
Abstract
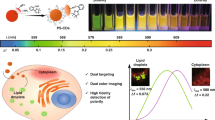
Polarity-sensitive fluorescent probes are powerful chemical tools for studying biomolecular structures and activities both in vitro and in vivo. However, the lack of “off-on” polarity-sensing probes has limited the accurate monitoring of biological processes that involve an increase in local hydrophilicity. Here, we design and synthesize a series of “off-on” polarity-sensitive fluorescent probes BP series consisting of the difluoroboron dippyomethene (BODIPY) fluorophore connected to a quaternary ammonium moiety via different carbon linkers. All these probes showed low fluorescence quantum yields in nonpolar solution but became highly fluorescent in polar media. BP-2, which contains a two-carbon linker and a trimethyl quaternary ammonium, displayed a fluorescence intensity and quantum yield that were both linearly correlated with solvent polarity. In addition, BP-2 exhibited high sensitivity and selectivity for polarity over other environmental factors and a variety of biologically relevant species. BP-2 can be synthesized readily via an unusual Mannich reaction followed by methylation. Using electrochemistry combined with theoretical calculations, we demonstrated that the “off-on” sensing behavior of BP-2 is primarily due to the polarity-dependent donor-excited photoinduced electron transfer (d-PET) effect. Live-cell imaging established that BP-2 enables the detection of local hydrophilicity within lysosomes under conditions of lysosomal dysfunction.
Similar content being viewed by others


Introduction
Polarity is an important environmental parameter that reflects the hydrophobicity or hydrophilicity of local environment. In biological systems, especially at the cellular level, polarity determines the interaction activities of a large number of proteins and controls the permeability of membrane compartments. However, polarity is a complex factor and encompasses a range of non–covalent interactions, including dipolarity/polarizability and hydrogen bonding1,2. Thus, realising its measurement in a straightforward manner in live cells is difficult, necessitating the development of new tools.
Polarity-sensitive fluorescent probes are powerful chemical tools for studying biomolecular structures and activities both in vitro and in vivo3,4,5,6,7,8,9,10,11,12,13,14. Numerous efforts have been devoted to the development of intramolecular-charge-transfer (ICT)-based probes for polarity sensing. They typically exhibit a marked red shift in their emission spectra and/or a decrease in their fluorescence intensities upon detecting an increase in surrounding polarity (“on-off” type). When conjugated to specific tags, such as amino acids4,5,6 and aliphatic chains7,8,9, these probes have been successfully applied in the study of structures and activities of hydrophobic proteins and membranes. However, the lack of “off-on” polarity-sensitive probes has limited the accurate monitoring of biological processes that involve an increase in local hydrophilicity, e.g., protein unfolding. Acridine15, pyrene-3-carboxaldehyde (PCA)16, and 7-methoxy-4-methylcoumarin (MMC)17 are fluorophores that are reported to emit fluorescence signals in polar media; unfortunately, they have limited applicability due to fluorescence quenching by protonation, difficulty of structural modification, and short excitation and emission wavelengths18. Other researchers have developed hydrogen-bonding-modulated fluorescent probes with strong fluorescence in protic solvents18,19, but their fluorescence is correlated with hydrogen-bonding donability rather than solvent polarity.
Lysosomes are sacs of hydrolytic enzymes within cells whose function is to break down unwanted macromolecules into basic molecules for cell recycling20,21. Lysosome dysfunctions, e.g., lysosome membrane permeabilization (LMP)22 and lysosome storage disorders (LSDs)23, are linked to cell death, several neurodegenerative disorders, cancer, cardiovascular diseases, and ageing-related diseases24,25,26. Depending on their functional state, lysosomes can vary considerably in structure, content, and internal environment27,28. Inherited LSDs are caused by a genetically determined deficiency of a lysosomal enzyme and are characterized by a significant augmentation of the lysosomal apparatus23 and an accumulation of polar lipids29 or water-soluble substrates30. The distinctive sign of LMP is the translocation of soluble lysosomal components from the lysosomal lumen to the cytosol22. These changes in lysosomal content are accompanied by alterations in the local polarity within cells. Thus, the determination of lysosomal polarity would be very helpful for the study of lysosomal physiology and pathology. Unfortunately, to the best of our knowledge, there are currently no polarity-sensitive fluorescent probes suitable for lysosomal applications.
Here, we report a rational design of a series of “off-on” polarity-sensitive fluorescent probes BP series that respond to highly polar media by emitting stronger fluorescence. Our approach relies on the polarity-dependent donor-excited photoinduced electron transfer (d-PET) mechanism that occurs between difluoroboron dippyomethene (BODIPY) fluorophore31,32,33,34,35,36 and the quaternary ammonium moiety. In addition, this study demonstrates that the novel polarity sensor BP-2 has significant potential for detecting local hydrophilicity in cell lysosomes.
Results
Probe design and synthesis
To achieve “off-on” polarity sensing, we introduced positively charged quaternary ammonium to the meso-position of BODIPY fluorophore. In nonpolar media, the quaternary ammonium cation possesses low negative reduction potential (i.e., reduction was favored) and can be served as an efficient electron acceptor for the BODIPY fluorophore, thus allowing it to “switch off” BODIPY fluorescence via a d-PET process. As the solvent polarity increases, the reduction potential of the acceptor decreases (i.e., reduction was unfavored)10. Finally, the fluorescence can be revived in polar solvents because of the suppression of d-PET. In addition, the introduction of the quaternary ammonium moiety mediates the hydrophilic-lipophilic balance of the BODIPY fluorophore and imparts lysosome-targeting capability to the probes37.
Figure 1 outlines the synthesis of these compounds. Briefly, amination of meso-chloromethyl-BODIPY 138 with dimethylamine furnished tertiary amine 2. Subsequent quaternization gave probe BP-1. BP-2 and BP-3 were synthesized via an unusual Mannich-type reaction39 of meso-methyl-BODIPY 340 with dimethylamine or diethylamine in dichloromethane (DCM) and subsequent quaternization. In the synthesis of BP-4, condensation of 2,4-dimethylpyrrole with 4-bromobutanoyl and BF3•OEt2 was expected to generate meso-bromopropyl-BODIPY 7. However, coupling of 4-bromobutanoyl and 2,4-dimethylpyrrole yielded dipyrromethene and water (see Supplementary Figure S1). Bromine was subsequently substituted by water under basic conditions (Et3N), followed by boron insertion with BF3•OEt2, which yielded meso-hydroxypropyl-BODIPY 6a. Parts of 6a were esterified with the remaining 4-bromobutanoyl to yield 6b. Bromination of 6a with PBr3 produced 7, which was then aminated and quaternized to yield BP-4.

Synthesis of probes BP-1, BP-2, BP-3, and BP-4.
Spectroscopic properties
We first investigated the spectroscopic properties of BP-1 (see Supplementary Figure S2), BP-2 (see Fig. 2 and Supplementary Table S1), BP-3 (see Supplementary Figure S3), and BP-4 (see Supplementary Figure S4) in several solvents that vary widely in polarity levels, as expressed by their orientation polarizability values (Δf, see Supplementary Equation S1). The absorption spectra of the probes showed almost no appreciable changes in their maxima and intensities with varying Δf. By contrast, the fluorescence spectra of the probes exhibited a prominent solvatofluoromism. All probes emitted weak fluorescence in apolar solvents but strong fluorescence in polar solvents. The emission intensities (I) and fluorescence quantum yields (Φf) were strongly correlated with the solvent polarity. Compared to BP-1 and BP-4, the two-carbon-linker probes BP-2 and BP-3 demonstrated higher linearity (R2 = 0.93–0.98) between I or Φf and Δf in the range of 0.199–0.320 and lower background fluorescence (in toluene). To quantify the sensitivity of the probes, the slope (x) of the fitted line was determined. BP-2 exhibited higher x values of 7.17 (I) and 4.27 (Φf) than BP-3. These results indicated that BP-2, with a two-carbon linker and a trimethyl quaternary ammonium moiety, showed the best linear response and highest sensitivity to solvent polarity among the BP probes. Thus, BP-2 was chosen for further investigation. The fluorescence lifetime (τ) of BP-2 did not vary significantly in different solvents (τ ranged from 3.42 to 5.14 ns in all solvents, see Supplementary Table S1). The radiative decay rate constant (kr) and non-radiative decay rate constant (knr) were calculated from Φf and τ using Supplementary Equation S2 and S341. With increasing solvent polarity, the kr value of BP-2 increased while the knr value decreased, suggesting that non-radiative processes were suppressed and BP-2 fluorescence was recovered in the polar solvents.

Absorption (a) and fluorescence (b) spectra of BP-2 in different solvents (EA, ethyl acetate; THF, tetrahydrofuran; MIPK, methyl isopropyl ketone). Normalized fluorescence intensities (c) and fluorescence quantum yields (d) of BP-2 as a function of the solvent orientational polarity parameter Δf. (e) Fluorescence spectra and intensities (inset) of BP-2 in THF with increasing amounts of water (0–10%). (f) Fluorescence spectra and intensities (inset) of BP-2 at various temperatures. λex = 480 nm.
The specific interactions of BP-2 with polar solvents were identified by examining its emission spectra in the binary solvents and temperature titration. Addition of a small amount of water to a solution of BP-2 in tetrahydrofuran (THF) resulted in substantial spectral changes (Fig. 2e and see Supplementary Figure S5), which is a hallmark of a specific solvent effect41. The emission intensity of BP-2 in aqueous solution decreased as the temperature increased (Fig. 2f), which is consistent with weaker fluorophore-solvent interactions at higher temperatures. Fortunately, BP-2 gave inconsiderable response to temperature within 35–40 °C which covers the range of general mammalian temperature (see Supplementary Figure S6a). In addition, the temperature-sensitive behavior of BP-2 was recyclable between 10 °C and 50 °C (see Supplementary Figure S6b). Given the quaternary ammonium in BP-2 is positively charged, the solvent effect between BP-2 and polar solvents is defined as the charge-dipole interactions42.
Studies of the d-PET process in BP-2
The feasibility of electron transfer between a fluorophore and a quencher can be assessed by the Rehm-Weller equation (Equation 1)43,

Differential pulse voltammetry (DPV) was performed to determine the redox potentials of BODIPY and the quaternary ammonium cation in different solvents, including toluene, THF, DCM, methanol (MeOH), acetonitrile (ACN), and water. The oxidation potential of BODIPY maintained an almost constant value (from 1.12 eV in toluene to 1.18 eV in water) regardless of solvent polarity, while the reduction potential of the quaternary ammonium moiety significantly decreased (from −1.00 eV in toluene to −1.79 eV in water) as the solvent polarity increased (Fig. 3a). This result indicates that it is easier for the quaternary ammonium moiety to be reduced (i.e., to accept electrons) in less polar solvents. For the other parameters in the Rehm-Weller Equation, ΔE0.0 was essentially constant at 2.45 ± 0.01 eV, and r was estimated to be 5 Å by geometry optimization of BP-2 using density functional theory (DFT) calculations. Taken together, the driving force (ΔGeT) in different solvents was calculated. As depicted in Fig. 3b, the ΔGeT value increased with increasing solvent polarity (from −1.57 eV in toluene to 0.49 eV in water), suggesting that electron transfer from BODIPY to the quaternary ammonium cation (d-PET) in BP-2 occurs more readily in solvents with lower polarity.

Electrochemical and theoretical studies of the d-PET process in BP-2.
(a) Plots of E(D+/D)ox and E(A/A−)red against solvent polarity; (b) changes in ΔGeT as a function of Δf; (c) frontier orbital energy representation of the d-PET process in BP-2 in different solvents.
Frontier orbital energy diagrams were also constructed according to the DFT method to investigate the fluorescence signal output of BP-2 in solvents with different polarities (Fig. 3c). The HOMO (−5.52 to −5.42 eV) and LUMO (−2.43 to −2.35 eV) levels of BODIPY remained almost unchanged in all solvents. For the quaternary ammonium cation, the LUMOs varied significantly with solvent polarity. In toluene and THF, d-PET was favored because the LUMOs of the cation (−3.75 eV for toluene and −2.48 eV for THF) were located between the HOMO and LUMO of BODIPY. However, in polar solvents, the LUMO of the cation rose far above that of BODIPY (0.72 eV for MeOH and ACN, 0.80 eV for water), thus inhibiting the d-PET process and restoring the fluorescence. The HOMO of the cation was not taken into account due to its much lower location compared to that of BODIPY (see Supplementary Figure S7). Taken above, we conclude that the “off-on” response of BP-2 fluorescence from apolar to polar solvents is primarily ascribed to the polarity-modulated d-PET process.
One may concern that the varied solubilities of BP-2 in different solvents may involve in its solvent-dependent fluorescence. As depicted in Supplementary Figure S8, the absorbance of BP-2 increased linearly with BP-2 concentration (R2 = 0.99) in the range of 1–10 μM in all solvents. No aggregation was detected by dynamic light scattering (DLS) at a concentration of 10 μM (data not shown), even in toluene and ACN, which are usually used to precipitate quaternary ammonium salts.
Evaluation of BP-2 selectivity
A set of assays was performed to evaluate BP-2 selectivity for solvent polarity (see Supplementary Figure S9). First, we investigated the sensitivity of BP-2 to solution viscosity in a binary solution of glycerol and ethanol, solvents with similar polarities (Δf = 0.274 and 0.290 for glycerol and ethanol, respectively). When the ratio of glycerol to ethanol was increased, only a slight increase in BP-2 fluorescence emission (0.3-fold) was observed, which may be attributed to inhibited vibration and rotation of the methyl groups on the BODIPY skeleton. A pH titration study revealed that BP-2 fluorescence maintained a constant maximum value in buffered aqueous solutions over a wide range of pH values (pH 4–10). Additionally, we evaluated the fluorescence response of BP-2 to a variety of biologically relevant species, including ions, amino acids, reactive oxygen species (ROS), nucleic acids, and proteins. The presence of these species did not significantly alter the fluorescence spectrum of BP-2. Altogether, these results indicate that BP-2 has potential as a specific polarity sensor in complicated biological environments.
Subcellular distribution of BP-2
Next, we turned our attention to evaluate BP-2 in live-cell imaging assays. After MCF-7 cells were incubated with 5 μM BP-2 for 20 min at 37 °C, BP-2 penetrated through the cell membrane and stained the cells. Punctate fluorescence was observed near perinuclear regions (see Supplementary Figure S10) in the green wavelength range (490 nm to 550 nm). The fluorescence spectra extracted from cell images was nearly identical to that of BP-2 measured in water. Subsequently, the subcellular distribution of BP-2 was determined by co-staining experiments (Fig. 4) with commercially available organelle markers. BP-2 fluorescence overlapped significantly with that of LysoTracker Red (yellow areas in merged images), whereas little overlap was observed with other markers (Fig. 4i–l). The changes in fluorescence intensities for BP-2 and LysoTracker Red were almost synchronized (Fig. 4m). From the correlation plots, a high Pearson’s coefficient (Rr = 0.95) was obtained for BP-2 with LysoTracker Red co-staining (Fig. 4n). These observations suggest that BP-2 can specifically localize to lysosomes in living cells.

Confocal fluorescence images of MCF-7 cells co-labeled with (e–h) BP-2 (5 μM) and commercial organelle markers: (a) ER-Tracker Red (1 μM); (b) MitoTracker Deep Red (500 nM); (c) BODIPY TR ceramide (1 μM); (d) LysoTracker Red (100 nM). (i–l) Merged images of (a,e), (b,f), (c,g), (d,h), respectively. (m) Intensity profile of the region of interest (ROI) across the cell in (l). (n) Correlation plot of BP-2 and LysoTracker Red fluorescence intensities.
Reporting changes in local hydrophilicity within lysosomes
We tested the utility of BP-2 in the visualization of chloroquine-induced LMP44. BP-2-prestained MCF-7 cells were treated with different concentrations of chloroquine (25, 50 and 100 μM) and the fluorescence images were recorded immediately. The lysosome-characteristic punctate fluorescence of BP-2 disappeared upon stimulation by chloroquine (25 μM) and was replaced by diffuse staining throughout the cytoplasm (Fig. 5b,e). The intensity profile (Fig. 5h) clearly displayed the substantial difference in the distribution of BP-2 fluorescence between before and after the addition of chloroquine in living cells: accumulating in specific regions (BP-2 only); almost uniformly distribution (in the presence of chloroquine). To quantify fluorescence intensity, ten regions of interest were selected for each image, and the average fluorescence over these areas was calculated. BP-2 fluorescence gradually decreased as the concentration of chloroquine increased (Fig. 5g). After treatment with 100 μM chloroquine, the relative emission intensity of BP-2 sharply decreased from 939 ± 226 to 79 ± 34. Note that the fluorescence of BP-2 was independent of chloroquine in the phosphate-buffered saline (PBS) buffer (see Supplementary Figure S11), which indicates that the decrease in local hydrophilicity was responsible for the observed variations in BP-2 fluorescence.

Confocal fluorescence images of MCF-7 cells stained with 5 μM BP-2 and stimulated with different concentrations of chloroquine: (a) blank; (b) 25 μM; (c) 50 μM; (f) 100 μM. (d,e) Enlarged images of representative cells (red squares) in (a,b). (g) Statistical analyses performed with the two-sample t-test (n = 10 fields of cells). *P < 0.05, **P < 0.01, ***P < 0.001; error bars are ± sem. (h) Intensity profile of the ROIs across the cell in (d,e).
Finally, we used BP-2 to characterize the mimic LSD induced by high sucrose. MCF-7 cells pre-labelled with BP-2 were loaded with 80 mM sucrose and then imaged with confocal fluorescence microscope immediately. Lysosomes swelled in the presence of sucrose (Fig. 6c,d), accompanied by a remarkable increase in BP-2 fluorescence. The relative fluorescence intensity of BP-2 increased from 825 ± 117 to 1857 ± 475 after sucrose treatment (Fig. 6e). No detectable change in fluorescence was observed when 80 mM sucrose was added to the buffer solution of BP-2 (see Supplementary Figure S11), demonstrating that the fluorescence enhancement was due to the increase in lysosomal local hydrophilicity. These results suggest that massive sucrose induces a more polar environment within lysosomes.

Confocal fluorescence images of MCF-7 cells stained with 5 μM BP-2 before (a,b) and after (c,d) the addition of 80 mM sucrose. (b,d) Enlarged images of representative cells (red squares) in (a,c). (e) Statistical analysis performed with the two-sample t-test (n = 10 fields of cells). ***P < 0.001; error bars are ± sem.
Discussion
Polarity-sensitive fluorescent probes offer a unique opportunity for non-invasive, in situ determination of microenvironmental polarity in biological systems3. Much effort has been devoted to the development of polarity-sensitive “on-off” probes to study hydrophobic interactions and structures4,5,6,7,8,9. However, there is a lack of “off-on” polarity-sensing probes for studying and imaging biological processes that involve in an increase in local hydrophilicity. In this work, we designed a series of polarity-sensitive fluorescent probes whose “off-on” mechanism operates based on the sensitivity of the redox potential of quaternary ammonium to solvent polarity. In apolar solvents, the quaternary ammonium is capable of quenching the fluorescence of BODIPY through a d-PET process due to its large electron deficiency and high reduction potential. As the environmental polarity increases, the charge-dipole interaction between the quaternary ammonium and polar solvent molecules decreases the reduction potential of quaternary ammonium. An increase in polarity reduces the ability of quaternary ammonium to accept electrons, thus blocking the d-PET process and recovering BODIPY fluorescence. We examined the effect of linker length and size of quaternary ammonium on the ability of the probes to sense changes in polarity. We found that a two-carbon linker is the optimal spacer for the electron transfer from BODIPY to the quaternary ammonium, and that the small size of trimethyl quaternary ammonium facilitates its specific interaction with polar molecules.
Lysosomes serve as the dispatch center for cell recycling21 and have significant functions in the pathogenesis of various disorders and diseases24,25,26. The morphology and contents of lysosomes are variable depending on their functional state27,28. Alterations in the internal environment of lysosomes under pathological conditions are reflected by changes in lysosomal polarity. Therefore, lysosomal polarity-sensing probes may be useful tools that can improve our understanding of lysosome-related activities and disorders. Although several existing polarity-sensitive probes show specific subcellular distributions9,10,11,12,14, none of these probes are designed to target lysosomes. In this study, BP-2 has been proven to be the first polarity-sensitive probe that specifically localized to lysosomes in living cells. Moreover, we utilized BP-2 in confocal fluorescence imaging to report the changes in local hydrophilicity within lysosomes under chloroquine-induced LMP (decrease) and mimic LSD (increase).
In summary, we have developed a series of “off-on” polarity-sensitive fluorescent probes (BP-1, BP-2, BP-3 and BP-4) that respond to highly polar media by emitting a higher fluorescence signal via a solvent-dependent d-PET mechanism. Among these probes, BP-2, with a two-carbon linker and a trimethyl quaternary ammonium moiety, exhibited the best polarity-sensing properties: linear response, high sensitivity and specificity. Significantly, using BP-2, we present the first fluorescence images that reflect local hydrophilicity within lysosomes under conditions of lysosomal dysfunction. Our work opens up a new arena for the design of “off-on” polarity-sensitive fluorescent probes and provides a novel probe (BP-2) that shows significant potential as a chemical tool for the study of lysosomal biology and the diagnosis of lysosome-related disorders.
Methods
General information
General chemicals were of analytical grade without further purification. All the solvents employed were of spectrometric grade. Solutions of ions were prepared from FeCl3 · 6H2O, CrCl3, MnCl2 · 5H2O, CoCl2 · 6H2O, NiCl2 · 6H2O, CuCl2 · 2H2O, KCl, NaCl, MgCl2 · 6H2O, CaCl2, Na2CO3, Na2SO3, Na3PO4, NaF, KBr, NaNO3, NaAc, NaClO4 dissolved in distilled water. Solutions or generation of ROS were prepared according to our previous report45. Amino acids, proteins, and nucleic acids were dissolved in distilled water to prepare stock solutions. Stock solutions (5 mM) of BP series were prepared in dimethyl sulphoxide (DMSO) and stored in a refrigerator for use. Organelle markers were purchased from Invitrogen (USA). Sucrose was obtained from Energy Chemical (China). Chloroquine was from Sigma-Aldrich (USA). 1H-NMR and 13C-NMR spectra were recorded on a VARIAN INOVA-400 spectrometer with chemical shifts (δ) reported as ppm (in CDCl3 or d6-DMSO, TMS as the internal standard). Mass spectrometry data were obtained with an HP1100LC/MSD mass spectrometer and an LC/Q-TOF MS spectrometer. Absorption spectra were measured on a Lambda 35 UV/VIS spectrophotometer (Perkin Elmer). Fluorescence measurements were performed on a VARIAN CARY Eclipse fluorescence spectrophotometer (Serial No. FL0812-M018). Excitation and emission slit widths were modified to adjust the fluorescence intensity to a suitable range. The fluorescence lifetimes were attained from a Horiba Jobin Yvon Fluoromax-4p. Slight pH variations in the solutions were achieved by adding the minimum volumes of NaOH or HCl (1 M). All pH measurements were made with a Model PHS-3C meter. For the viscosity sensitivity experiment34, BP-2 was added to the solvent mixtures of ethanol and glycerol in different proportions. The final solutions were sonicated for 5 min to eliminate air bubbles. After standing for 1 h at r. t., the fluorescence measurements were performed.
Live-cell imaging experiments
MCF-7 cells were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen). Cells were seeded in 24-well flat-bottomed plates and incubated for 24 h at 37 °C under 5% CO2. BP-2 (5 μM) was added (the concentration of DMSO was maintained to be less than 0.2%) and cells were further incubated for 20 min, followed by washing thrice with phosphate-buffered saline (PBS). The fluorescence imaging was performed with OLYMPUS FV-1000 inverted fluorescence microscope with 100× objective lens. Under the confocal fluorescence microscope, BP-2 was excited at 488 nm and emission was collected at 490–550 nm. Co-localization experiments were conducted by co-staining the cells with combinations of BP-2 and ER-Tracker Red (1 μM, λex = 559 nm, λem = 570–650 nm)/MitoTracker Deep Red (500 nM, λex = 635 nm, λem = 655–755 nm)/BODIPY TR ceramide (1 μM, λex = 559 nm, λem = 570–650 nm)/LysoTracker Red (100 nM, λex = 559 nm, λem = 570–650 nm) for 20 min. For the LMP experiment, MCF-7 cells internalized with BP-2 (5 μM, 20 min) were treated with chloroquine (25, 50 and 100 μM). For the sucrose-stimulation experiment, MCF-7 cells were incubated with BP-2 (5 μM, 20 min) and then sucrose (80 mM) was added.
Additional Information
How to cite this article: Zhu, H. et al. d-PET-controlled “off-on” Polarity-sensitive Probes for Reporting Local Hydrophilicity within Lysosomes. Sci. Rep. 6, 35627; doi: 10.1038/srep35627 (2016).
References
Szczupak, B. et al. Polarity Assessment of Thermoresponsive Poly(NIPAM-co-NtBA) Copolymer Films Using Fluorescence Methods. J. Fluoresc. 20, 719–731 (2010).
Suppan, P. Solvent effects on the energy of electronic transitions: experimental observations and applications to structural problems of excited molecules. J. Chem. Soc. A 3125–3133 (1968).
Yang, Z. et al. Macro-/micro-environment-sensitive chemosensing and biological imaging. Chem. Soc. Rev. 43, 4563–4601 (2014).
Summerer, D. et al. A genetically encoded fluorescent amino acid. Proc. Natl. Acad. Sci. 103, 9785–9789 (2006).
Cohen, B. E. et al. Probing Protein Electrostatics with a Synthetic Fluorescent Amino Acid. Science 296, 1700–1703 (2002).
Venkatraman, P. et al. Fluorogenic probes for monitoring peptide binding to class II MHC proteins in living cells. Nat. Chem. Biol. 3, 222–228 (2007).
Parasassi, T., Krasnowska, E., Bagatolli, L. & Gratton, E. Laurdan and Prodan as Polarity-Sensitive Fluorescent Membrane Probes. J. Fluoresc. 8, 365–373 (1998).
Kim, H. M. et al. Two-Photon Fluorescent Turn-On Probe for Lipid Rafts in Live Cell and Tissue. J. Am. Chem. Soc. 130, 4246–4247 (2008).
Kucherak, O. A. et al. Switchable Nile Red-Based Probe for Cholesterol and Lipid Order at the Outer Leaflet of Biomembranes. J. Am. Chem. Soc. 132, 4907–4916 (2010).
Sunahara, H., Urano, Y., Kojima, H. & Nagano, T. Design and Synthesis of a Library of BODIPY-Based Environmental Polarity Sensors Utilizing Photoinduced Electron-Transfer-Controlled Fluorescence ON/OFF Switching. J. Am. Chem. Soc. 129, 5597–5604 (2007).
Signore, G., Nifosì, R., Albertazzi, L., Storti, B. & Bizzarri, R. Polarity-Sensitive Coumarins Tailored to Live Cell Imaging. J. Am. Chem. Soc. 132, 1276–1288 (2010).
Jiang, N. et al. Ratiometric Fluorescence Imaging of Cellular Polarity: Decrease in Mitochondrial Polarity in Cancer Cells. Angew. Chem. Int. Ed. 54, 2510–2514 (2015).
Tainaka, K. et al. PRODAN-Conjugated DNA: Synthesis and Photochemical Properties. J. Am. Chem. Soc. 129, 4776–4784 (2007).
Yang, Z. et al. A Nile Red/BODIPY-based bimodal probe sensitive to changes in the micropolarity and microviscosity of the endoplasmic reticulum. Chem. Commun. 50, 11672–11675 (2014).
Kellmann, A. Intersystem crossing and internal conversion quantum yields of acridine in polar and nonpolar solvents. J. Phys. Chem. 81, 1195–1198 (1977).
Kalyanasundaram, K. & Thomas, J. K. Solvent-dependent fluorescence of pyrene-3-carboxaldehyde and its applications in the estimation of polarity at micelle-water interfaces. J. Phys. Chem. 81, 2176–2180 (1977).
Seixas de Melo, J. S., Becker, R. S. & Macanita, A. L. Photophysical Behavior of Coumarins as a Function of Substitution and Solvent: Experimental Evidence for the Existence of a Lowest Lying 1(n,pi.*) State. J. Phys. Chem. 98, 6054–6058 (1994).
Uchiyama, S., Takehira, K., Yoshihara, T., Tobita, S. & Ohwada, T. Environment-Sensitive Fluorophore Emitting in Protic Environments. Org. Lett. 8, 5869–5872 (2006).
Han, F. et al. Environment sensitive phenothiazine dyes strongly fluorescence in protic solvents. J. Photochem. Photobiol. A 196, 10–23 (2008).
Kornfeld, S. & Mellman, I. The Biogenesis of Lysosomes. Annu. Rev. Cell Biol. 5, 483–525 (1989).
Appelqvist, H., Wäster, P., Kågedal, K. & Öllinger, K. The lysosome: from waste bag to potential therapeutic target. J. Mol. Cell Biol. 5, 214–226 (2013).
Boya, P. & Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 27, 6434–6451 (2008).
Neufeld, E. F. Lysosomal Storage Diseases. Annu. Rev. Biochem. 60, 257–280 (1991).
Boya, P. et al. Lysosomal Membrane Permeabilization Induces Cell Death in a Mitochondrion-dependent Fashion. J. Exp. Med. 197, 1323–1334 (2003).
Platt, F. M., Boland, B. & van der Spoel, A. C. Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell Biol. 199, 723–734 (2012).
He, L.-q., Lu, J.-h. & Yue, Z.-y. Autophagy in ageing and ageing-associated diseases. Acta Pharmacol. Sin. 34, 605–611 (2013).
Lüllmann-Rauch, R. In Lysosomes 1–16 (Springer: US,, 2005).
Wang, L., Xiao, Y., Tian, W. & Deng, L. Activatable Rotor for Quantifying Lysosomal Viscosity in Living Cells. J. Am. Chem. Soc. 135, 2903–2906 (2013).
Lüllmann-Rauch, R., Matzner, U., Franken, S., Hartmann, D. & Gieselmann, V. Lysosomal sulfoglycolipid storage in the kidneys of mice deficient for arylsulfatase A (ASA) and of double-knockout mice deficient for ASA and galactosylceramide synthase. Histochem. Cell Biol. 116, 161–169 (2001).
Stinchi, S. et al. Targeted Disruption of the Lysosomal α-Mannosidase Gene Results in Mice Resembling a Mild form of Human α-Mannosidosis. Hum. Mol. Genet 8, 1365–1372 (1999).
Boens, N., Leen, V. & Dehaen, W. Fluorescent indicators based on BODIPY. Chem. Soc. Rev. 41, 1130–1172 (2012).
Zhu, H., Fan, J., Wang, J., Mu, H. & Peng, X. An “Enhanced PET”-Based Fluorescent Probe with Ultrasensitivity for Imaging Basal and Elesclomol-Induced HClO in Cancer Cells. J. Am. Chem. Soc. 136, 12820–12823 (2014).
Zhu, H. et al. Ratiometric fluorescence imaging of lysosomal Zn2+ release under oxidative stress in neural stem cells. Biomater. Sci. 2, 89–97 (2014).
Zhu, H. et al. A “Distorted-BODIPY”-Based Fluorescent Probe for Imaging of Cellular Viscosity in Live Cells. Chem. Eur. J 20, 4691–4696 (2014).
Zhao, C. et al. Förster Resonance Energy Transfer Switchable Self-Assembled Micellar Nanoprobe: Ratiometric Fluorescent Trapping of Endogenous H2S Generation via Fluvastatin-Stimulated Upregulation. J. Am. Chem. Soc. 137, 8490–8498 (2015).
Wang, F. et al. A dual-response BODIPY-based fluorescent probe for the discrimination of glutathione from cystein and homocystein. Chem. Sci. 6, 2584–2589 (2015).
Zhang, X., Wang, C., Han, Z. & Xiao, Y. A Photostable Near-Infrared Fluorescent Tracker with pH-Independent Specificity to Lysosomes for Long Time and Multicolor Imaging. ACS Appl. Mater. Interfaces 6, 21669–21676 (2014).
Wu, Y. et al. Boron dipyrromethene fluorophore based fluorescence sensor for the selective imaging of Zn(ii) in living cells. Org. Biomol. Chem. 3, 1387–1392 (2005).
Yang, L. & Yan, H. A Mannich-Type Reaction at the meso-Methyl Position of the BODIPY Fluorophore. Synlett 23, 1526–1528 (2012).
Guo, B. et al. Synthesis and spectral properties of new boron dipyrromethene dyes. Dyes Pigm. 73, 206–210 (2006).
Lakowicz, J. R. Principles of Fluorescence Spectroscopy, 3rd (Springer, 2006).
Neuwald, A. F. The Charge-dipole Pocket: A Defining Feature of Signaling Pathway GTPase On/Off Switches. J. Mol. Biol. 390, 142–153 (2009).
Rehm, D. & Weller, A. Kinetics of Fluorescence Quenching by Electron and H-Atom Transfer. Isr. J. Chem. 8, 259–271 (1970).
Boya, P. et al. Mitochondrial membrane permeabilization is a critical step of lysosome-initiated apoptosis induced by hydroxychloroquine. Oncogene 22, 3927–3936 (2003).
Chen, G. et al. FRET spectral unmixing: a ratiometric fluorescent nanoprobe for hypochlorite. Chem. Commun. 48, 2949–2951 (2012).
Acknowledgements
We thank Prof. Shiguo Sun for assistance in electrochemical measurements. This work was financially supported by NSF of China (21136002, 21576037, 21422601, 21406028 and 21421005), National Basic Research Program of China (2013CB733702).
Author information
Authors and Affiliations
Contributions
H.Z. performed all the compound synthesis and charaterization. H.M. performed cellular imaging and DFT calculations. T.Z. and Z.Z performed spectra measurements. All authors contributed to data analysis. H.Z., J.F., J.D. and X.P. conceived the idea and co-wrote the paper.
Ethics declarations
Competing interests
Dalian University of Technology is in the process of filing a patent protection on the work described in this manuscript.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhu, H., Fan, J., Mu, H. et al. d-PET-controlled “off-on” Polarity-sensitive Probes for Reporting Local Hydrophilicity within Lysosomes. Sci Rep 6, 35627 (2016). https://doi.org/10.1038/srep35627
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep35627
This article is cited by
-
An off-on fluorescence probe targeting mitochondria based on oxidation-reduction response for tumor cell and tissue imaging
Journal of Nanoparticle Research (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
