and tobacco genomes using Cpf1 from Francisella novicida. Sci. Rep. 6, 38169; doi: 10.1038/srep38169 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Lee, J., Chung, J.-H., Kim, H. M., Kim, D.-W. & Kim, H. Designed nucleases for targeted genome editing. Plant Biotechnol. J. 14, 448–462 (2016).
Osakabe, Y. & Osakabe, K. Genome editing with engineered nucleases in plants. Plant Cell Physiol. 56, 389–400 (2015).
Voytas, D. F. Plant genome engineering with sequence-specific nucleases. Annu. Rev. Plant Biol. 64, 327–350 (2013).
Khatodia, S., Bhatotia, K., Passricha, N., Khurana, S. M. P. & Tuteja, N. The CRISPR/Cas Genome-Editing Tool: Application in Improvement of Crops. Front. Plant Sci. 7, 506 (2016).
Kumar, V. & Jain, M. The CRISPR-Cas system for plant genome editing: advances and opportunities. J. Exp. Bot. 66, 47–57 (2015).
Ran, F. A. et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 (2015).
Fonfara, I. et al. Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Res. 42, 2577–2590 (2014).
Makarova, K. S. et al. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 13, 722–736 (2015).
Jinek, M. et al. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science (80-.). 337, 816–821 (2012).
Hou, Z. et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc. Natl. Acad. Sci. U. S. A. 110, 15644–15649 (2013).
Steinert, J., Schiml, S., Fauser, F. & Puchta, H. Highly efficient heritable plant genome engineering using Cas9 orthologues from Streptococcus thermophilus and Staphylococcus aureus. Plant J. 84, 1295–1305 (2015).
Kaya, H., Mikami, M., Endo, A., Endo, M. & Toki, S. Highly specific targeted mutagenesis in plants using Staphylococcus aureus Cas9. Sci. Rep. 6, 26871 (2016).
Zetsche, B. et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771 (2015).
Budman, J. & Chu, G. Processing of DNA for nonhomologous end-joining by cell-free extract. EMBO J. 24, 849–860 (2005).
Altpeter, F. et al. Advancing Crop Transformation in the Era of Genome Editing. Plant Cell 28, 1510–1520 (2016).
Schaeffer, S. M. & Nakata, P. A. CRISPR/Cas9-mediated genome editing and gene replacement in plants: Transitioning from lab to field. Plant Sci. 240, 130–142 (2015).
Mikami, M., Toki, S. & Endo, M. Comparison of CRISPR/Cas9 expression constructs for efficient targeted mutagenesis in rice. Plant Mol. Biol. 88, 561–572 (2015).
Kawalleck, P., Somssich, I. E., Feldbrugge, M., Hahlbrock, K. & Weisshaar, B. Polyubiquitin gene expression and structural properties of the ubi4-2 gene in Petroselinum crispum. Plant Mol. Biol. 21, 673–684 (1993).
Fauser, F., Schiml, S. & Puchta, H. Both CRISPR/Cas-based nucleases and nickases can be used efficiently for genome engineering in Arabidopsis thaliana. Plant J. 79, 348–359 (2014).
Toki, S. et al. Expression of a Maize Ubiquitin Gene Promoter-bar Chimeric Gene in Transgenic Rice Plants. Plant Physiol. 100, 1503–1507 (1992).
Norris, S. R., Barrette, T. R. & DellaPenna, D. Genetic dissection of carotenoid synthesis in arabidopsis defines plastoquinone as an essential component of phytoene desaturation. Plant Cell 7, 2139–2149 (1995).
McHale, N. A. & Marcotrigiano, M. LAM1 is required for dorsoventrality and lateral growth of the leaf blade in Nicotiana. Development 125, 4235–4243 (1998).
Tadege, M. et al. STENOFOLIA regulates blade outgrowth and leaf vascular patterning in Medicago truncatula and Nicotiana sylvestris. Plant Cell 23, 2125–2142 (2011).
Nagasawa, N. et al. SUPERWOMAN1 and DROOPING LEAF genes control floral organ identity in rice. Development 130, 705–718 (2003).
Yamaguchi, T. et al. The YABBY gene DROOPING LEAF regulates carpel specification and midrib development in Oryza sativa. Plant Cell 16, 500–509 (2004).
McCourt, J. A. & Duggleby, R. G. Acetohydroxyacid synthase and its role in the biosynthetic pathway for branched-chain amino acids. Amino Acids 31, 173–210 (2006).
Tan, S., Evans, R. R., Dahmer, M. L., Singh, B. K. & Shaner, D. L. Imidazolinone-tolerant crops: history, current status and future. Pest Manag. Sci. 61, 246–257 (2005).
Tan, B. C. et al. Molecular characterization of the Arabidopsis 9-cis epoxycarotenoid dioxygenase gene family. Plant J 35, 44–56 (2003).
Hirano, K. et al. Comprehensive transcriptome analysis of phytohormone biosynthesis and signaling genes in microspore/pollen and tapetum of rice. Plant Cell Physiol. 49, 1429–1450 (2008).
Toki, S. et al. Early infection of scutellum tissue with Agrobacterium allows high-speed transformation of rice. Plant J. 47, 969–976 (2006).
Mikami, M., Toki, S. & Endo, M. Parameters affecting frequency of CRISPR/Cas9 mediated targeted mutagenesis in rice. Plant Cell Rep. 34, 1807–1815 (2015).
Endo, M., Mikami, M. & Toki, S. Multigene knockout utilizing off-target mutations of the CRISPR/Cas9 system in rice. Plant Cell Physiol. 56, 41–47 (2015).
Nishizawa-Yokoi, A. et al. A Defect in DNA Ligase4 Enhances the Frequency of TALEN-Mediated Targeted Mutagenesis in Rice. Plant Physiol. 170, 653–666 (2016).
Mao, Y. et al. Development of germ-line-specific CRISPR-Cas9 systems to improve the production of heritable gene modifications in Arabidopsis. Plant Biotechnol. J. 14, 519–532 (2016).
Hyun, Y. et al. Site-directed mutagenesis in Arabidopsis thaliana using dividing tissue-targeted RGEN of the CRISPR/Cas system to generate heritable null alleles. Planta 241, 271–284 (2015).
Osakabe, K., Osakabe, Y. & Toki, S. Site-directed mutagenesis in Arabidopsis using custom-designed zinc finger nucleases. Proc. Natl. Acad. Sci. 107, 12034–12039 (2010).
Mikami, M., Toki, S. & Endo, M. Precision Targeted Mutagenesis via Cas9 Paired Nickases in Rice. Plant Cell Physiol. 57, 1058–1068 (2016).
Kim, Y., Kweon, J. & Kim, J.-S. TALENs and ZFNs are associated with different mutation signatures. Nature methods 10, 185 (2013).
Lieber, M. R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79, 181–211 (2010).
Budman, J., Kim, S. A. & Chu, G. Processing of DNA for nonhomologous end-joining is controlled by kinase activity and XRCC4/ligase IV. J. Biol. Chem. 282, 11950–11959 (2007).
Chen, X. et al. Probing the impact of chromatin conformation on genome editing tools. Nucleic Acids Res. 44, 6482–6492 (2016).
Hilton, I. B. et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 33, 510–517 (2015).
Hendel, A. et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 33, 985–989 (2015).
Doench, J. G. et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol. 32, 1262–1267 (2014).
Fonfara, I., Richter, H., Bratovic, M., Le Rhun, A. & Charpentier, E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 532, 517–521 (2016).
Kleinstiver, B. P. et al. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat. Biotechnol. 34, 869–74 (2016).
Kleinstiver, B. P. et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523, 481–485 (2015).
Hirano, S., Nishimasu, H., Ishitani, R. & Nureki, O. Structural Basis for the Altered PAM Specificities of Engineered CRISPR-Cas9. Mol. Cell 61, 886–894 (2016).
Anders, C., Bargsten, K. & Jinek, M. Structural Plasticity of PAM Recognition by Engineered Variants of the RNA-Guided Endonuclease Cas9. Mol. Cell 61, 895–902 (2016).
Slaymaker, I. M. et al. Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88 (2016).
Kim, D. et al. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat. Biotechnol. 34, 863–868 (2016).
Kim, Y. et al. Generation of knockout mice by Cpf1-mediated gene targeting. Nat. Biotechnol. 34, 808–810 (2016).
Hajdukiewicz, P., Svab, Z. & Maliga, P. The small, versatile pPZP family of Agrobacterium binary vectors for plant transformation. Plant Mol. Biol. 25, 989–994 (1994).
Acknowledgements
We thank Drs. M. Endo, A. Nishizawa-Yokoi, H. Saika, S. Hirose, K. Abe and N. Ohtsuki for valuable discussions and suggestions, and K. Amagai, R. Aoto, C. Furusawa, A. Mori, A. Nagashii, A. Nakano, F. Suzuki and R. Takahashi for general support. This work was supported by Cabinet Office, Government of Japan, the Cross-ministerial Strategic Innovation Promotion program (SIP).
Author information
Authors and Affiliations
Contributions
A.E., M.M. and S.T. designed the experiments. A.E. and M.M. performed the experiments and wrote the main manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Endo, A., Masafumi, M., Kaya, H. et al. Efficient targeted mutagenesis of rice and tobacco genomes using Cpf1 from Francisella novicida. Sci Rep 6, 38169 (2016). https://doi.org/10.1038/srep38169
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep38169
This article is cited by
-
Efficient genome editing in grapevine using CRISPR/LbCas12a system
Molecular Horticulture (2023)
-
Applications of CRISPR/Cas genome editing in economically important fruit crops: recent advances and future directions
Molecular Horticulture (2023)
-
Research progress on nucleic acid detection and genome editing of CRISPR/Cas12 system
Molecular Biology Reports (2023)
-
Tailoring crops with superior product quality through genome editing: an update
Planta (2023)
-
Reverse genetic approaches for breeding nutrient-rich and climate-resilient cereal and food legume crops
Heredity (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
