
Abstract
Neisseria meningitidis contains a very potent hexa-acylated LPS that is too toxic for therapeutic applications. We used systematic molecular bioengineering of meningococcal LPS through deletion of biosynthetic enzymes in combination with induction of LPS modifying enzymes to yield a variety of novel LPS mutants with changes in both lipid A acylation and phosphorylation. Mass spectrometry was used for detailed compositional determination of the LPS molecular species, and stimulation of immune cells was done to correlate this with endotoxic activity. Removal of phosphethanolamine in lipid A by deletion of lptA slightly reduces activity of hexa-acylated LPS, but this reduction is even more evident in penta-acylated LPS. Surprisingly, expression of PagL deacylase in a penta-acylated lpxL1 mutant increased LPS activity, contradicting the general rule that tetra-acylated LPS is less active than penta-acylated LPS. Further modification included expression of lpxP, an enzyme known to add a secondary 9-hexadecenoic acid to the 2’ acyl chain. The LpxP enzyme is temperature-sensitive, enabling control over the ratio of expressed modified hexa- and penta-acylated LPS by simply changing the growth temperature. These LPS derivatives display a broad range of TLR4 activity and differential cytokine induction, which can be exploited for use as vaccine adjuvant or other TLR4-based therapeutics.
Similar content being viewed by others

Introduction
Lipopolysaccharides (LPS), also known as bacterial endotoxin, are an abundant component of the outer membrane of Gram-negative bacteria. During infection with Gram-negative bacteria, LPS or more precisely the lipid A part of LPS activates the host’s innate immune system1,2,3. This activation occurs through binding of LPS to the pattern recognition receptor Toll-like receptor 4/myeloid differentiation factor 2 (TLR4/MD-2) complex, which starts a signaling cascade leading to cytokine production necessary to clear the infection3,4. However, overstimulation of this signaling cascade and overproduction of the inflammatory cytokines is detrimental to the host and can lead to life-threatening conditions such as septic shock5,6.
For complete activation of the TLR4/MD-2 complex, a lipid A structure with six acyl chains and two phosphate groups is critical7. However, many bacterial species carry enzymes that can modify their lipid A structure either by changing the number of acyl chains or phosphate groups resulting in altered activation of the TLR4/MD-2 complex8, even to the point of being an antagonist instead of an agonist as is observed for the tetra-acylated E. coli lipid IVa structure7,9.
The TLR4/MD-2 complex is unique among the TLR family of receptors because it can signal through both the MyD88 as well as the TRIF pathway. Modified lipid A structures can induce select signaling by preferential recruitment of the MyD88 or TRIF adaptor molecules. Preferential signaling through the TRIF pathway, which triggers production of type I interferons, is thought to be important for vaccine adjuvants10,11. Monophosphoryl lipid A (MPLA) is an example of a modified lipid A that triggers a TRIF-biased signaling11. MPLA is a heterogeneous lipid A mixture from Salmonella Minnesota, which has been chemically detoxified and is approved for the use as adjuvant in some vaccines12. The main component of MPLA consists of a hexa-acylated 4′-monophosphoryl lipid A. Use of MPLA has the disadvantage that for its production a chemical treatment is needed in addition to LPS isolation from the bacteria and it only consists of the lipid A portion of LPS, making it completely water insoluble, whereas complete LPS can be administered in water.
Neisseria meningitidis typically produces hexa-acylated LPS with phosphate and phosphoethanolamine groups appended to the 1 and 4′ position of the lipid A13,14. Heterologous expression of LPS modifying enzymes such as PagL or deletion of lipid A biosynthesis enzymes such as LpxL1 and LpxL2 has been used to detoxify the highly active meningococcal LPS13,15. Deletion of LpxL1 was shown to be an advantageous method for detoxifying meningococcal LPS when making meningococcal outer membrane vesicle vaccines, without the need to use a detergent to reduce excess LPS-related reactogenicity16. However, the activity of this modified LPS has been reduced to the extent that it barely induces any activation of the TLR4/MD-2 complex on human cells, making it less applicable as a stand-alone vaccine adjuvant17. Heterologous expression of pagL in N. meningitidis results in a different attenuated penta-acylated LPS structure, which is still capable of inducing TLR4 activation and induces a TRIF-biased cytokine production on a human monocytic cell line13. The challenge of LPS-based adjuvants is finding the optimal balance between retaining a sufficient amount of immune activation while limiting toxic side effects. In the present study, we have used heterologous expression of LPS modifying enzymes in combination with targeted deletion of lipid A biosynthesis genes to collect a diverse set of meningococcal LPS structures with a broad range of TLR4/MD-2 activation capacities.
Results
Bioengineering of modified LPS structures
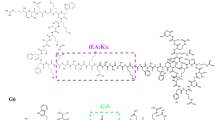
LPS mutants in N. meningitidis were constructed in the HB-1 derivative of strain H44/76. Mass spectrometric analysis demonstrated that HB-1 expresses a hexa-acylated, tri-phosphate, bis-phosphoethanolamine lipid A structure (see below). To construct a diverse set of LPS mutants in strain HB-1, we inactivated the autologous genes encoding for the LPS enzymes LptA, LpxL1 and LpxL2 and heterologously expressed the LpxE, LpxP and PagL LPS enzymes (Fig. 1A,B). In addition, combinations of deletion of autologous genes and expression of heterologous enzymes were constructed. This approach resulted in 10 LPS mutant strains as depicted in Fig. 1C.

Applied lipid A modification enzymes.
The structural changes made by the various combinations of modifying enzymes are depicted in color (A). LpxL1 (green), LpxL2 (blue), LpxP (pink) and LptA (brown) all add the corresponding group to the molecule, whereas PagL (red) and LpxE (orange) remove the group. The abbreviation of the enzymes, organism source and activity are presented (B) and the acylation pattern and number of phosphate and phosphoethanolamine groups for each strain are indicated (C).
For the expression of LpxE (Protein ID: CAE41138.1) we initially cloned an lpxE homologue from Bordetella pertussis. However, expression of this gene in HB-1 or its lptA mutant derivative did not result in any LPS structural changes as determined by mass spectrometry. As an alternative the lpxE (Genbank accession number: WP_003809405.1) homologue from Bordetella bronchiseptica, which exists as a pseudogene in B. pertussis, was cloned and expressed in a ΔlptA mutant strain. This resulted in the loss of a phosphate group in the lipid A and was included in our panel of LPS mutant strains (Fig. 2L).

Charge deconvoluted ESI-FT mass spectra of LPS.
The charge deconvoluted ESI-FT mass spectra of the LPS isolated from twelve different strains of Neisseria meningitidis are shown as follows: parent HB-1 strain (A), ΔlpxL1 (B), ΔlpxL2 (C), pagL (D), ΔlpxL1-pagL (E), ΔlpxL2-pagL (F), ΔlpxL1-lpxP cultured at 30 °C for 5 h (G) or at 25 °C overnight (H), ΔlptA (I), ΔlptA-ΔlpxL1 (J), ΔlptA-pagL (K) and ΔlptA-lpxE (L). A simplified representation of the LPS structure assigned to the ion of 3408.507 u is included in mass spectrum (A). The vertical line at a mass of 3408.514 u, which corresponds to the calculated molecular mass of this latter LPS species, is used as a reference to indicate LPS composition assigned to other ion signals. See Supplementary Table 1 for detailed LPS composition proposals. All annotations refer to monoisotopic masses of the neutral molecules.
LpxP (Genbank accession number: U49787.1), an enzyme known to add a secondary 9-hexadecenoic acid (C16:1) to the 2′ acyl chain in E. coli18, was expressed in the N. meningitidis ΔlpxL1 mutant strain. The LpxL1 enzyme adds a secondary acyl chain to the same position as LpxP, so we reasoned that its absence could increase the efficiency of any C16:1 addition. This modification was expected to create a hexa-acylated lipid A structure different from the original by carrying the longer, mono-unsaturated C16:1 secondary acyl chain in the 2′ position instead of C12. When LpxP was expressed in the ΔlpxL1 mutant strain at 37 °C this resulted in a very faint addition of C16:1. However, C16:1 is added onto E. coli LPS only at 12 °C, so for this reason we grew the bacteria at lower temperatures. Cultivation of meningococci below 25 °C is, unlike in E. coli, not possible, but at 25 °C and 30 °C we already found a much higher relative abundance of the LpxP hexa-acylated lipid A structure carrying the additional C16:1, with 25 °C resulting in the highest efficiency (at least 50% relative abundance) (Fig. 2H).
Mass spectrometric characterization of modified LPS
The charge-deconvoluted ESI-FT mass spectra of intact LPS isolated from the constructed N. meningitidis mutants are shown in Fig. 2. The mass spectrum of LPS of the HB-1 (galE-) parent strain (Fig. 2A), displayed an ion signal of 3408.507 u consistent with LPS comprised of wild-type hexa-acyl lipid A carrying three phosphate (P) and two phosphoethanolamine (PEA) groups and an L3-immunotype oligosaccharide structure substituted with a glycine (Gly) residue and truncated at the proximal galactose (Gal) of its alpha chain due to inactivation of the galE gene (Mcalc. = 3408.514 u, see Supplementary Table 1 for LPS composition proposals). Accompanying ion peaks of 3351.488, 3285.501 and 3228.480 u (Fig. 2A) corresponded to LPS species which lack Gly (Δmeas. = −57.019 u), carry one less PEA group in the lipid A (Δmeas. = −123.006 u) or both (Δmeas. = −180.027 u), respectively. Therefore, the chemical heterogeneity of the LPS from HB-1 (galE-) strain was caused by variation of lipid A phosphorylation and oligosaccharide non-stoichiometric substitution with glycine. Composition proposals based on mass spectra of intact LPS were additionally supported by FT-MS analysis of LPS fragment ions corresponding to lipid A and oligosaccharide moieties, which were generated by in-source collision induced dissociation (SID) of intact LPS. For instance, SID FT mass spectra of LPS from HB-1 (galE-) strain displayed fragment ions of 1916.098 and 2039.106 u corresponding to hexa-acyl lipid A species with 2 and 3 PEA groups (Mcalc. = 1916.100 and 2039.109 u, respectively) and a fragment ion of 1369.404 u corresponding to the dehydrated derivative of the oligosaccharide moiety described above (Mcalc. = 1369.406 u). Fragmentation analyses of LPS derived from other strains of N. meningitidis described here showed that different types of LPS carry the same oligosaccharide moieties (PEA1•Hex1•Hep2•HexNAc1•Kdo2•Gly1), with the exception of some LPS species, which lack a glycine or carry a second hexose residue (Hex) (Supplementary Table 2). Consequently, other differences observed between the LPS species, such as in the number of PEA and P groups, were attributed to changes in the composition of the lipid A (Supplementary Table 2).
Analysis of the intact LPS from the ΔlpxL1 mutant revealed that he main ion peaks of the mass spectrum (3046.315, 3103.336, 3169.324 and 3226.342 u, Fig. 2B) had shifted compared to the 4 main ion signals of the LPS from the parent HB-1 (galE-) strain (Fig. 2A) by -182.165 u. This is in agreement with the lack of a dodecanoic acid (C12) (Δcalc. = −182.167 u) in the lipid A after deletion of the lpxl1 gene.
The mass spectrum of the LPS from the ΔlpxL2 mutant displayed ion peaks of 3023.367, 2966.348, 3185.419 and 3128.398 u (Fig. 2C), which are consistent with the loss of a C12 fatty acyl chain together with PPEA from the lipid A (Δcalc. = −385.142 u) in combination with non-stoichiometric substitution of the oligosaccharide with Gly (Δcalc. = 57.021 u) or a second hexose (Δcalc. = 162.053 u). This is in agreement with effective deletion of the lpxL2 gene. It is worthy to note that deletion of the lpxL2 gene not only led to the loss a C12 fatty acyl chain, as observed earlier upon deletion of the lpxL1 gene, but also resulted in the loss of a P and a PEA group from the lipid A.
The ion peaks in the mass spectrum of the LPS from the pagL mutant (3210.345, 3153.325, 3087.338 and 3030.318 u, Fig. 2D) were found to be shifted by −198.163 u from the 4 main ion peaks of the LPS from the parent HB-1 strain. This is in agreement with efficient removal of a 3-hydroxy-dodecanoic acid (C12OH) (Δcalc. = −198.162 u) from the lipid A by the PagL enzyme. Nonetheless, display of minor ion peaks of 3408.505 and 3351.485 u (Fig. 2D) corresponding to unmodified hexa-acyl LPS species indicated that LPS 3-O-deacylation activity of the PagL enzyme could not fully exhaust the hexa-acyl lipid A substrate.
The 4 main ion signals in the mass spectrum of the LPS from the ΔlpxL1-pagL mutant (3028.180, 2971.160, 2905.173 and 2848.152 u, Fig. 2E) differed by −380.328 u from the 4 main ion signals of the LPS from the HB-1 strain. This is accordance with lack of a C12 and a C12OH in the lipid A of the ΔlpxL1-pagL mutant (Δcalc. = −380.329 u). The absence of ion signals corresponding to LPS carrying two C12 acyl chains indicates that the deletion of the lpxL1 gene resulted in complete removal of a single C12 from the lipid A (see Supplementary Table 1 for detailed LPS composition proposals). In contrast, minor ion signals of 3226.339 and 3169.319 u were present in the mass spectrum of the LPS from the ΔlpxL1-pagL mutant, which correspond to penta-acyl LPS species carrying two C12OH acyl chains. This indicates that a low level of LPS molecules was not 3-O-deacylated by the PagL enzyme.
The mass spectrum of the LPS from the ΔlpxL2-pagL mutant showed an ion peak of 2825.206 u (Fig. 2F) that was shifted by −583.301 u from the ion signal of 3408.507 u of the mass spectrum of the LPS from the parent HB-1 strain (Fig. 2A). This fits the expected loss of a C12OH, a C12 and PPEA from the lipid A (Δcalc. = −583.304 u). Other ion signals of 2768.187, 2930.236 and 2987.257 u (Fig. 2F) are consistent with non-stoichiometric substitution of the oligosaccharide with Gly or a second Hex.
Comparison of the mass spectrum of the LPS from the ΔlpxL1-lpxP mutant grown at 30 °C (Fig. 2G) with that of the LPS from the ΔlpxL1 mutant (Fig. 2B) revealed that the LPS from the ΔlpxL1-lpxP mutant contained not only the main LPS species that were present in the LPS from the ΔlpxL1 mutant (3046.315, 3103.333, 3169.322 and 3226.340 u, Fig. 2G), corresponding to penta-acyl LPS lacking a C12, but also LPS species (3282.524, 3339.543, 3405.533 and 3462.553 u, Fig. 2G) that shifted in the spectrum to higher mass values by 236.211 u. This is in agreement with incorporation of a 9-hexadecenoic acid (C16:1) to the lipid A. Therefore, this preparation comprised a mixture of penta-acyl LPS that lacks a C12 and hexa-acyl LPS that lacks a C12 and additionally carry a C16:1.
The mass spectrum of the LPS from the ΔlpxL1-lpxP mutant cultured at 25 °C (Fig. 2H) showed ion signals corresponding to hexa-acyl LPS lacking a C12 and carrying additionally a C16:1 (3282.526, 3339.546, 3405.535 and 3462.554 u, Fig. 2H), which were of a higher relative abundance as compared to the same signals in the spectrum of the LPS from the ΔlpxL1-lpxP mutant grown at 30 °C. Furthermore, other ion peaks corresponding to hexa-acyl LPS carrying a C16:1 were displayed which arose from elongation of the oligosaccharide with a second Hex (3624.608 u) or the latter in combination with the loss of Gly substitution (3567.586 u) and the loss of a PEA group from the lipid A (3501.596 u) (Fig. 2H).
The ion peak of 3162.489 u in the mass spectrum of the LPS from the ΔlptA mutant (Fig. 2I) differed by −246.018 u from the ion signal of 3408.507 u of the mass spectrum of the LPS from the parent HB-1 strain (Fig. 2A). This points to the loss of two PEA groups from the lipid A (Δcalc. = −246.017 u). Other ion signals corresponded to LPS species that in addition to lacking PEA in the lipid A either lacked Gly in the oligosaccharide (3105.471), contained a second Hex in the oligosaccharide (3324.541) or contained a second Hex and lacked Gly in the oligosaccharide (3267.521 u) (Fig. 2I).
The mass spectrum of the LPS from the ΔlptA-ΔlpxL1 mutant displayed ion peaks of 2980.324, 2923.307, 3142.375 and 3085.354 u indicating the loss of 2PEA and a C12 from the lipid A (Δcalc. = −428.184 u) combined with non-stoichiometric substitution of the oligosaccharide with Gly or a second Hex (Fig. 2J).
The main ion signals of the mass spectrum of the LPS from the ΔlptA-pagL mutant (2964.328 and 2907.311 u, Fig. 2K) are consistent with the loss of 2PEA and a C12OH from the lipid A (Δcalc. = −444.179 u) together with non-stoichiometric substitution of the oligosaccharide with Gly (Δcalc. = 57.021 u). Minor ion peaks of 3105.468 and 3162.488 u were observed corresponding to hexa-acyl LPS species which lost only 2PEA from the lipid A, indicating a low level of incomplete LPS 3-O-deacylation by the PagL enzyme.
Finally, the mass spectrum of the LPS from the ΔlptA-lpxE mutant showed 2 main ion peaks of 3082.525 and 3025.508 u consistent with loss of 2PEA and P from the lipid A (Δcalc. = −325.983 u) in combination with non-stoichiometric substitution of the oligosaccharide with Gly (Fig. 2L). In addition, MS/MS spectra of the main lipid A fragment ion produced by in-source collision-induced dissociation of LPS were consistent with the presence of a P group at both the 1 and 4′ positions of the lipid A (data not shown). Therefore, the activity of the LpxE enzyme consisted in removal of one of the three P groups present in lipid A producing bisphosphorylated lipid A species with a P group on each side of the diglucosamine backbone.
TLR4 stimulation by the LPS mutant strains
To determine the scope of TLR4 activation by the entire set of lipid A mutant structures, an initial screening was done using HEK-Blue human TLR4 cells. These cells express human TLR4, MD-2, and CD14 and contain a nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and activator protein 1 (AP-1) dependent secreted embryonic alkaline phosphatase (SEAP) reporter gene. Stimulation of cells with serial dilutions of the different LPS mutants yielded a wide range of TLR4 activities (Fig. 3), with HB-1 inducing strongest TLR4 activation and ΔLpxL2 bacteria yielding lowest levels of activation. The other LPS mutants showed intermediate TLR4 stimulating activity (Fig. 3). A particularly notable result was that the absence of phosphoethanolamine in the ΔlptA strain resulted in reduced TLR4 activation in both the hexa-acylated wild type strain and the penta-acylated ΔlpxL1 and pagL backgrounds. Induction of LpxE in the ΔlptA strain showed similar TLR4 activation as ΔlptA strain, which was slightly less than the HB-1 wild type strain. The expression of LpxE in the ΔlptA strain did not show major additional effects compared to the ΔlptA mutant strain thus indicating that the reduction of three phosphates to two in the lipid A structure did not have a major effect on TLR4 signaling.

TLR4 activation by N. meningitidis strains.
HEK-blue hTLR4 cells were stimulated with 5-fold serial dilutions of heat-inactivated N. meningitidis for 20 h. TLR4 activation was measured by detection of secreted alkaline phosphatase. Results of serial dilutions are depicted in a line graph (A) and for a single OD600 nm of 0.0004 in a bar graph (B). Data are expressed as mean values or mean ± SD of three independent experiments. Statistical significance was determined with an ANOVA test comparing against HB-1. *p < 0.05.; **p < 0.001.
Expression of LpxP at 25 °C in combination with deletion of LpxL1 resulted in a heterogeneous hexa- and penta-acylated structure-LPS expressing strain with a slightly reduced TLR4 activating potential compared to the wild type bacteria. Cultivation of this strain at 30 °C resulted in less hexa-acylated lipid A and even slightly less TLR4 activity.
Surprisingly, when the ΔlpxL1 strain was combined with expression of PagL, reducing the penta-acylated lipid A structure to a tetra-acylated form, an increase of TLR4 activity was obtained. This was unexpected as tetra-acylated lipid A structures typically act as a TLR4 antagonists as reported for E. coli lipid IVa7,9,19. Interestingly, PagL-mediated deacylation of wildtype LPS instead reduced its activity.
Human TLR4 stimulation using purified mutant LPS
We also purified LPS from all strains and used the molecules to stimulate HEK-Blue TLR4 cells to confirm our initial findings with whole bacteria. Purified LPS generally yielded similar results as those obtained with intact bacteria although purified LPS from ΔlpxL1, ΔlptA-ΔlpxL1, ΔlpxL2 and ΔlpxL2-pagL mutants showed almost no induction of TLR4 activity and were barely distinguishable from each other (Fig. 4), whereas the corresponding bacteria displayed low but distinct TLR4 activities above the background. In addition, a higher concentration of purified penta-acylated pagL LPS was needed for activation of TLR4 than with all the hexa-acylated LPS derivatives, but with whole bacteria stimulation, a lower optical density was necessary for the pagL strain to induce TLR4 activity than the other hexa-acylated mutant strains (Figs 3 and 4). However, the maximum amount of alkaline phosphatase secretion was still lower for the pagL mutant strain compared to the hexa-acylated mutant strains. Of note, the three LPS mutants ΔlpxL1-pagL, pagL and ΔlptA-pagL had substantially reduced activating capabilities when compared to the wild type LPS, but still induced activation above the background level of unstimulated cells (Fig. 4).

TLR4 activation by purified LPS.
HEK-blue hTLR4 cells were stimulated with 10-fold serial dilutions of 12 different LPS mutants. TLR4 activation was measured by detection of secreted alkaline phosphatase. Data shown are from one representative experiment out of three independent experiments with similar outcomes and are depicted as the mean ± SEM of triplicates.
Cytokine induction by the purified mutant LPS
The cytokine induction profile of the modified LPS structures was investigated in the human monocytic cell line Mono Mac 6 (MM6). The concentration of secreted MyD88 dependent cytokines IL-6 (Fig. 5A) and IL-1β (Fig. 5B) and TRIF dependent cytokines interferon gamma-induced protein 10 (IP-10) (Fig. 5C) and monocyte chemotactic protein-1 (MCP-1) (Fig. 5D) were determined after 20 h of stimulation with purified LPS (Fig. 5). The possible contribution of minor protein contamination in LPS samples to the observed responses was excluded as activation of a HEK-hTLR2 cell line by the LPS samples was negligible in the range of LPS concentrations tested (data not shown).

Cytokine release of MM6 cells stimulated with purified LPS.
MM6 cells were incubated with 10-fold serial dilution of different LPS mutants for 20 h. IL-6 (A), IL-1β (B), IP-10 (C), MCP-1 (D) production was measured by ELISA. IL-6 and IL-1β are considered MyD88 dependent cytokines and IP-10 and MCP-1 are more TRIF dependent. Cytokine levels of MM6 cells stimulated with 5 ng/ml LPS are also presented as percentages of the HB-1 strain (E) and cytokine ratios in concentration (F) and percentages (G). For the cytokine ratios (F,G) the background without LPS stimulation was subtracted. Data shown are depicted as the mean values of two independent experiments. Statistical significance was determined with a 2-way ANOVA test comparing against HB-1. *p < 0.05.
A wide variety of cytokine levels was determined from the different LPS structures, with the highest levels being produced by the HB-1 wild type hexa-acylated LPS and all other LPS ranging from close to wild type until virtually zero cytokine induction as seen for ΔlpxL2 LPS. Besides quantitative differences in cytokine induction, we also observed qualitative differences with LPS structures causing reduced levels of certain cytokines, but still capable of producing others. Some examples are pagL and ΔlptA-pagL LPS, which displayed a reduced capacity to induce the production of MyD88 dependent pro-inflammatory cytokines IL-6 and IL-1β (10% or less of the levels induced by wild-type LPS, respectively), but retained most of the ability to induce the secretion of TRIF dependent IP-10 (~50%) and MCP-1 (~90%). Interestingly, differences were observed between ΔlpxL1-lpxP grown at 30 °C and 25 °C, with ΔlpxL1-lpxP grown at 30 °C producing 20–30% IL-6 and IL-1β and 60–80% of those cytokines at 25 °C, whereas IP-10 and MCP-1 induction were similar. These results emphasize how LPS bioengineering can provide a wide range of agonists to fine-tune cytokine release.
Discussion
Although LPS has great potential as an adjuvant, adverse effects keep being a concern. Finding the optimal balance between adjuvant activity and minimal toxic effects requires the development of new LPS derivatives. Here we report a collection of novel meningococcal LPS structures inducing a broad range of TLR4 responses and differential cytokine patterns. These combinatorial bioengineered LPS mutants can be used as part of a whole cell vaccine, OMV vaccine or as purified LPS or lipid A molecule. OMVs of N. meningitidis are being actively investigated as potential vaccines and have been already approved for use in humans as a component of the Bexsero vaccine against serogroup B meningococcal disease20,21. Attenuated ΔlpxL1 LPS is under investigation as constituent of meningococcal OMV vaccines and combinatorial bioengineering of LPS is a safe method to detoxify OMVs16,22. In addition, in an immunization study in mice purified ΔlpxL1 LPS retained similar adjuvant activity compared to wild type meningococcal LPS, but with reduced toxicity15.
The modified LPS molecules ΔlpxL1, ΔlpxL2 and pagL all result in a reduced TLR4 activity compared to the parent strain13,15. This was expected because of the reduction of the number of acyl chains in the LPS from hexa to penta. Surprisingly, the expectation that tetra-acylated LPS is always less active than penta-acylated LPS is challenged by our results. Tetra-acylated lipid IVa of E. coli is a known antagonist of the human TLR4/MD-2 complex7,9,19. Yet, we show that meningococcal tetra-acylated ΔlpxL1-pagL LPS is more active than the penta-acylated ΔlpxL1 LPS, whereas tetra-acylated ΔlpxL1-ΔlpxL2 LPS did not yield detectable activity (data not shown). Stimulation with ΔlpxL2-pagL whole bacteria that also carry a tetra-acylated LPS again increased TLR4/MD-2 activity compared to its penta-acylated ΔlpxL2 parent strain, although purified LPS from both the ΔlpxL2-pagL and ΔlpxL2 were inactive. Together these findings indicate that removal of C12OH from the 3-position by PagL in combination with deletion of a secondary acyl chain resulting in tetra-acylated lipid A yields a higher TLR4 activity compared to sole removal of the secondary acyl chain or both secondary acyl chains. By contrast, PagL deacylation in wildtype LPS results in a decrease of TLR4 activity. LPS structures engineered in E. coli by Needham et al.23 included a tetra-acylated LPS consistently being more active than its penta-acylated parent strain. Although this structure had a different acyl chain distribution and chain length than our tetra-acylated structure ΔlpxL1-pagL, through expression of PagL and deletion of LpxM it also resulted in removal of C12OH from the 3-position and deletion of a secondary acyl chain, respectively.
Interestingly, introduction of LpxP from E. coli into the N. meningitidis lpxL1 strain conferred a temperature-sensitive lipid A modification to N. meningitidis, with a secondary C12 being replaced by C16:1. Since conservation of temperature-sensitive gene expression signals is unlikely, this means that the enzyme itself is most active at lower temperatures. Selection of a temperature of 25 or 30 °C for culture of the ΔlpxL1-lpxP strain influenced the amount of hexa-acylated LPS species present in the mixture of penta- and hexa-acylated LPS produced by this mutant, with the lower temperature leading to the highest degree of substitution. The temperature sensitivity of the LpxP enzyme thus enables the preparation of penta- and hexa-acylated LPS mixtures in a controlled manner. By selecting the time and/or temperature that the mutant strain is grown, it is feasible to increase or decrease the amount of hexa-acylated lipid A structure and thereby the TLR4 activity and cytokine profile. This provides a new approach of fine-tuning the immunological properties of meningococcal OMV vaccines.
In addition, we have obtained new insight in the specificity of the LpxE enzyme. Previously, the lpxE gene from Francisella tularensis or Francisella novicida expressed in E. coli was shown to be specific for the removal of the P group in the 1-position23,24. We have found that the lpxE homologue from B. bronchiseptica removed only one P group from the total of three present in the lipid A of N. meningitidis. MS/MS spectra of the lipid A from ΔlptA-lpxE mutants were consistent with the presence of a P group at both the 1 and 4′ positions of the lipid A. In addition, removal of the P group was only seen in double ΔlptA-lpxE mutants, therefore only in the absence of PEA substitution of the lipid A. Thus, it is likely that the presence of PEA prevents LpxE from removing the P group. Most likely, the newly described LpxE enzyme is a pyrophosphatase, only catalyzing hydrolysis between two phosphate groups. The absence of PEA in the lipid A through deletion of the lptA gene resulted in a reduced TLR4/MD-2 activity. This concurs with earlier observations by John et al.25 that show a significant reduction of TNFα release by THP-1 cells upon stimulation with LptA lacking strains. Here we showed that reduction of the activity is even more apparent when stimulated with penta-acylated ΔlptA-pagL LPS or whole bacteria.
Interestingly, our results indicate that the absence of a C12 fatty acyl chain by deletion of LpxL2 is accompanied by loss of a single P group and PEA group. Possibly, loss of this secondary acyl chain may interfere with efficient P or PEA addition. This was not observed in previous structural analysis of the lpxL2 mutant, due to isolation of lipid A by an acid hydrolysis method before mass spectrometric analysis, which can result in the loss of P groups from the lipid A15. In the present study, we used complete LPS molecules without introducing any deleterious chemical modifications for mass spectrometric analysis, giving us the possibility to observe new phosphorylation changes of the lipid A.
Several of the constructed attenuated LPS structures did not only need a higher concentration to induce TLR4 stimulation, but also did not reach the maximum level of activation observed for the parent strain. This was most apparent for pagL LPS. The reason for this phenomenon is unclear, but could be due to instable dimerization of the LPS-TLR4-MD2 receptor complex at the cell surface but stable dimerization inside the cell, and/or to a less stable dimerization with high concentrations of the particular LPS. In addition, certain LPS species showed no activation at all and could potentially have antagonistic features, and might therefore serve as a TLR4 blocking drug. Indeed, meningococcal ΔlpxL1, ΔlpxL2 and pagL penta-acylated LPS can block the TLR4 response when administered together with hexa-acylated wild type meningococcal or E. coli LPS13,26.
In the present study, we have used combinatorial bioengineering in meningococci to produce a range of LPS species with a broad array of TLR4 activity and cytokine profile. Together, they cover the whole range of activity from wildtype maximum to practically zero (Fig. 4). The application of these structures can be very broad, from inclusion into vaccines as adjuvants to their use in various forms of immunotherapy. OMVs from the mutant strains can be isolated, using the modified LPS as an improved internal adjuvant. In addition, they can enable innovative forms of immunotherapy which have been described or suggested for LPS, such as cancer therapy, Alzheimer’s disease treatment or generalized immune stimulation to prevent diverse infections3,27,28,29.
Methods
Bacterial strains and plasmids
All mutants were created in a N. meningitidis strain HB-1, a capsule-deficient derivative of strain H44/76 obtained by transformation with plasmid pMF121, resulting in deletion of the capsular biosynthesis locus including the galE gene. N. meningitidis strains were grown on GC medium base (Difco) plates supplemented with IsoVitaleX, in a humid atmosphere containing 5% CO2 at 37 °C. For liquid culture, strains were grown in 36 mg/mL tryptic soy broth medium (Difco) in a conical flask at 37 °C, shaken at 140 RPM. Required antibiotics were added to plate and liquid cultures (kanamycin 100 μg/ml, chloramphenicol 3 μg/ml). The lpxL1 and lpxL2 mutants were obtained by transformation with a linearized pCRII plasmid (Invitrogen) carrying the genes disrupted with a kanamycin resistance cassette described by van der Ley et al.15 or a pGem-T easy plasmid (Promega) with the lpxL1 gene that has a deleted section replaced with a chloramphenicol (CAM) cassette. For construction of the lptA mutant the gene was amplified by PCR from H44/76, cloned into a pGem-T easy plasmid (Promega) and a kanamycin cassette was inserted at the MunI restriction site. The resulting plasmid was linearized and used for transformation of N. meningitidis strain HB-1. N. meningitidis derivatives carrying the heterologous genes pagL, lpxP and lpxE were created using the pEN11 shuttle plasmid previously described for the expression of the Bordetella bronchiseptica pagL gene13,30. To obtain lpxP and lpxE derivatives, the genes were amplified by PCR from E. coli and B. bronchiseptica, respectively, and subsequently cloned into pEN11 by replacing the pagL gene using restriction enzymes. Expression of heterologous genes cloned in pEN11 was induced by addition of 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) to the liquid culture medium. Primers are listed in Table 1.
LPS isolation
LPS from bacterial mutants was extracted with hot phenol-water31 and purified further by solid phase extraction (SPE) on reverse phase cartridges. In short, cells from 50 ml of bacterial culture with an OD600 nm of 1.4 (or 100 ml of the ΔlpxL1-lpxP mutant grown at 30 °C) were collected by centrifugation at 2,739 × g for 1 h at 20 °C. Then, bacteria were suspended in 20 ml of water and centrifuged at 2,739 × g for 25 min at 20 °C. For hot phenol-water extraction, bacterial pellets were suspended with 4 ml of water, heated to 70 °C, mixed with 3.2 ml of phenol at the same temperature and kept under agitation for 10 min at 70 °C. The aqueous phase was separated from the phenolic phase by centrifugation at 2,739 × g for 15 min at 20 °C. After transferring the aqueous phase to a new vial, the phenolic phase was extracted again by adding 3 ml of water at 70 °C and repeating the extraction procedure. The aqueous phases from two consecutive extractions were pooled (∼6.5 ml) and prepared for SPE by adding 5 ml of 0.356 M triethylammonium acetate (TEAA) pH 7 (solvent A) and 3.8 ml of 2-propanol:water:triethylamine:acetic acid (70:30:0.03:0.01, v/v) pH 8.7 (solvent B). In total, ten LPS extracts each from a different bacterial mutant could be purified simultaneously by SPE on reverse phase Sep-Pak C18 cartridges (1 ml syringe-barrel-type Vac cartridge, 50 mg of C18 resin, Waters) using a 20-position vacuum manifold (Waters). Cartridges were conditioned for SPE by applying consecutively 1 ml of 2-propanol:water:triethylamine:acetic acid (85:15:0.015:0.005, v/v) pH 8.7 (solvent C), 0.07 mM TEAA pH 7 (solvent D) and solvent A under vacuum. Then, samples were split into two aliquots of equal volume and each aliquot was applied into a different cartridge. Next, cartridges were washed once with 1 ml of solvent A and twice with 1 ml of 20% (v/v) solvent B in solvent D. LPS was eluted from the columns by applying 0.6 ml of solvent C. Eluates from the same sample were combined (1.2 ml per sample in total) and dried in a centrifugal vacuum concentrator (Concentrator plus, Eppendorf) at room temperature. LPS concentration in isolated samples was determined by the 3-deoxy-d-manno-oct-2-ulosonic acid (Kdo) assay32. In addition, the purity and integrity of purified samples were judged by Tricine-SDS-PAGE using 1 mm-thick, 16% precast Novex® mini-gels (Thermo Fisher Scientific Inc.), LPS silver staining33 and protein visualization with ImperialTM Protein Stain (Thermo Scientific).
Mass spectrometry
Electrospray ionization Fourier transform mass spectrometry (ESI-FT-MS) was performed on an LTQ Orbitrap XL instrument (Thermo Scientific) in negative ion mode. LPS samples were dissolved in a mixture of water, 2-propanol and triethylamine (50:50:0.001, by volume) pH 8.5 and infused into the mass spectrometer by static nano-ESI34,35. The MS instrument was calibrated with a Pierce Negative Ion Calibration Solution (Thermo Scientific) and internally with taurocholic acid following standard procedures provided by the manufacturer (Thermo Scientific). Fragmentation analysis of intact LPS was carried out by in-source collision-induced fragmentation (SID). Y- and B-type fragment ions, corresponding to the lipid A and oligosaccharide moieties of LPS, respectively, were generated by SID at a potential difference of 100 V. Fragment ions are annotated according to the nomenclature of Domon and Costello36. Mass spectra were charge-deconvoluted using the Xtract tool of Thermo Xcalibur 3.0 software (Thermo Scientific). All mass values given refer to monoisotopic molecular masses. Proposed LPS compositions are based on the general chemical structure of the L3 immunotype LPS from N. meningitidis reported previously37,38.
Cell stimulation
Mono Mac 6 cells were seeded at 1 × 105 cells per well in 96 well microtiter plates in 100 μl Iscove’s modified Dulbecco’s medium (IMDM) (Gibco, ThermoFisher scientific) medium supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, 292 μg/ml l-glutamine (Gibco, ThermoFisher scientific), and 10% fetal calf serum (Gibco, ThermoFisher scientific). Hek blue-hTLR4 cells (Invivogen), a HEK293 cell line stably expressing human TLR4, MD-2 and CD14, were seeded at 3.5 × 104 cells per well in 96-well microtiter plates in 100 μl DMEM (Gibco, ThermoFisher scientific) medium supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, 292 μg/ml l-glutamine (Gibco, ThermoFisher scientific) and 10% fetal calf serum (Gibco, ThermoFisher scientific). Cells were stimulated with 10-fold serial dilutions of LPS in IMDM (MM6 cells) or DMEM (HEK blue-hTLR4 cells) for 18–20 h at 37 °C in a humid atmosphere containing 5% CO2. HEK-blue-hTLR4 cells were also stimulated with serial dilution of heat-inactivated (1 h at 56 °C) whole bacterial cells. Cytokine concentration in the supernatants of MM6 cells was determined by enzyme-linked immunosorbent assay (ELISA). All cytokine (IL-6, IL-1β, IP-10, MCP-1) concentrations were determined using a DUOset ELISA development kit (R&D systems) following the manufacturer’s instructions. To quantify alkaline phosphatase secreted by HEK-blue-hTLR4 cells, 20 μl of the supernatant from each well was added to 200 μl Quanti-blue (Invivogen) and incubated at 37 °C for 2–3 hours. Read out was done on a spectrophotometer at 649 nm. Statistically significant differences were determined by the one-way (alkaline phosphatase secretion) or two-way (cytokine release) ANOVA test by using GraphPad Prism 6.04 statistical software (GraphPad Software, Inc.).
Additional Information
How to cite this article: Zariri, A. et al. Modulating endotoxin activity by combinatorial bioengineering of meningococcal lipopolysaccharide. Sci. Rep. 6, 36575; doi: 10.1038/srep36575 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Aderem, A. & Ulevitch, R. J. Toll-like receptors in the induction of the innate immune response. Nature 406, 782–787, doi: 10.1038/35021228 (2000).
Loppnow, H. et al. IL-1 induction-capacity of defined lipopolysaccharide partial structures. Journal of immunology 142, 3229–3238 (1989).
Zariri, A. & van der Ley, P. Biosynthetically engineered lipopolysaccharide as vaccine adjuvant. Expert review of vaccines 14, 861–876, doi: 10.1586/14760584.2015.1026808 (2015).
Akira, S. & Takeda, K. Toll-like receptor signalling. Nature reviews. Immunology 4, 499–511, doi: 10.1038/nri1391 (2004).
Raetz, C. R. & Whitfield, C. Lipopolysaccharide endotoxins. Annual review of biochemistry 71, 635–700, doi: 10.1146/annurev.biochem.71.110601.135414 (2002).
Salomao, R. et al. Bacterial sensing, cell signaling, and modulation of the immune response during sepsis. Shock 38, 227–242, doi: 10.1097/SHK.0b013e318262c4b0 (2012).
Park, B. S. et al. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458, 1191–1195, doi: 10.1038/nature07830 (2009).
Raetz, C. R., Reynolds, C. M., Trent, M. S. & Bishop, R. E. Lipid A modification systems in gram-negative bacteria. Annual review of biochemistry 76, 295–329, doi: 10.1146/annurev.biochem.76.010307.145803 (2007).
Ohto, U., Fukase, K., Miyake, K. & Shimizu, T. Structural basis of species-specific endotoxin sensing by innate immune receptor TLR4/MD-2. Proceedings of the National Academy of Sciences of the United States of America 109, 7421–7426, doi: 10.1073/pnas.1201193109 (2012).
Gandhapudi, S. K., Chilton, P. M. & Mitchell, T. C. TRIF is required for TLR4 mediated adjuvant effects on T cell clonal expansion. PloS One 8, e56855, doi: 10.1371/journal.pone.0056855 (2013).
Mata-Haro, V. et al. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science 316, 1628–1632, doi: 10.1126/science.1138963 (2007).
Casella, C. R. & Mitchell, T. C. Putting endotoxin to work for us: monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cellular and molecular life sciences : CMLS 65, 3231–3240, doi: 10.1007/s00018-008-8228-6 (2008).
Pupo, E., Hamstra, H. J., Meiring, H. & van der Ley, P. Lipopolysaccharide engineering in Neisseria meningitidis: structural analysis of different pentaacyl lipid A mutants and comparison of their modified agonist properties. The Journal of biological chemistry 289, 8668–8680, doi: 10.1074/jbc.M114.554345 (2014).
Zughaier, S. M. et al. Physicochemical characterization and biological activity of lipooligosaccharides and lipid A from Neisseria meningitidis. Journal of endotoxin research 13, 343–357, doi: 10.1177/0968051907084435 (2007).
van der Ley, P. et al. Modification of lipid A biosynthesis in Neisseria meningitidis lpxL1 mutants: influence on lipopolysaccharide structure, toxicity, and adjuvant activity. Infection and immunity 69, 5981–5990, doi: 10.1128/IAI.69.10.5981-5990.2001 (2001).
van de Waterbeemd, B. et al. Improved OMV vaccine against Neisseria meningitidis using genetically engineered strains and a detergent-free purification process. Vaccine 28, 4810–4816, doi: 10.1016/j.vaccine.2010.04.082 (2010).
Steeghs, L. et al. Differential activation of human and mouse Toll-like receptor 4 by the adjuvant candidate LpxL1 of Neisseria meningitidis. Infection and immunity 76, 3801–3807, doi: 10.1128/IAI.00005-08 (2008).
Carty, S. M., Sreekumar, K. R. & Raetz, C. R. Effect of cold shock on lipid A biosynthesis in Escherichia coli. Induction at 12 degrees C of an acyltransferase specific for palmitoleoyl-acyl carrier protein. The Journal of biological chemistry 274, 9677–9685 (1999).
Golenbock, D. T., Hampton, R. Y., Qureshi, N., Takayama, K. & Raetz, C. R. Lipid A-like molecules that antagonize the effects of endotoxins on human monocytes. The Journal of biological chemistry 266, 19490–19498 (1991).
Findlow, J. et al. Multicenter, open-label, randomized phase II controlled trial of an investigational recombinant meningococcal serogroup B vaccine with and without outer membrane vesicles, administered in infancy. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America 51, 1127–1137, doi: 10.1086/656741 (2010).
Snape, M. D. et al. Immunogenicity of two investigational serogroup B meningococcal vaccines in the first year of life: a randomized comparative trial. The Pediatric infectious disease journal 29, e71–e79, doi: 10.1097/INF.0b013e3181f59f6d (2010).
Keiser, P. B. et al. A phase 1 study of a meningococcal native outer membrane vesicle vaccine made from a group B strain with deleted lpxL1 and synX, over-expressed factor H binding protein, two PorAs and stabilized OpcA expression. Vaccine 29, 1413–1420, doi: 10.1016/j.vaccine.2010.12.039 (2011).
Needham, B. D. et al. Modulating the innate immune response by combinatorial engineering of endotoxin. Proceedings of the National Academy of Sciences of the United States of America 110, 1464–1469, doi: 10.1073/pnas.1218080110 (2013).
Wang, X., Karbarz, M. J., McGrath, S. C., Cotter, R. J. & Raetz, C. R. MsbA transporter-dependent lipid A 1-dephosphorylation on the periplasmic surface of the inner membrane: topography of francisella novicida LpxE expressed in Escherichia coli. The Journal of biological chemistry 279, 49470–49478, doi: 10.1074/jbc.M409078200 (2004).
John, C. M. et al. Lack of lipid A pyrophosphorylation and functional lptA reduces inflammation by Neisseria commensals. Infection and immunity 80, 4014–4026, doi: 10.1128/IAI.00506-12 (2012).
Sprong, T. et al. Neisseria meningitidis lipid A mutant LPSs function as LPS antagonists in humans by inhibiting TLR 4-dependent cytokine production. Innate immunity 17, 517–525, doi: 10.1177/1753425910383999 (2011).
Michaud, J. P. et al. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer’s disease-related pathology. Proceedings of the National Academy of Sciences of the United States of America 110, 1941–1946, doi: 10.1073/pnas.1215165110 (2013).
Peri, F. & Piazza, M. Therapeutic targeting of innate immunity with Toll-like receptor 4 (TLR4) antagonists. Biotechnology advances 30, 251–260, doi: 10.1016/j.biotechadv.2011.05.014 (2012).
Adams, S. Toll-like receptor agonists in cancer therapy. Immunotherapy 1, 949–964, doi: 10.2217/imt.09.70 (2009).
Geurtsen, J. et al. Expression of the lipopolysaccharide-modifying enzymes PagP and PagL modulates the endotoxic activity of Bordetella pertussis. Infection and immunity 74, 5574–5585, doi: 10.1128/IAI.00834-06 (2006).
Westphal, O. & Jann, K. In Methods in Carbohydrate Chemistry Vol. 5 (eds R. L. Whistler & M. L. Wolfan ) 83–91 (Academic Press, Inc., 1965).
Karkhanis, Y. D., Zeltner, J. Y., Jackson, J. J. & Carlo, D. J. A new and improved microassay to determine 2-keto-3-deoxyoctonate in lipopolysaccharide of Gram-negative bacteria. Analytical biochemistry 85, 595–601 (1978).
Tsai, C. M. & Frasch, C. E. A sensitive silver stain for detecting lipopolysaccharides in polyacrylamide gels. Analytical biochemistry 119, 115–119 (1982).
Kondakov, A. & Lindner, B. Structural characterization of complex bacterial glycolipids by Fourier transform mass spectrometry. European journal of mass spectrometry 11, 535–546, doi: 10.1255/ejms.721 (2005).
Wilm, M. S. & Mann, M. Electrospray and Taylor-Cone theory, Dole’s beam of macromolecules at last? Int J Mass Spectrom Ion Proc 136, 167–180, doi: 10.1016/0168-1176(94)04024-9 (1994).
Domon, B. & Costello, C. E. A Systematic Nomenclature for Carbohydrate Fragmentations in FAB-MS/MS Spectra of Glycoconjugates. Glycoconjugate J 5, 397–409, doi: 10.1007/BF01049915 (1988).
Pavliak, V., Brisson, J. R., Michon, F., Uhrin, D. & Jennings, H. J. Structure of the sialylated L3 lipopolysaccharide of Neisseria meningitidis. The Journal of biological chemistry 268, 14146–14152 (1993).
van der Ley, P., Kramer, M., Martin, A., Richards, J. C. & Poolman, J. T. Analysis of the icsBA locus required for biosynthesis of the inner core region from Neisseria meningitidis lipopolysaccharide. FEMS microbiology letters 146, 247–253 (1997).
Author information
Authors and Affiliations
Contributions
A.Z. and P.L. conceived and designed the study. A.Z. designed and performed the experiments and prepared the figures for publication. E.P. purified LPS and did the mass spectrometry analysis. E.R., J.P.M.P. and P.L. helped analyze the data and contributed ideas for the research. A.Z. and E.P. wrote the manuscript. P.L., J.P.M.P., E.R. and E.P. revised the manuscript and all authors approved the final version of the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zariri, A., Pupo, E., van Riet, E. et al. Modulating endotoxin activity by combinatorial bioengineering of meningococcal lipopolysaccharide. Sci Rep 6, 36575 (2016). https://doi.org/10.1038/srep36575
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep36575
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
