
Abstract
Among Candida species, the opportunistic fungal pathogen Candida glabrata has become the second most common causative agent of candidiasis in the world and a major public health concern. Yet, few molecular tools and resources are available to explore the biology of C. glabrata and to better understand its virulence during infection. In this study, we describe a robust experimental strategy to generate loss-of-function mutants in C. glabrata. The procedure is based on the development of three main tools: (i) a recombinant strain of C. glabrata constitutively expressing the CRISPR-Cas9 system, (ii) an online program facilitating the selection of the most efficient guide RNAs for a given C. glabrata gene, and (iii) the identification of mutant strains by the Surveyor technique and sequencing. As a proof-of-concept, we have tested the virulence of some mutants in vivo in a Drosophila melanogaster infection model. Our results suggest that yps11 and a previously uncharacterized serine/threonine kinase are involved, directly or indirectly, in the ability of the pathogenic yeast to infect this model host organism.
Similar content being viewed by others


Introduction
Due to the growing population of patients with weakened immune systems and to modern medical practices, such as the widespread use of catheters or of antibacterials and antifungals1,2,3, the incidence of fungal infections has increased at an alarming rate in the past two decades. Recent estimates suggest that about 1.2 billion people worldwide suffer from a fungal infection4. Human fungal infections account for more deaths annually than either tuberculosis or malaria, most of them caused by species belonging to four genera of fungi: Aspergillus, Candida, Cryptococcus, and Pneumocystis5. Among Candida species, the opportunistic fungal pathogen Candida glabrata, a nondimorphic, haploid budding yeast, has become the second most common causative agent of candidiasis in the world. Whereas C. albicans ranks first in isolation frequency, C. glabrata is more resistant to antifungal therapy and is associated with higher mortality6,7. As a result, C. glabrata has become a major public health concern during the past two decades. This reinforces the need to develop innovative therapeutic strategies to circumvent its high rates of resistance to azole antifungal compounds5.
The development of novel strategies requires a heightened understanding of C. glabrata infection. Indeed, whereas C. albicans aggressively attacks the host, C. glabrata seems to have developed a process based on a ‘stealth-like’ approach. Actually, key features of C. albicans pathogenicity, such as the formation of hyphae, mating, and the secretion of proteinases, have no equivalent in C. glabrata. Since its sequenced genome places it phylogenetically closer to S. cerevisiae than to C. albicans8, it has been proposed that the ability of C. glabrata to infect humans has evolved independently from that of other Candida species9,10. C. glabrata presents peculiar features related to cell wall organization such as an exceptionally high number of genes encoding adhesion-like glycosylphosphatidylinositol (GPI)-anchored proteins11 or the implication of GPI-anchored aspartyl proteases (a.k.a. yapsins) in the infection process12,13. These features represent key virulence factors, with roles in the high tolerance to azole drugs7, in the adhesion to host cells or in survival inside macrophages (reviewed in14). The functional annotation of the genome of C. glabrata has also revealed the loss of specific genes, such as those needed for galactose, phosphate, nicotinic acid or allantoin metabolism9,15,16.
Despite the characterization of these specific traits, the molecular mechanisms underlying C. glabrata infectivity are far from being completely understood. Unfortunately, few molecular tools and resources are available to explore its biology in comparison to those accessible for the S. cerevisiae model organism17. Among the techniques available to investigate gene function in living organisms, genetic approaches (gene disruption/replacement, RNA interference…) have been extensively employed. Systems like homologous recombination or Tn7-based genome-wide random insertion mutagenesis have already been successfully applied to C. glabrata18,19. The recent discovery of the CRISPR-Cas9 system in bacteria and archaea has provided a new and extremely efficient approach to inactivate gene function20. In this system, a Cas9 endonuclease is guided to specific genomic locations by a short single-guide RNA (sgRNA). Cas9 cleaves double-stranded DNA in a site-specific manner, and thereby activates the double strand break repair machinery (a.k.a. Non Homologous End Joining repair (NHEJ)), resulting in insertions or deletions (indels) that ultimately disrupt the targeted locus. Whereas the CRISPR-Cas9 system has initially been selected during evolution to perform gene disruption in bacteria, several groups have extended its application to RNA interference or in situ labeling21,22,23. Today, the CRISPR-Cas9 system is successfully applied to numerous organisms, from bacteria24 to human25 and animals26 through protozoan parasites27 and fungal pathogens28,29,30.
Here, we have established an efficient CRISPR-Cas9 system in C. glabrata to generate loss-of-function mutants through the NHEJ repair pathway. The system relies on a plasmid that expresses Cas9 from a S. cerevisiae or a C. glabrata promoter, which is transformed into strains that express sgRNAs under the control of either S. cerevisiae or C. glabrata promoters. We further demonstrate that this system can be used to increase the efficiency of homologous recombination in C. glabrata. Some of the mutant strains generated by this approach were further used to study colonization and infection in an invertebrate model31,32. Namely, the targeted genes were: (i) a GPI-anchored aspartyl protease involved in the infection process of C. glabrata and (ii) a putative serine/threonine kinase of C. glabrata33. Interestingly, we report that the corresponding mutants were less virulent in the D. melanogaster model and thus that these two genes should be involved, directly or indirectly, in the infection process of C. glabrata in vivo. Accordingly, this CRISPR-Cas9 system is powerful to assess the potential role of candidate genes in C. glabrata infectivity in this mini-host model and presumably in other infection models.
Results
Engineering of a CRISPR-Cas9 strain
For the development of the CRISPR-Cas9 system in C. glabrata, we took advantage of the well studied ∆HTL strain (Supplementary Table S1)34. This triple auxotrophic strain (his3, trp1 and leu2) has been extensively studied for persistence in mice, for the identification of novel antifungal tolerance genes, for testing antifungal compounds, and for investigating its phagocytosis by immune cells18,34,35. Although RNA polymerase II promoters from S. cerevisiae can be easily used in C. glabrata36,37, RNA expression is prone to species-specific regulations due to discrepancies in A- and B-boxes location in the upstream region of RNA gene38. Therefore, for sgRNAs expression, we used a pRS315 vector developed for S. cerevisiae expression under S. cerevisiae SNR52 promoter (pSNR52)39 and we also engineered another pRS315 vector bearing the RNA polymerase III promoter of the nuclear C. glabrata RNAH1 gene (pRNAH1) followed by the tRNA Tyr 2 terminator (tTY2) (Fig. 1A, Supplementary Fig. S1 and Supplementary Table S2).

Design of CRISPR-Cas9 in C. glabrata and schematic of ADE2 disruption.
(A) CAS9 expression was done either with the p414 vector engineered by DiCarlo and colleagues (39) under the S. cerevisiae TEF1 promoter (pTEF1) or with the pRS314 under the C. glabrata CYC1 promoter (pCYC1). sgRNAs were cloned in a pRS315 and expressed either under the RNA Pol III promoters of S. cerevisiae SNR52 (pSNR52) or C. glabrata RNAH1 (pRNAH1). (B) Simplified scheme of adenine biosynthesis. Cells deprived of ADE2 display a red phenotype due to the accumulation of a red pigment. (C) Two target sequences localized in 5′ of the ADE2 were chosen for gene disruption: sgADE2.1 and sgADE2.3. Arrowheads indicate Cas9 cleavage sites. (D) Workflow of the sequential transformations done for sgRNA and CAS9 expression in C. glabrata and description of the following experiments.
For CAS9 expression, we used the CRISPR-Cas9 system developed by DiCarlo and colleagues in S. cerevisiae39: CAS9 was expressed from a centromeric p414 vector under the S. cerevisiae translational elongation factor EF-1 alpha (TEF1) promoter (plasmid named p414-CAS9(TEF1), Supplementary Table S2). We first checked whether CAS9 expression under the S. cerevisiae TEF1 promoter (pTEF1) was affecting C. glabrata fitness. After the transformation of C. glabrata with the p414-CAS9(TEF1) vector, we monitored its average generation time in SCGlc 2% liquid media by measuring its growth over a period of 48 hours. The comparison of the doubling time of the ∆HTL + CAS9(TEF1) strain to that of the ∆HTL strain revealed that the constitutive expression of CAS9 in C. glabrata delays its average generation time by a factor of 3.5 (323.6 min vs 91.2 min; p < 0.0001) (Supplementary Fig. S3). In yeasts, a longer generation time and loss of fitness can be attributed to many factors including nuclear accumulation of a protein, metabolic disturbance or nutrient limitation. In the case of the ∆HTL + CAS9(TEF1) strain, we hypothesized that CAS9 expression under the pTEF1 promoter might be too strong for C. glabrata fitness and that this caused nuclear accumulation of the protein Cas9. To overcome this, we decided to substitute the pTEF1 promoter with the endogenous promoter of the cytochrome C isoform1 protein of C. glabrata (plasmid named pRS314 − CAS9(CYC1), Supplementary Table S2). In S. cerevisiae, this promoter has been shown to induce a weak and ubiquitous expression of genes under its dependency, with up to 48 times less expression as compared to the pTEF1 promoter40. However, this alternative approach only led to a small reduction of the generation time, as compared to the ∆HTL + CAS9(TEF1) strain (292.8 min vs 323.6 ; p 0.264), which was still 3.2 times slower than the ∆HTL strain (292.8 min vs 91.2 min; p 0.009) (Supplementary Fig. S3). This indicates that CAS9 expression hampers C. glabrata fitness. To circumvent this problem, we generated the ∆HTL strain expressing sgRNA prior to pRS314-CAS9(CYC1) transformation in every further assay, since expression of sgRNA alone does not impede C. glabrata fitness (data not shown).
CRISPR-Cas9 gene disruption by indels
We then tested the ability to create indels in C. glabrata by targeting the disruption of the ADE2 gene, known to be a good phenotypic marker because of the red phenotype of ade2 cells (Fig. 1B). With the help of our bioinformatics tool CASTING, we designed two sgRNAs targeting the ADE2 locus: sgADE2.1 and sgADE2.3 (Fig. 1A,C, Supplementary Fig. S2 and Mat & Met). We have tested all possibilities by combining sgRNAs expressed either under a S. cerevisiae (pSNR52) or a C. glabrata promoter (pRNAH1) with CAS9 expressed either from a S. cerevisiae heterologous promoter (pTEF1) or from a C. glabrata endogenous promoter (pCYC1) (Fig. 1D). As expected, when yeasts were plated on appropriate synthetic media, a strong red phenotype was observed upon ADE2 disruption (Fig. 2A), whereas strains expressing CAS9 or sgRNA alone displayed the expected white phenotype. To further assess ADE2 disruption, we performed a Surveyor assay on strains expressing CAS9 alone or in combination with sgADE2.1 or sgADE2.3. To better detect indels, we designed two primers (see Supplementary Table S3); the ADE2-Fwd primer is respectively localized 271nt and 198nt upstream of the sgADE2.1 and sgADE2.3 disruption sites. Likewise, the ADE2-Rev primer is respectively localized 339nt and 412nt downstream of the sgADE2.1 and sgADE2.3 target sites. No indels were detected when CAS9 was expressed alone in C. glabrata (Fig. 2B,C left lane). In contrast, we observed efficient indels in each strain co-expressing CAS9, with any of the two sgRNAs targeting ADE2 (Fig. 2B,C middle and right lanes). To characterize at the nucleotide level the events that led to ADE2 disruption, we sequenced the region in which indels were expected to occur. We then made two interesting observations: (i) when disruption was induced by sgADE2.1 under the control of a C. glabrata promoter, NHEJ in C. glabrata mostly lead to insertion of only one nucleotide (essentially a T) at the cutting site (whether CAS9 was expressed under a C. glabrata or a S. cerevisiae promoter; Fig. 2D upper panel), and (ii) NHEJ elicited by sgADE2.3 expressed under a S. cerevisiae promoter often generated large insertions of nucleotides, and this was predominantly observed when co-expressed with CAS9(TEF1) which mimics a S. cerevisiae system (Fig. 2D lower panel). These results suggest that for efficient gene disruption by NHEJ repair in C. glabrata, CAS9 should be expressed under the pCYC1 promoter in combination with sgRNAs expression under the pRNAH1 promoter.

Efficient CRISPR-Cas9 gene disruption of the ADE2 locus.
(A) Drop test of ∆HTL, ∆HTL + sgADE2.1 or sgADE2.3, ∆HTL + CAS9 (TEF1 or CYC1), ∆HTL + sgADE2.1 or sgADE2.3 + CAS9 (TEF1 or CYC1) on SCGlc 2% and SCGlc 2% without leucine (-L) or tryptophan (-W) or both -L-W. Plates were incubated 2 days at 30 °C. (B,C) Indels in ADE2 cells were monitored by Surveyor assay in ∆HTL strain compared to the ∆HTL + CAS9 (CYC1), ∆HTL + sgADE2.1 + CAS9 (CYC1) or ∆HTL + sgADE2.3 + CAS9 (CYC1). (D) Events leading to ADE2 disruption were monitored by sequencing each strain and by comparing to the in silico constructs. Insertions and deletions are shown in green. For each deleted strain, 6 different clones were tested.
CRISPR-Cas9 targeted homologous recombination
To get effective homologous recombination (HR) in C. glabrata routinely, inserted DNA fragments need to bear a minimum of 500 bp of homology domain (HD) at both 5′ and 3′ ends of the sequence18. We asked whether HD length could be decreased by disrupting the ADE2 locus through the co-expression of CAS9(CYC1) and sgADE2.1 in the presence of a template repair DNA. We tested two cassettes: one of 57 bp composed of three TAG stop codons in the three reading frames (XTAG) and another one of 1,202 bp encompassing 385nt of the HIS3 promoter and the HIS3 gene of C. glabrata (cgHIS3) (Fig. 3A,B and Supplementary Fig. S4A,B). Twenty or 200 bp of ADE2 HD were added at the 5′ and 3′ ends of each cassette to study the effect of HD length on HR (see Supplementary Methods). We transformed C. glabrata strains expressing sgADE2.1 with either CAS9 alone or CAS9 supplemented with 1 μg of XTAG or HIS3 PCR products and we measured the recombination frequency in each case. After screening and sequencing, we observed a better recombination frequency for XTAG insertion with 4 times more recombination events taking place when disruption was elicited by coexpression of CAS9 with sgADE2.1, compared to expression of CAS9 alone. The recombination rate was even more efficient (8-fold higher) when 200 bp of HD were used, as compared to 20 bp (Fig. 3C). To ensure that XTAG was indeed inserted at the disruption site in ADE2, we sequenced clones after insertion and documented efficient HR (Supplementary Fig. S4A). We also observed an increase in HR for the HIS3 cassette, with up to a 3-fold increase when 20 bp HD were used. Unexpectedly, the frequency was not different whether we used 20 or 200 bp of HD and was similar to the frequency of XTAG with only 20 bp of HD after Cas9 cleavage (Fig. 3C, Supplementary Fig. S4B). This suggests that directed gene disruption facilitates HR and that the length of the homology region required for HR seems to have a greater impact with short inserts. In any case, we have demonstrated here that the CRISPR-Cas9 system allows achieving efficient recombination in C. glabrata with only 20 or 200 bp of HD, as compared to the recommended 500 bp.

Recombination efficiency in C. glabrata using targeted gene disruption with CRISPR-Cas9.
(A) Workflow of C. glabrata expressing sgADE2.1 transformation with CAS9 (CYC1) and either XTAG or HIS3 dsDNA bearing either 20 or 200 bp of homology domain (HD). (B) For homologous recombination, XTAG and HIS3 cassettes were inserted at the sgADE2.1 cut site in ADE2. Sequences of XTAG and HIS3 are listed in Supplementary Fig. 3. For each cassette, we appended either 20 or 200 bp of HD corresponding to the sequence around sgADE2.1 cutting site. (C) ∆HTL strain expressing the sgADE2.1 was transformed with a dsDNA cassette XTAG or HIS3 with or without the pRS314-CAS9 (CYC1). To evaluate efficient recombination at the ADE2 locus, we checked integration of XTAG and HIS3 by PCR after selection of mutants on SCGlc 2% -L-W plates. In each experiment, we tested HR with cassettes bearing 20 or 200 bp-long flanking regions. *p < 0,05, **p < 0,1, nd: not different, each bar represents mean values of n > 200 clones, SEM are shown.
C. glabrata Cas9-generated mutant strain to study C. glabrata infection in D. melanogaster
Recent studies highlighted the use and advantages of Drosophila melanogaster as a model organism to study virulence and host adaptation17,31,32. This model is based on two D. melanogaster strains, a WT immuno-competent strain (A5001) in which the Toll pathway senses fungal infections, and an immuno-compromised strain which lacks the MyD88 Toll adapter, MyD8841. In WT flies, the response mediated by the Toll pathway acts as a barrier against fungal infection and controls yeast development, independently of the known antifungal peptides Drosomycin and Metchnikowin and may involve other Drosophila immune induced molecules31,42. In contrast, MyD88 mutant flies succumb rapidly after fungal infection and this is correlated to fungal burden. Except for phagocytosis, no adequate immune response is mounted in these mutant flies31. We used this infection model to test two mutants of C. glabrata generated by Cas9 (Supplementary Methods and Supplementary Figs S2 and S5A,B). The first is the yps11 strain in which the YPS11 gene was disrupted. This gene encodes an aspartyl protease that was shown to be upregulated during macrophage internalization12. The second strain is mutant for the CAGL0K04301g gene, which codes for a putative serine/threonine kinase in C. glabrata. The latter is homologous to the mitochondrial protein FMP48 in S. cerevisiae43 and to a putative kinase implicated in virulence and mouse kidney colonization HSL1 in C. albicans33,44. Since the Candida Genome Database describes this gene as an “Uncharacterized ORF”45, we decided to rename it VPK1 for Validated Protein by Knock-out 1. We tested the subsequent vpk1 and yps11 strains for in vitro culture and infection analysis.
Prior to any experiment, C. glabrata cells were grown in YPD for 24h at 30 °C in order to get rid of both vectors that allow expressing the sgRNAs or CAS9 (data not shown). We first monitored the behavior of the ∆HTL C. glabrata strain (Supplementary Table S1) grown in liquid culture until the exponential phase was reached and compared it with the two different strains we engineered.
Many C. glabrata strains deleted in genes implicated in the cell wall integrity pathway display sensitivity to caffeine, CaCl2 and other salts at pH7.5 or pH5.512,13,46,47,48. We therefore tested the ade2, yps11 and vpk1 strains along with the ∆HTL strain for cell wall-related phenotypes. In any of the tested conditions, we did not observe differences in growth nor in colony size for the ∆HTL, yps11 or vpk1 strains, which suggests that YPS11 and VPK1 do not play major role in cell wall integrity maintenance (Fig. 4A). Likewise, we did not detect any significant discrepancy in the average generation time of these four strains grown in SCGlc 2% media when compared to that of the ∆HTL strain (Fig. 4B). To our surprise, the ade2 strain displayed susceptibility to caffeine in every condition tested and to ZnCl2 at pH7.5 at 30 and 37 °C, although to a lesser extent. Growth of ade2 was slightly altered on YPD, NaCl and CaCl2 at pH5.5 and 7.5 both at 30 and 37 °C. This emphasizes that the quadruple auxotrophy (∆HTLA) might impact either the Mpk1-mediated cell wall integrity pathway or more deeply the metabolism of amino acids.

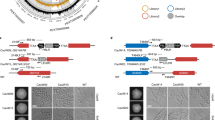
Phenotypic characterization of the ∆HTL, ade2, yps11 and vpk1 strains.
(A) Drop test of each strain engineered on YPD plates at pH5.5 and 7.5 supplemented in NaCl (500 mM), ZnCl2 (10 mM), CaCl2 (100 mM) or caffeine (7.5 mM). Plates were stored for 1 day at 30 or 37 °C except for YPD plates supplemented with ZnCl2 stored for 3 days. (B) Average generation time of ∆HTL, ade2, yps11 and vpk1 strains monitored during growth in SCGlc 2% liquid medium. Numbers are mean generation time for each strain. n = 3, SEM are shown.
To test the possibility to use Cas9-engineered C. glabrata strains to study the infection process in Drosophila, our strategy was to disrupt YPS11 or VPK1, to get rid of both expressing plasmids, and then to sequence deleted loci prior to infection (Fig. 5A). In vivo, we first ascertained that clean injury was not affecting fly survival; as expected, no colonies grew on YPD plates supplemented with streptomycin when fly extracts were plated (Fig. 5B,C). Then, we asked to what extent fly survival was affected by infection with ∆HTL or ∆HTL + Cas9. After 96 hours of infection, 75 and 71.4% of MyD88 flies succumbed by C. glabrata ∆HTL and ∆HTL + Cas9 respectively (Fig. 5B). This correlates with a constant increase in fungal load as observed by colony-forming units (CFU) realized at 0–24–48 and 72 hours post-infection (Fig. 5C). Infection of WT A5001 flies with ∆HTL and ∆HTL + Cas9 yielded similar results as a mock-infection (clean injury after 96 hours) and was not associated with any fungal growth (data not shown). Next, we compared fly survival after infection with ∆HTL, yps11 or vpk1 cells. MyD88 flies infected with the ∆HTL strain succumbed faster to the infection (time for death of 50% of the flies [LT50] was about 110 hours), while flies infected with the yps11 strain were killed slower (LT50 about 130 hours), but nevertheless faster than vpk1 flies (LT50 about 180 hours) (Fig. 5D). Inoculation of ∆HTL, yps11 or vpk1 in A5001 WT flies however, led roughly to 80% of survival which is in accordance with what is expected in immuno-competent flies (Fig. 5B,D). To confirm that mortality was associated with increasing fungal load, we also measured CFUs at different time points. vpk1 grew as efficiently as the ∆HTL strain in the host MyD88 flies, whereas yps11 appeared to proliferate somewhat slower (Fig. 5E).

Study of C. glabrata infection in D. melanogaster after genome engineering by the CRISPR-Cas9 system.
(A) Scheme of Cas9-directed gene disruption in C. glabrata prior to D. melanogaster infection. After sequential transformation of sgRNA- and CAS9 (CYC1)-expressing vectors, targeted gene disruption was checked by sequencing. Vectors were then lost by growing yeasts overnight in liquid SCGlc 2% media. (B) Survival curves of immuno-competent A5001 flies and immuno-compromised MyD88 flies after infection with clean prick (cp), ∆HTL strain or ∆HTL + Cas9. Survival are shown as percentage of surviving flies. **p < 0,05 when compared to survival curve of MyD88 + cp or A5001 + cp. (C) Colony Forming Units (CFU) of cp and C. glabrata ∆HTL or ∆HTL + Cas9 after infection of D. melanogaster A5001 and MyD88 flies. (D) Infection of A5001 and MyD88 flies with the ∆HTL strain and strains disrupted for YPS11 and VPK1. ****p < 0,0001 when compared to survival cure of MyD88 + ∆HTL; ns, not significant. (E) CFU experiments related to infections performed in (D). Drosophila drawing is the work of B. Nuhanen, and was obtained from https://commons.wikimedia.org/wiki/File:Drosophila-drawing.svg, and is licensed under a CC BY-SA license (https://creativecommons.org/licenses/by-sa/3.0/deed.en). (F) Infection of latex (LTX) beads-injected A5001 and MyD88 flies with the ∆HTL strain or the NHEJ-vpk1 mutant.
To further confirm these results, we sequenced the yps11 and vpk1 strains after infection to ensure that the observed survival rates were indeed resulting from infection by the different disrupted strains. In every case, we retrieved the same mutations, thus indicating that the strains used for infection were indeed responsible for the differing survival rates we observed (Supplementary Fig. S5C,D). The results reported here are unlikely to be caused by second-site mutations, as we used three independently-generated mutants for each targeted gene in survival experiments. Finally, we also generated an additional mutant strain for vpk1 by homologous recombination, which yielded an insertion of the HIS3 gene in the vpk1 locus. This mutant strain (vpk1::HIS3) displayed a decreased virulence in survival assays, although the NHEJ-generated CRISPR-Cas9 mutants were somewhat even more resistant to the infection (Supplementary Fig. S6). This latter difference might be accounted for by the fact that the HR mutant is no longer auxotrophic for histidine.
A major antifungal host defense remaining in the MyD88 flies is the cellular arm, which can be impaired by saturating the phagocytic apparatus with injected non-degradable latex beads (LTX). As expected, LTX-injected MyD88 flies succumbed much more rapidly to ∆HTL infection (LT50 about 24 hours; Fig. 5F). Interestingly, the NHEJ vpk1 strain also killed flies faster (LT50 about 48 hours), yet not as rapidly as the ∆HTL strain. However, the difference in the killing rate of vpk1 was reduced with respect to the ∆HTL strain, and was never higher than 48 hours, instead of 72 hours in non LTX-injected MyD88 flies. Thus, vpk1 appears to be somewhat more virulent when the cellular defenses of the host are ablated.
Discussion
In this study, we have developed a CRISPR-Cas9 system in C. glabrata and have shown easy and efficient gene disruption by indels, as well as gene recombination by directed homologous recombination. Using D. melanogaster as a model of infection, we have demonstrated that CRISPR-Cas9 is a suitable tool to engineer C. glabrata, disrupt any specific gene and then study its impact during the infection process. This is, to our knowledge, the first example of the CRISPR-Cas9 genetic tool applied to the opportunistic pathogen C. glabrata.
Currently, the C. glabrata toolbox is being expanded, but still lacks several techniques such as Tet-regulable library of essential genes, GFP-tagged or yeast two-hybrid strains libraries17. The CRISPR-Cas9 engineering system was part of these missing tools. This is of great importance since C. glabrata has a dominant NHEJ DNA repair pathway that makes gene replacement difficult49. Nevertheless, C. glabrata gene replacement or gene deletion by insertion of cassette can be achieved via homologous recombination with large flanking regions. Schwarzmuller and colleagues have recently set up a gene deletion library using 500 bp HD18, and were able to disrupt roughly 13% of all C. glabrata genes. To increase the number of deleted genes, we hypothesized that generating a double strand DNA cut with Cas9 endonuclease might favor the homology-directed DNA repair. Using two types of disrupting cassettes (XTAG and HIS3) and two different HD (20 and 200 bp) (Fig. 3 and Supplementary Fig. 4), we showed that HR can be efficiently accomplished both with short and large fragments. Our results also showed that the size of flanking regions is important for short but not large fragments (Fig. 3).
To test the efficiency of the homologous recombination in C. glabrata, two options were conceivable: (i) the ability to get revertants through the reinsertion of the gene to its original locus after Cas9 cut, or (ii) the ability to knock-out the target gene through the insertion of a selection marker. We chose the second option for two main reasons. First, we wanted to compare the efficiency of this recombination with the NHEJ assays. Second, it was in our interest to test the feasibility of an alternative way to knock-out genes, which was helpful to further characterize mutants during Drosophila infections, and with the clear advantage to positively select mutants.
We have shown that ∆HTL strain expressing CAS9 has an increased generation time as compared to the ∆HTL strain (Supplementary Fig. S3). When Drosophila flies were challenged with ∆HTL + CAS9 (CYC1), survival rates were similar to the ∆HTL strain (Fig. 5B). This is in contradiction with the slow doubling time displayed in liquid SCGlc 2% -W media. CFU tests after infection clearly demonstrated that ∆HTL + CAS9 was growing at the same rate as a ∆HTL strain in the host fly (Fig. 5C). This can be explained by the fact that tryptophan is available in Drosophila and thus no selection pressure is exerted to maintain the CAS9 expressing plasmid; in these conditions, yeasts recovered their native ∆HTL generation time.
Among the numerous models available to study infection by yeasts, the most common ones are macrophages, mice and Drosophila12,31,32,34,35,50,51,52. Stress tolerance and adaptation can be monitored in any of these three models, and in vitro as well. Up to now, Drosophila, and to a lower extent mouse, seem to be more suitable to study systemic infection. This is partly due to the fact that fungal infection kills immunosuppressed Drosophila, in contrast to mice in which only fungal colonization of organs can be assessed32,34,52. In addition, there are no ethical or cost-related concerns when working with Drosophila. Accordingly, the Drosophila model described by Quintin and colleagues31 seemed to be the adequate model to study C. glabrata pathogenesis with our CRISPR-Cas9 system.
We note that we did not observe a perfect correlation between fungal titer in vivo and virulence, as measured in survival assays. For instance, the vpk1 NHEJ-CRISPR-Cas9 mutant is less virulent than the ∆HTL strain, yet displays a similar fungal load. By the same token, yps11 appears to be more virulent than vpk1 even though its titer in flies is lower. One possibility is that C. glabrata mutants may differentially adhere or colonize distinct organs. It will be interesting to determine whether vpk1 apparent proliferation is restricted to a specific organ. Alternatively, one significant difficulty lies in the interpretation of CFU counts when survival curves display the shape observed in our experiments. Flies start to succumb early on, significantly already at 48 hours. Our experience with CFU counts once the flies have started dying from the infection is that these counts may be misleading, with errors introduced by sampling: flies about to die often yield high titers whereas flies bound to survive will display a low titer, as they may have managed to control the infection. Thus, a definitive conclusion will be reached only when this experiment has been repeated much more extensively than in the preliminary characterization reported here.
Several studies have focused on the yapsin genes and their role in C. glabrata pathogenicity12,13,51,53,54. Yapsins form a family of 11 genes encoding putative GPI-linked aspartyl proteases, and named because of their structural similarities with YPS genes of S. cerevisiae12. Some yapsin genes have been shown to be upregulated during internalization in macrophages (YPS 2-4-8-9-10-11) or in neutrophils (YPS 1-2-5-6-8-9-10-11)12,54. In addition, yeasts lacking YPS1 displayed a reduced dissemination into kidneys, liver and spleen during a 7-day infection in mice and were less associated with macrophages after 24 hours, whereas ∆yps7 strain displayed a normal association with macrophages and dissemination into mice kidneys, liver and spleen12. Pathogenicity was more reduced when YPS1 and -7 or when YPS1-11 were deleted. Nevertheless, these observations are tempered by the fact that ∆yps1, ∆yps1-∆yps7 or ∆yps1-11 cells are sensitive to NaCl at pH7.5, ZnCl2 at pH5.5 and 7.5, CaCl2 and KCl at pH7.512,13. In liquid stationary phase, loss of viability was observed for 3 of these strains and the presence of depressions on ∆yps1-11 cell surfaces was a striking evidence for cell wall defects12,13. Altogether, these observations point toward a more general defect at the level of cell wall composition, and the inability of these mutants to respond to environmental and intracellular stresses, rather than directly implicating yapsin genes in the infection process. In our analysis, we could not detect any cell wall-defect phenotype during growth test assays or generation time using the yps11 strain (Fig. 4A,B). CFU assays showed a constant increase of the fungal burden during yps11 cells infection and a reduced virulence in survival assays (Fig. 5D,E). This suggests that YPS11 might not be involved in the regulation of cell wall composition, but more likely hinders the maturation or incorporation of proteases or other proteins at the cell wall that would be important for pathogenicity.
We named the CAGL0K04301g gene Validated Protein by Knock-out 1 (VPK1), because the Candida Genome Database describes it as uncharacterized ORF, and because our results validate it as a gene whose product impacts the infection process in D. melanogaster (Fig. 5D–F). Having in mind that the understanding of the role of VPK1 needs further investigation, we would like to emphasize several observations that would suggest a direct or indirect role in the virulence of C. glabrata. Indeed, the vpk1 mutant does regain some virulence in phagocytosis-impaired flies, which suggests that one of its function may be to counteract or circumvent the cellular immune response, as shown for Pseudomonas aeruginosa rhlR gene55. FMP48, its homolog in S. cerevisiae, was reported to be a mitochondrial protein43, the expression of which was induced by UV radiation and 8-methoxypsoralen treatment56. HSL1, its homolog in C. albicans, is implicated both in the determination of morphology during the cell cycle33, and in virulence and kidney colonization in a mouse model of systemic infection44. With respect to VPK1 in C. glabrata, caution should be taken in interpreting its function in the light of its yeasts counterparts, as the role of proteins can vary between organisms. Although sometimes homologs do share the same role in phylogenetically-related organisms like YPS1 or MSN2/4 in S. cerevisiae, C. albicans and C. glabrata9,57, roles can be dramatically different in some cases, as exemplified by SCH9 that regulates ribosome biogenesis, translation and entry into the G0 phase of S. cerevisiae, but controls chromosome segregation in C. albicans58. Furthermore VPK1 does not seem to be a mitochondrial protein, as we failed to identify a mitochondrial targeting sequence (data not shown).
To conclude, virulence-gene studies in relevant infection models is crucial to understanding fungal pathogenesis. The use of technologies such as CRISPR-Cas9, in combination with an invertebrate infection model like Drosophila melanogaster, will now allow simultaneous genes disruption in genome-wide genetic screens using sgRNA libraries. Such large-scale approaches will lead to the identification of the factors required directly or indirectly for C. glabrata virulence in vivo.
Material and Methods
Identification of CRISPR-Cas9 targets in the C. glabrata genome
To characterize the best CRISPR-Cas9 targets for a given C. glabrata gene, we have developed a dedicated bioinformatics tool. Named CASTING (for « CAS9 Targets IN Genomes »), it has been implemented in Python 2.7 and is mainly based on the library PyRNA provided by the RNA-Science-Toolbox project (https://github.com/fjossinet/RNA-Science-Toolbox). Using the complete C. glabrata CBS138 genome (NCBI BioProject PRJNA12376), extended with the new genes identified by Linde and colleagues59, CASTING takes as input a gene name, a genomic location, or a user-defined sequence. In a first step, CASTING searches for all PAM motifs of the form NGG or NAG. In a second step, each motif is extended by 20 nt on its 5′-end. Among all these potential 23nt-long sequences, CASTING filters out suboptimal sequences containing k-mers as described by Xie and colleagues60. In a third step, CASTING restricts the candidates to the sequences with a GC content between 35% and 75%. For each remaining on-target sequence, CASTING delegates to the tool SeqMap the identification of its off-target sites61. All the on-target sites are then ranked according to the sgRNA activity predictive model proposed by Doench and colleagues62. CASTING has been made available online at http://charn-ibmc.u-strasbg.fr:8080/casting.html. The website is based on the Python microframework Flask (http://flask.pocoo.org) and on the JavaScript libraries jQuery (https://jquery.com) and Bootstrap (http://getbootstrap.com).
Plasmids construction
All single-guide RNA sequences (sgRNA, see Supplementary Material S2) were cloned in a centromeric pRS315 vector under the control of S. cerevisiae SNR52 (pSNR52) or C. glabrata RNAH1 (pRNAH1) constitutive promoters (Supplementary Table S2 and Fig. 1A). Using the HindIII cloning site of pRS315, we cloned a cassette containing either the pSNR52 or pRNAH1 promoters, followed by a 46 bp sequence harboring two AarI restriction sites, the structural guiding RNA and a scTY2 or cgTY2 terminator respectively (sequences are listed in Supplementary Fig. S1). Each sgRNA sequence was synthesized by Integrated DNA Technologies (IDT) as two 20 bp complementary single strand DNA oligomers flanked by 20 bp of homology to the AaRI-digested pRS315 (sgRNA sequences are shown in Supplementary Material S2). They were used at a final concentration of 10μM in Duplex buffer (100 mM potassium acetate, 30 mM HEPES pH7.5) and 50 pmoles of each strand were mixed and incubated at 94 °C for 2 min, then cooled 2 min at room temperature and finally kept at 4 °C before use. The dsDNA was then diluted 10 times in deionized water, 1 pmole was used with 1 μg of AarI-digested pRS315 and assembly was achieved by the Gibson assembly method (New England BioLabs).
Streptococcus pyogenes CAS9 was used in two different vectors. First, the p414-CAS9(TEF1) (AddGene) expresses CAS9 under the S. cerevisiae TEF1 promoter (pTEF1)39 (Fig. 1A and Supplementary Table S2). Second, we also produced our own CAS9 expressing vector based on a C. glabrata promoter (plasmid named pRS314-CAS9(CYC1)). To do so, we first cloned the CAS9 gene from the p414-CAS9(TEF1) plasmid into the pRS314 after a KpnI/XbaI digestion. We then amplified the C. glabrata CYC1 promoter (pCYC1) flanked with SpeI restriction sites and cloned it into the pRS314-CAS9 by the Gibson assembly method and by following manufacturer’s instructions (Fig. 1A and Supplementary Table S2).
Fly strains
Stocks were raised on rich standard cornmeal-agar medium at 25 °C. w A5001 flies were used as wild-type throughout the whole experiments and MyD88 mutant flies in the A5001 genetic background have been described previously41.
Survival experiments and statistical analysis
Female A5001 (WT) or MyD88 mutant flies aged between 7 and 16 days were isolated in batches of 20–25 flies. WT and MyD88 mutant flies were challenged by septic injury with a needle previously dipped into a solution of various C. glabrata strains grown to stationary phase (∆HTL and derivatives) concentrated at an OD600nm15 in YPD. Vials containing infected flies were put in an incubator for 240 hours at 29 °C. After 96 h of incubation, flies were moved to new vials. Results are expressed as percentage of surviving flies at different time points post-infection. Each survival experiment is representative of at least two replicates done with three independent yeast colonies obtained by two independent disruption experiments. Statistical significance during survival experiments was calculated with the method of Mantel and Cox using the log-rank test (GraphPad PRISM6 software), and p < 0.05 was considered significant.
CFU experiments
During flies infection, three flies were taken at each time point post-infection in order to monitor C. glabrata replication. Flies were crushed in an Eppendorf using a 0.5 mL grinder pestles in 150 μL of PBS 1x and 0.1% Tween 20. Appropriate dilutions were made for each time point in YPD, plated on YPD supplemented with streptomycin (50 μg/mL) and incubated at 30 °C for 2 days.
Additional Information
How to cite this article: Enkler, L. et al. Genome engineering in the yeast pathogen Candida glabrata using the CRISPR-Cas9 system. Sci. Rep. 6, 35766; doi: 10.1038/srep35766 (2016).
References
Almirante, B. et al. Epidemiology and predictors of mortality in cases of Candida bloodstream infection: results from population-based surveillance, barcelona, Spain, from 2002 to 2003. J. Clin. Microbiol. 43, 1829–1835 (2005).
Lin, M. Y. et al. Prior antimicrobial therapy and risk for hospital-acquired Candida glabrata and Candida krusei fungemia: a case-case-control study. Antimicrob. Agents Chemother. 49, 4555–4560 (2005).
Sandven, P. et al. Candidemia in Norway (1991 to 2003): results from a nationwide study. J. Clin. Microbiol. 44, 1977–1981 (2006).
Brown, G. D. et al. Hidden killers: human fungal infections. Sci Transl Med 4, 1–9 (2012).
Denning, D. W. & Bromley, M. J. Infectious Disease. How to bolster the antifungal pipeline. Science 347, 1414–1416 (2015).
Krcmery, V. & Barnes, A. J. Non-albicans Candida spp. causing fungaemia: pathogenicity and antifungal resistance. J. Hosp. Infect. 50, 243–260 (2002).
Vale-Silva, L. A. & Sanglard, D. Tipping the balance both ways: drug resistance and virulence in Candida glabrata. 15, 1–8 (2015).
Dujon, B. et al. Genome evolution in yeasts. Nature 430, 35–44 (2004).
Roetzer, A., Gabaldón, T. & Schüller, C. From Saccharomyces cerevisiae to Candida glabrata in a few easy steps: important adaptations for an opportunistic pathogen. FEMS Microbiology Letters 314, 1–9 (2010).
Gabaldón, T. & Carreté, L. The birth of a deadly yeast: tracing the evolutionary emergence of virulence traits in Candida glabrata. FEMS Yeast Res. 16, fov110 (2016).
de Groot, P. W. J. et al. The cell wall of the human pathogen Candida glabrata: differential incorporation of novel adhesin-like wall proteins. Eukaryotic Cell 7, 1951–1964 (2008).
Kaur, R., Ma, B. & Cormack, B. P. A family of glycosylphosphatidylinositol-linked aspartyl proteases is required for virulence of Candida glabrata. Proc. Natl. Acad. Sci. USA. 104, 7628–7633 (2007).
Bairwa, G., Rasheed, M., Taigwal, R., Sahoo, R. & Kaur, R. GPI (glycosylphosphatidylinositol)-linked aspartyl proteases regulate vacuole homoeostasis in Candida glabrata. Biochem. J. 458, 323–334 (2014).
Kasper, L., Seider, K. & Hube, B. Intracellular survival of Candida glabrata in macrophages: immune evasion and persistence. FEMS Yeast Res. 15, fov042 (2015).
Kaur, R., Domergue, R., Zupancic, M. L. & Cormack, B. P. A yeast by any other name: Candida glabrata and its interaction with the host. Curr. Opin. Microbiol. 8, 378–384 (2005).
Gabaldón, T. et al. Comparative genomics of emerging pathogens in the Candida glabrata clade. BMC Genomics 14, 623–638 (2013).
Ho, H.-L. & Haynes, K. Candida glabrata: new tools and technologies-expanding the toolkit. FEMS Yeast Res. 15, fov066 (2015).
Schwarzmüller, T. et al. Systematic phenotyping of a large-scale Candida glabrata deletion collection reveals novel antifungal tolerance genes. PLoS Pathog. 10, e1004211 (2014).
Castano, I. et al. Tn7-based genome-wide random insertional mutagenesis of Candida glabrata. Genome Res. 13, 905–915 (2003).
Hsu, P. D., Lander, E. S. & Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278 (2014).
Qi, L. S. et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013).
Anton, T., Bultmann, S., Leonhardt, H. & Markaki, Y. Visualization of specific DNA sequences in living mouse embryonic stem cells with a programmable fluorescent CRISPR/Cas system. Nucleus 5, 163–172 (2014).
Deng, W., Shi, X., Tjian, R., Lionnet, T. & Singer, R. H. CASFISH: CRISPR/Cas9-mediated in situ labeling of genomic loci in fixed cells. Proc. Natl. Acad. Sci. USA. 112, 11870–11875 (2015).
Jiang, W., Bikard, D., Cox, D., Zhang, F. & Marraffini, L. A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 31, 233–239 (2013).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013).
Nelson, C. E. et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351, 403–407 (2015).
Vinayak, S. et al. Genetic modification of the diarrhoeal pathogen Cryptosporidium parvum. Nature 523, 477–480 (2015).
Fuller, K. K., Chen, S., Loros, J. J. & Dunlap, J. C. Development of the CRISPR/Cas9 System for Targeted Gene Disruption in Aspergillus fumigatus. Eukaryotic Cell 14, 1073–1080 (2015).
Schuster, M., Schweizer, G., Reissmann, S. & Kahmann, R. Genome editing in Ustilago maydis using the CRISPR–Cas system. Fungal Genetics and Biology 89, 3–9 (2015).
Vyas, V. K., Barrasa, M. I. & Fink, G. R. A Candida albicans CRISPR system permits genetic engineering of essential genes and gene families. Science Advances 1, e1500248 (2015).
Quintin, J., Asmar, J., Matskevich, A. A., Lafarge, M.-C. & Ferrandon, D. The Drosophila Toll pathway controls but does not clear Candida glabrata infections. J. Immunol. 190, 2818–2827 (2013).
Brunke, S. et al. Of mice, flies--and men? Comparing fungal infection models for large-scale screening efforts. Dis Model Mech 8, 473–486 (2015).
Wightman, R., Bates, S., Amornrrattanapan, P. & Sudbery, P. In Candida albicans, the Nim1 kinases Gin4 and Hsl1 negatively regulate pseudohypha formation and Gin4 also controls septin organization. J. Cell Biol. 164, 581–591 (2004).
Jacobsen, I. D. et al. Candida glabrata persistence in mice does not depend on host immunosuppression and is unaffected by fungal amino acid auxotrophy. Infection and Immunity 78, 1066–1077 (2010).
Mota, S. et al. Candida glabrata susceptibility to antifungals and phagocytosis is modulated by acetate. Front Microbiol 6, 919 (2015).
Macreadie, I., Castelli, L., Mehra, R. & Winge, J. T. A. Heterologous gene expression and protein secretion from Candida glabrata. Biotechnology and Applied Biochemistry 19, 265–269 (1994).
Zordan, R. E. et al. Expression plasmids for use in Candida glabrata. G3 (Bethesda) 3, 1675–1686 (2013).
Marck, C. et al. The RNA polymerase III-dependent family of genes in hemiascomycetes: comparative RNomics, decoding strategies, transcription and evolutionary implications. Nucl. Acids Res. 34, 1816–1835 (2006).
DiCarlo, J. E. et al. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucl. Acids Res. 41, 4336–4343 (2013).
Mumberg, D., Müller, R. & Funk, M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156, 119–122 (1995).
Tauszig-Delamasure, S., Bilak, H., Capovilla, M., Hoffmann, J. A. & Imler, J.-L. Drosophila MyD88 is required for the response to fungal and Gram-positive bacterial infections. Nature Immunology 3, 91–97 (2001).
Clemmons, A. W., Lindsay, S. A. & Wasserman, S. A. An effector Peptide family required for Drosophila toll-mediated immunity. PLoS Pathog. 11, e1004876 (2015).
Reinders, J., Zahedi, R. P., Pfanner, N., Meisinger, C. & Sickmann, A. Toward the complete yeast mitochondrial proteome: multidimensional separation techniques for mitochondrial proteomics. J. Proteome Res. 5, 1543–1554 (2006).
Umeyama, T. et al. Candida albicans protein kinase CaHsl1p regulates cell elongation and virulence. Mol. Microbiol. 55, 381–395 (2005).
Binkley, J. et al. The Candida Genome Database: the new homology information page highlights protein similarity and phylogeny. Nucl. Acids Res. 42, D711–D716 (2014).
Martín, H., Rodríguez-Pachón, J. M., Ruiz, C., Nombela, C. & Molina, M. Regulatory mechanisms for modulation of signaling through the cell integrity Slt2-mediated pathway in Saccharomyces cerevisiae. J. Biol. Chem. 275, 1511–1519 (2000).
Krysan, D. J., Ting, E. L., Abeijon, C., Kroos, L. & Fuller, Rs. S. Yapsins are a family of aspartyl proteases required for cell wall integrity in Saccharomyces cerevisiae. Eukaryotic Cell 4, 1364–1374 (2005).
Levin, D. E. Regulation of cell wall biogenesis in Saccharomyces cerevisiae: the cell wall integrity signaling pathway. Genetics 189, 1145–1175 (2011).
Zhang, C., Meng, X., Wei, X. & Lu, L. Highly efficient CRISPR mutagenesis by microhomology-mediated end joining in Aspergillus fumigatus. Fungal Genet. Biol. 86, 47–57 (2015).
Kasper, L. et al. Identification of Candida glabrata genes involved in pH modulation and modification of the phagosomal environment in macrophages. PLoS ONE 9, e96015 (2014).
Bairwa, G. & Kaur, R. A novel role for a glycosylphosphatidylinositol-anchored aspartyl protease, CgYps1, in the regulation of pH homeostasis in Candida glabrata. Mol. Microbiol. 79, 900–913 (2011).
Brieland, J. et al. Comparison of pathogenesis and host immune responses to Candida glabrata and Candida albicans in systemically infected immunocompetent mice. Infection and Immunity 69, 5046–5055 (2001).
Rai, M. N., Balusu, S., Gorityala, N., Dandu, L. & Kaur, R. Functional Genomic Analysis of Candida glabrata-Macrophage Interaction: Role of Chromatin Remodeling in Virulence. PLoS Pathog. 8, e1002863 (2012).
Fukuda, Y., Tsai, H.-F., Myers, T. G. & Bennett, J. E. Transcriptional profiling of Candida glabrata during phagocytosis by neutrophils and in the infected mouse spleen. Infection and Immunity 81, 1325–1333 (2013).
Limmer, S. et al. Pseudomonas aeruginosa RhlR is required to neutralize the cellular immune response in a Drosophila melanogaster oral infection model. Proc. Natl. Acad. Sci. USA. 108, 17378–17383 (2011).
Dardalhon, M. L., Lin, W., Nicolas, A. & Averbeck, D. Specific transcriptional responses induced by 8-methoxypsoralen and UVA in yeast. 7, 866–878 (2007).
Gagnon-Arsenault, I., Tremblay, J. & Bourbonnais, Y. Fungal yapsins and cell wall: a unique family of aspartic peptidases for a distinctive cellular function. FEMS Yeast Res. 6, 966–978 (2006).
Varshney, N. et al. A surprising role for the Sch9 protein kinase in chromosome segregation in Candida albicans. Genetics 199, 671–674 (2015).
Linde, J. et al. Defining the transcriptomic landscape of Candida glabrata by RNA-Seq. Nucl. Acids Res. 43, 1392–1406 (2015).
Xie, S., Shen, B., Zhang, C., Huang, X. & Zhang, Y. sgRNAcas9: a software package for designing CRISPR sgRNA and evaluating potential off-target cleavage sites. PLoS ONE 9, e100448 (2014).
Jiang, H. & Wong, W. H. SeqMap: mapping massive amount of oligonucleotides to the genome. Bioinformatics 24, 2395–2396 (2008).
Doench, J. G. et al. Rational design of highly active sgRNAs for CRISPR-Cas9–mediated gene inactivation. Nat. Biotechnol. 32, 1262–1267 (2014).
Acknowledgements
The research in this paper has been published under the framework of the LABEX: ANR-10-LABX-0036_NETRNA. It was supported by the University of Strasbourg Institute for Advanced Study (USIAS) as part of a USIAS Fellowship (to F.J.), the CNRS and the Investissement d’Avenir Program Grant ANR-11-EQPX-0022. We would also like to thank Dr. Adrien Franchet, Miriam Yamba, Catherine Socha and Dr. Samuel Liégeois for their technical help with Drosophila.
Author information
Authors and Affiliations
Contributions
L.E. designed CRISPR-Cas9 system. A.M. made the CASTING bioinformatics tool. D.R. set up fly infection. L.E. and D.R. realized all experiments. L.E., F.J. and D.F. designed the experiments and F.J. wrote the paper. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Enkler, L., Richer, D., Marchand, A. et al. Genome engineering in the yeast pathogen Candida glabrata using the CRISPR-Cas9 system. Sci Rep 6, 35766 (2016). https://doi.org/10.1038/srep35766
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep35766
This article is cited by
-
Applications of CRISPR/Cas gene-editing technology in yeast and fungi
Archives of Microbiology (2022)
-
Lessons from the Nakaseomyces: mating-type switching, DSB repair and evolution of Ho
Current Genetics (2021)
-
Genome-wide piggyBac transposon-based mutagenesis and quantitative insertion-site analysis in haploid Candida species
Nature Protocols (2020)
-
A glance at genome editing with CRISPR–Cas9 technology
Current Genetics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.

{kind=link}