
Abstract
In brains of Alzheimer’s disease (AD), reactive oxygen species (ROS) levels are significantly higher than that of healthy brains. Evidence suggests that, during AD onset and progression, a vicious cycle revolves around amyloid beta (Aβ) production, aggregation, plaque formation, microglia/immunological responses, inflammation, and ROS production. In this cycle, ROS species play a central role, and H2O2 is one of the most important ROS species. In this report, we have designed a fluorescent imaging probe CRANAD-88, which is capable of cascade amplifying near infrared fluorescence (NIRF) signals at three levels upon interacting with H2O2 in AD brains. We demonstrated that the amplification was feasible in vitro and in vivo. Remarkably, we showed that, for the first time, it was feasible to monitor the changes of H2O2 concentrations in AD brains before and after treatment with an H2O2 scavenger. Our method opens new revenues to investigate H2O2 in AD brains and can be very instructive for drug development.
Similar content being viewed by others

Introduction
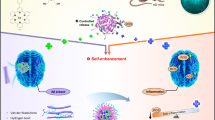
Alzheimer’s disease (AD) is a terminal neurodegenerative disease, and no effective treatment is available so far. Based on the hallmarks of amyloid beta (Aβ) plaques and neurofibrillary Tau tangles, the so-called amyloid hypothesis and tau hypothesis have been proposed for AD pathology. However, the prevalent drug development strategies, which are primarily built on these hypotheses, have not been successful in delivering effective treatment1,2,3,4. This strongly indicates that other factors should also be taken into account in AD drug development. It has long been believed that AD is closely associated with oxidative stress, and evidence shows that in AD brains the reactive oxygen species (ROS) level is significantly higher than that in healthy control brains5,6,7. The increased ROS level can be attributed to multiple factors, such as over-accumulation of metal ions, aggregation of Aβs, inflammation, and microglia activation.
During the formation and growth of Aβ plaques, numerous ROS species are generated, and H2O2 can be considered to be one of the most important ROS species. H2O2 is produced through the Fenton reaction of Aβs with metal ions8,9,10,11. The generated ROS/H2O2 can further induce over-accumulation of inflammatory cytokines such as TNFα12,13, which can then attract microglia to encircle the active plaques13,14,15. The surrounding microglia release more ROS/H2O2, and contribute to the neuronal loss characteristic of this disease15. Studies show that interactions between microglia cells and the plaques can activate numerous receptors and enzymes. Among them, oxidative stress related NAD(P)H oxidase plays a crucial role15,16. Upon activation, NAD(P)H oxidase produces highly reactive ROS/H2O215. This production of ROS/H2O2 further initiates production and crosslinking/aggregation of Aβs17,18,19, which in turn can lead to the generation of ROS/H2O28,9,10,11. Moreover, recent data indicates that TNFα can also increase the production of Aβs18,19, which can additionally strengthen Aβ aggregation and crosslinking. According to the above facts, it is abundantly clear that a vicious cycle revolves around plaque formation, microglia/immunological responses, inflammation, ROS production, Aβ production and aggregation/crosslinking. As illustrated in Fig. 1a, ROS/H2O2 species play a central role. Therefore, detection of ROS/H2O2 in the AD brain in vivo is highly desirable. In this report, we have concentrated our efforts on the detection of H2O2 in AD brains.

(a) The vicious cycle revolves around Aβ species and plaques; (b) Design of CRANAD-88 and the proposed three levels of cascade signal amplification.
Although H2O2 plays critical roles in numerous normal and abnormal biological processes, few references are available for the concentration of H2O2 in the brain of living beings, including human and mice, and it is not clear how the concentration of H2O2 changes under disease conditions such as AD20. Numerous imaging probes have been reported for detecting H2O2 in cells and in peripheral targets; however, none has been reported to detect H2O2 in brains under different disease status21,22,23,24,25,26,27. Clearly, it is an urgent need to provide an in vivo imaging tool to detect the H2O2 concentration in the living brain, particularly AD brains.
Several strategies for detecting H2O2 have been developed, and Chang’s boronate method and Nagano’s diketone method are the most validated approaches25,26,27. Although the reported H2O2 imaging probes are great for peripheral application and cell imaging25,26,27, the visualization of H2O2 in the brain is not very straightforward. Therefore it is still challenging to use them for in vivo imaging of H2O2 in AD brains.
In this report, we designed a curcumin analogue CRANAD-88, which has a capacity of three levels of cascade signal amplification. Remarkably, we demonstrated for the first time, that the changes of H2O2 concentrations from AD brains could be monitored before and after H2O2 scavenging. We believe that our method will be a valuable tool for investigating H2O2 in AD brains, and for AD drug development and treatment regime design.
Results and Discussion
Design of fluorescence imaging probe CRANAD-88
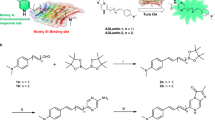
Other researchers have reported various strategies to design H2O2 responsive probes25,26,27. Among them, boronate oxidation is one of the most validated approaches for the design of imaging probes that can increase fluorescent intensity after reaction with H2O225,26. However, utilizing this method for cascade signal amplification has not been explored. In our previous studies, we demonstrated that CRANAD-58 was a “smart” NIRF probe for Aβs, and displayed about a 100-fold intensity increase after binding to Aβs28,29,30,31. To achieve cascade signal amplification, we integrated our previous Aβ “smart” probe strategy into the design of an H2O2 responsive probe. Through the integration, CRANAD-88 was designed by linking a close mimic of CRANAD-58 (Aβ responsive moiety) to a boronate (H2O2 responsive moiety) via a carbamate moiety (Fig. 1b). The benefit of the carbamate linker is that it leads the designed CRANAD-88 to be “invisible” in the near infrared spectrum. This invisibility is due to the carbamate’s electron-withdrawing effect, which shortens the probe’s excitation/emission wavelength. However, it becomes “visible” once it reacts with H2O2, because the boronate and carbamate moieties are removed to generate an electron-rich amino group for red-shifting. The probe’s reaction with H2O2, and the subsequent binding of the reaction intermediate with Aβs, as well as the red-shift all contribute to the cascade amplification of the NIRF signal (Fig. 1b).
Synthesis and spectral characterization
As shown in Fig. 2a, we first prepared the boronate-attached benzaldehyde via Curtius reaction with DPPA (Diphenylphosphoryl azide) in a high yield (67%). CRANAD-88 was obtained based on our previous published procedures28,29,30,31. The excitation spectrum showed that its excitation peak is around 580 nm and emission was about 690 nm in PBS buffer with 20% DMSO (SI Fig. 1).

(a) The synthetic route for CRANAD-88; (b) Fluorescence intensity changes of CRANAD-88 (black line) with H2O2 (blue line, 1st amplification and red-shift), and with H2O2 + Aβ aggregates (red line, 2nd amplification). (c) Cascade signal amplification validation of CRANAD-88 with imaging parameters (Ex = 640 nm, Em = 700 nm) on a plate that was loaded with CRANAD-88 only (column 1), CRANAD-88 with H2O2 (column 2), and CRANAD-88 with H2O2 and Aβs (column 3), and (d) quantitative analysis of the image in (c).
In vitro solution testing for validating cascade fluorescence amplification
The emission peak of CRANAD-88 is around 690 nm, but its excitation peak is around 580 nm, therefore it is “invisible” for NIRF imaging (Ex > 640 nm required). To investigate whether CRANAD-88 is H2O2-responsive, we incubated CRANAD-88 in PBS buffer with various concentrations of H2O2. We found that the fluorescence intensity of CRANAD-88 was increased 3-fold after incubation with H2O2 (100 μM) at 37 °C (the first signal amplification) (Fig. 2b). We also observed significant red-shifts for both excitation and emission (Ex = 630 nm, Em = 730 nm) after the incubation (Fig. 2b). These red-shifts are very important for in vivo imaging, since a longer emission will allow imaging at greater depths and reduce background signal. Furthermore, these red-shifts convert “invisible” CRANAD-88 into a “visible” intermediate, which allows the probe to be visualized in vivo with NIRF imaging (the indirect signal amplification). To investigate whether the generated intermediate from the above incubation was responsive towards Aβs, we incubated the above solution with Aβ40 aggregates. As we expected, the fluorescence intensity of the intermediate was dramatically increased (2.5-fold) upon exposure to the aggregates (the second signal amplification) (Fig. 2b). To investigate whether the amplifications could be observed with parameters for in vivo imaging (Ex = 640 nm, Em = 700 nm), we conducted imaging (Ex = 640 nm, Em = 700 nm) with a plate that contained 1) CRANAD-88 only, 2) CRANAD-88 with H2O2, and 3) CRANAD-88 with H2O2 and Aβ40 aggregates. Consistent with solution spectral tests, significant increases could be observed from wells containing H2O2 (4-fold), and H2O2 with Aβs (16-fold) (Fig. 2c,d). Remarkably, amplifications from the plate imaging were significantly higher than that from solution spectra at 700 nm, due to the “invisibility” of CRANAD-88 with the imaging parameters. Our in vitro data suggested that three levels of cascade fluorescence signal amplification were attainable through H2O2-reponsive interactions, Aβ-responsive interactions, and transformation of the probe from “invisible” to “visible” in the near infrared spectrum.
Consistent with other reported boronate imaging probes25,26, CRANAD-88 showed higher H2O2 selectivity over other ROS species that include NO•, O2, ClO− (SI Fig. 2a). It is also very sensitive to H2O2, a significant increase could be seen with 10.0 μM H2O2 (SI Fig. 2b). In addition, CRANAD-88 exhibited quick responses towards H2O2, and a significant increase was observed only after 10 minutes of incubation (SI Fig. 2c).
Fluorescence responses of CRANAD-88 with Aβs without H2O2
When we incubated CRANAD-88 with various Aβs, but without H2O2, increases in fluorescence intensity were observed. However, we also observed significant blue-shifts to 640 nm (SI Fig. 3). The data indicates that, without H2O2, CRANAD-88 was “invisible” in the presence of Aβs under conventional NIRF imaging parameters (Ex > 640 nm and Em > 650 nm required), due to its short excitation and emission.
Phantom imaging with mouse brain homogenate
To validate whether CRANAD-88 can be used in a biologically relevant environment, we used brain homogenates from a wild type mouse to investigate the changes of CRANAD-88 after exposure to H2O2, Aβs, and both H2O2 and Aβs. We used an IVIS imaging system to collect data (Ex = 640 nm, Em = 700 nm). We observed an apparent signal increase after 30 minutes of incubation with H2O2 (500 nM), but no significant signal increase was observed after incubation with Aβs alone (Fig. 3a,b). This was because the probe was “invisible” with the imaging parameters until it was “turned on” by reaction with H2O2 (Fig. 3a,b). To confirm the cascade signal amplification in the homogenate, we further incubated the H2O2 containing homogenate with Aβ40 aggregates, and another significant increase of intensity could be observed (Fig. 3a,b). Taken together, the data suggested that the cascade signal amplification could be achieved in a biologically relevant environment.

(a) Phantom imaging with brain homogenate. CRANAD-88 with Aβ 40 aggregates, with H2O2, with H2O2 + Aβ40 aggregates (upper panel), and brain homogenate blank, and CRANAD 88 only (low panel). (b) Quantification of (a) by normalizing with the signal from the blank brain homogenate. (c–f) Brain slice staining test with CRANAD-88. (c) Background of a brain slice; (d) Treated with CRANAD-88; (e) Treated with CRANAD-88 and H2O2 (100 μM); Plaques are indicated by white arrows. Scale Bar: 200 μm. (f) Quantitative analysis of intensity ratio of a plaque and vicinity background.
In vitro mouse brain slice microscopic imaging
To investigate whether we can observe the fluorescence intensity changes of CRANAD-88 in AD mouse brain slices, we first incubated the probe with a brain slice of an 11-month old APP/PS1 mouse for 30 minutes. There was no significant intensity increase compared to the slice before probe treatment. However, we found that several plaques lit up once the same brain slice was incubated with H2O2 at 37 °C for 30 minutes (Fig. 3c,d), and the lit-up plaques were well co-localized with thioflavin S staining (SI Fig. 4a,b). We quantified the signal using the fluorescence intensity ratio of the plaque of interest vs. an ROI of the same size of the vicinity of the plaque. We found that the amplification of the signal was about 2-fold with Aβ plaques and H2O2 when compared with the background of pre-probe treatment or CRANAD-88 only (Fig. 3f). These results strongly indicated that signal amplification could be observed in an AD brain slice, and is also Aβ specific.
BBB penetrating studies
To investigate whether CRANAD-88 can cross BBB, we injected the probe into a wild type mouse, which was sacrificed and perfused at 30 minutes after the injection. The dissected brain was homogenized and extracted with ethyl acetate. Fluorescent spectral testing and LC-MS data from the brain extraction confirmed that CRANAD-88 was able to cross the BBB (SI Fig. 4c–e).
In vivo imaging with CRANAD-88
Few references are available for the concentration of H2O2 in the brain of living beings. It is not clear how the concentration of H2O2 changes under disease conditions such as AD. Studies suggest that the concentrations of H2O2 in AD brains are higher than that of normal brains5,6,7. However, direct evidence is still lacking. To prove the existence of elevated H2O2 in the AD mouse brain, we used CRANAD-88 to conduct NIRF imaging with transgenic AD model APP/PS1 mice, a well-studied transgenic model for AD research32,33. Age-matched wild type mice were used as the control group. We monitored the NIRF signal at 15-, 30-, 60-, 120-, and 240-minutes post injection. We found that the APP/PS1 group showed significantly higher fluorescence signal at all of the time points (1.56-, 1.62-, 1.51-, 2.32-, and 10.91-folds) than WT mice. This data indicated that the difference between APP/PS1 and WT reflected by CRANAD-88 was likely due to the cascade NIRF signal amplification (Fig. 4a,b).

In vivo imaging study with CRANAD-88.
(a) Representative images of WT and APP mice (0.1 mg/mice) at pre-injection, 15-, 30-, 60-, 120- and 240 min after injection. (b) The time course of CRANAD-88 with APP and WT mice. (c) Representative images of WT and APP mice, which both were treated with sodium pyruvate, d) the NIRF signal differences of CRANAD-88 between AD and WT mice (F(APP) − F(WT)) treated without (blue bar) and with sodium pyruvate, and re-appearance of H2O2 one month after pyruvate scavenging (green bar). P value: *<0.05, **<0.01, and ***<0.005.
Monitoring H2O2 scavenging in vivo
To further confirm whether the above amplification is due to the existence of H2O2 in the brain, we used sodium pyruvate, which is a widely used H2O2 scavenger for cell culture34, and is capable of penetrating the BBB35. To conduct the experiments, we injected a mixture of CRANAD-88 and sodium pyruvate intravenously. As expected, the difference between the signals from the APP/PS1 group and the control group is significantly decreased (Fig. 4c,d, red bar), strongly indicating that amplification in vivo is due to H2O2 triggering and engagement with Aβs. In order to further investigate whether H2O2 would be re-produced after the clearance of sodium pyruvate, we imaged the same groups of mice after one month. We found that the difference between APP/PS1 and WT was restored (Fig. 4d, green bar), suggesting that H2O2 can be re-produced if toxic Aβ species are still existing.
Longitudinally monitoring the changes of H2O2 concentration could be very important for investigating the effects of H2O2 on the progression of AD pathology. From the design of CRANAD-88, it is clear that the total NIRF signal originates from the interaction with H2O2 and Aβs, and it is known that both Aβ and H2O2 concentrations change with ageing; therefore, the NIRF signal changes from CRANAD-88 can’t be used to quantitatively reflect the concentration change of H2O2 in the longitudinal studies. To quantitatively calculate the relative H2O2 changes during ageing and longitudinal therapy monitoring, we will use the ratio between CRANAD-88 and CRANAD-3 (RH2O2 = R(CRANAD-88)/R(CRANAD-3)). For CRANAD-3, our previous data showed that it could reflect the changes in Aβs during ageing and treatment31. Using the ratio of the two probes, we will be able to reliably calculate relative H2O2 changes in the longitudinal studies.
Summary
H2O2 plays important roles for AD pathology. However, no method is available to detect the changes of H2O2 in the brain. In this report, results from in vitro spectral studies, plate imaging, phantom imaging, and brain slice imaging consistently suggested that three levels of cascade signal amplification was feasible. Remarkably, for the first time, we demonstrated that the changes of H2O2 concentrations in AD brain could be monitored with a NIRF imaging probe capable of cascade signal amplification. Our study is not only crucial for the proof of the existence of H2O2, but also points out several important questions that are needed to be addressed in terms of seeking effective treatments for AD. Our study strongly indicated that H2O2 can be reproduced if the Aβ plaques or other Aβs are still existing, and this pointed out that AD treatment is not only related to the removal of Aβs, but also related to ROS. Our study also pointed out another important question: whether Aβ-lowering treatment can lead to the decrease of ROS such as H2O2. If the Aβ-removal cannot reduce the ROS level, the toxic ROS will continuously damage the neurons. Therefore, ROS reduction should be considered to be one type of treatment outcome to be evaluated. In summary, our method can be used to monitor the changes of H2O2 in brains of AD mice, and this technology will be an important tool for AD drug development and treatment regime management.
Materials and Methods
Reagents used for the synthesis were purchased from Sigma-Aldrich and used without further purification. The pH of the PBS buffer was 7.4. Column chromatography was performed on silica gel (SiliCycle Inc., 60 Å, 40–63 mm) slurry packed into glass columns. Synthetic Aβ peptides (1–40/42) were purchased from rPeptide (Bogart, GA, 30622). Aggregates for in vitro studies were generated by the slow stirring of Aβ40 in PBS buffer for 3 days at room temperature. CRANAD-88 was dissolved in DMSO to prepare a 25.0 μM stock solution. 1H and 13C NMR spectra were recorded at 300 and 125 MHz, respectively, and reported in ppm downfield from tetramethylsilane. Fluorescence measurements were carried out using an F-4500 fluorescence spectrophotometer (Hitachi). Transgenic female APP-PS1 mice and age matched wild-type female mice were purchased from Jackson Laboratory. All animal experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Massachusetts General Hospital, and carried out in accordance with the approved guidelines.
Synthesis of CRANAD-88
4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl) benzyl 4-formyl-phenylcarbamate
4-Formylbenzoic acid (1.0 g, 6.7 mmol) was dissolved in 1,4-dioxane (14 mL), followed by the addition of triethylamine (0.93 mL, 6.7 mmol), DPPA (1.6 mL, 7.3 mmol), and 4-(hydroxymethyl) phenylboronic pinacol ester (1.7 g, 7.3 mmol). The resulting mixture was heated to 100 °C for 2 hours. After evaporating off the solvent, the residue was purified with a silica gel flash column chromatography (Hexanes/ethyl acetate = 3/2) to afford the intermediate as a pale yellow solid (1.7 g, yield: 66.7%).
1H NMR (CDCl3): δ(ppm) 1.34 (s, 12H), 5.23 (s, 2H), 7.06 (br, s, 1H), 7.37 (d, 2H, J = 7.8 Hz), 7.54 (d, 2H, J = 8.4 Hz), 7.81–7.84 (m, 4H), 9.89 (s, 1H). 13C NMR (CDCl3): δ(ppm) 191.9, 152.6, 143.4, 138.4, 135.1, 135.0, 131.7, 131.2, 127.4, 118.0, 83.9, 67.3, 24.8 ESI-MS (M + H) m/z = 382.1.
CRANAD-88
To a solution of the above intermediate (209 mg, 0.54 mmol) in acetonitrile (3.0 mL), 2,2-Difluoro-1,3-dioxaboryl-pentadione crystals (60 mg, 0.54 mmol) was added, followed by the additions of acetic acid (80 μL), tetrahydroisoquinoline (16 μL), and 4-(dimethylamino)-benzaldehyde (71.3 mg, 0.54 mmol). The resulting solution was stirred at 60 °C overnight. A black residue was obtained after removing the solvent and further purified with a flash column (Hexane/Ethyl acetate = 1/1) to give a black power CRANAD-88 (8.0 mg, yield: 2.2%).
1H NMR (CDCl3) δ (ppm) 1.23–1.25 (t, 6H), 1.34 (s, 12H), 3.56–3.63 (q, 4H), 5.22(s, 2H), 5.95 (s, 1H), 6.41 (d, J = 15.3 Hz, 1H), 6.51 (d, J = 9.3 Hz, 2H), 6.56 (d, J = 15.6 Hz, 1H), 6.86 (s, 1H), 7.38 (d, J = 8.1 Hz, 2H), 7.44 (d, J = 8.4 Hz, 2H), 7.53 (d, J = 8.7 Hz, 2H), 7.67 (dd, J1 = 2.4 Hz, J2 = 9 Hz, 1H), 7.81 (d, J = 8.1 Hz, 2H), 7.88 (d, J = 15.6 Hz, 1H), 7.95 (d, J = 15.3 Hz, 1H), 8.35 (d, J = 2.1 Hz, 1H); 13C NMR (CDCl3) δ (ppm) 12.88, 24.81, 43.13, 67.2, 83.87, 101.46, 106.17, 114.48, 118.08, 118.54, 119.44, 127.35, 129.49, 130.10, 135.06, 135.73, 138.57, 140.67, 144.83, 145.96, 152.71, 153.49, 158.78, 177.5, 179.72; 19F NMR (CDCl3) δ (ppm) 141.44, 141.38; ESI-MS (M + H) m/z = 672.2.
Fluorescence spectral testing of CRANAD-88 with H2O2, Aβ aggregates and H2O2+ Aβ aggregates
To record the fluorescence response of CRANAD-88 with different solutions, we utilized the following procedures. Step 1: 1.0 mL of PBS buffer with 20% DMSO was added to a quartz cuvette as a blank control and its fluorescence was recorded with the same parameters as for CRANAD-88. Step 2: The fluorescence emission spectrum of a CRANAD-88 solution (1.0 mL, 250 nM) was recorded with excitation at 560 nm and emission from 580 to 900 nm. Step 3: To the above incubated CRANAD-88 solution, 10 μL H2O2 was added (final concentration 100 μM). The emission spectra at different time points were recorded (Ex = 560 nm, Em = 580–900 nm). Step 4: To the above solution of CRANAD-88 and H2O2, 10 μL of Aβ 40 aggregates were added to make the final Aβ concentration of 250 nM. The emission spectra of the resulting mixture at different time points were recorded as described in Step 2. The final spectra from steps 2 and 3 were corrected using the blank control from Step 1.
BBB penetration of CRANAD-88
Similar procedure from ref. 30 was followed. The ethyl acetate extraction was subjected to fluorescence spectra recording and LC-MS analysis.
Brain phantom studies
A 4-month old balb/c mouse was perfused and sacrificed, and its brain was dissected. The brain was homogenized with 2.0 ml PBS, and the homogenate was equally divided into five eppendorf tubes and diluted to 1 ml with PBS. Every tube had added 20 μL CRANAD-88 (final concentration 500 nM) except the control of homogenate blank. No. 1: CRANAD-88 only; No. 2: CRANAD-88 and 20 μL H2O2 (final concentration 10 μM); No. 3: CRANAD-88 and H2O2 (final concentration 10 μM) incubated for 30 minutes at 37 °C, and then 20 μL Aβ40 aggregates (final concentration 500 nM) was added; No. 4: CRANAD-88 and 20 μL Aβ40 aggregates (final concentration 500 nM). The tubes were imaged with EX/EM = 640/700 nm on an IVIS Spectrum imaging system.
Brain slice testing
A brain slice (25 micron) of an 11-month old APP/PS1 mouse was washed with double-distilled water for 5 minutes, then fixed with 4% formalin for 5 minutes, and washed with double-distilled water. First, a background image of a brain slice was taken using a fluorescence microscope before probe treatment. Second, an image was taken after the slice was incubated with CRANAD-88 (25 μM) for 30 minutes. Third, an image was taken after the above slice was incubated with H2O2 (100 μM) at 37 °C for 30 minutes.
In Vivo NIRF Imaging
In vivo NIRF imaging was performed using an IVIS Spectrum animal imaging system (Caliper LifeSciences, Perkin-Elmer, Hopkinton, MA). Images were acquired with a 640 nm excitation filter and a 700 nm emission filter. Data analysis was performed using Living Image 4.2.1 software. 15-month old transgenic APP-PS1 mice (female, n = 3–4) and age-matched wild-type control mice (female, n = 3–4) were shaved to remove the fur from the brain area before background imaging. An injection solution of CRANAD-88 (4.0 mg/kg) was freshly prepared in 15% DMSO, 15% cremorphor, and 70% PBS, and the solution was stabilized for 20 min before injection. Each mouse was intravenously injected with 100 μL of CRANAD-88 via tail vein. Fluorescence signals from the brain areas were recorded at 0-, 15-, 30-, 60-, 120-, and 240-min after the intravenous injection. To quantify the NIRF signal, an equal size ROI was drawn around each brain region.
Monitoring H2O2 scavenging in vivo
The same mice from the above experiments were used after waiting for one week. In vivo imaging data indicated that the injected CRANAD-88 was totally washed out. For scavenging H2O2, 100 mM sodium pyruvate solution (sigma) was used. Both WT and APP/PS1 mice were i.v. injected with 100 μL of CRANAD-88 (same dose and formulation as the above experiments) and 100 μL of sodium pyruvate at the same time. After injection, the mice were imaged using the same imaging protocol as the above experiments.
Additional Information
How to cite this article: Yang, J. et al. Imaging hydrogen peroxide in Alzheimer’s disease via cascade signal amplification. Sci. Rep. 6, 35613; doi: 10.1038/srep35613 (2016).
References
Brookmeyer, R., Johnson, E., Ziegler-Graham, K. & Arrighi, M. H. Forecasting the global burden of Alzheimer’s disease. Alzheimer’s and Dementia 3 186–191 (2007).
Melnikova, I. Therapies for Alzheimer’s disease. Nature Rev Drug Discov. 6, 341–342 (2007).
Selkoe, D. J. Resolving controversies on the path to Alzheimer’s therapeutics. Nature medicine 17, 1060–1065 (2011).
Van Eldik, L. J., Koppal, T. & Watterson, D. M. Barriers to Alzheimer disease drug discovery and development in academia. Alzheimer disease and associated disorders 16 Suppl 1, S18–S28 (2002).
Perry, G., Cash, A. D. & Smith, M. A. Alzheimer Disease and Oxidative Stress. J Biomed Biotechnol 2, 120–123 (2002).
Markesbery, W. R. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med 23, 134–147 (1997).
Pratico, D. Oxidative stress hypothesis in Alzheimer’s disease: a reappraisal. Trends Pharmacol Sci 29, 609–615 (2008).
Behl, C., Davis, J. B., Lesley, R. & Schubert, D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 77, 817–827 (1994).
Anderton, B. Free radicals on the mind. Hydrogen peroxide mediates amyloid beta protein toxicity. Hum Exp Toxicol 13, 719 (1994).
Huang, X. et al. Cu(II) potentiation of alzheimer abeta neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J Biol Chem 274, 37111–37116 (1999).
Atwood, C. S. et al. Dramatic aggregation of Alzheimer abeta by Cu(II) is induced by conditions representing physiological acidosis. J Biol Chem 273, 12817–12826 (1998).
Nakao, N. et al. Hydrogen peroxide induces the production of tumor necrosis factor-alpha in RAW 264.7 macrophage cells via activation of p38 and stress-activated protein kinase. Innate Immun 14, 190–196 (2008).
Huang, Y., Erdmann, N., Peng, H., Zhao, Y. & Zheng, J. The role of TNF related apoptosis-inducing ligand in neurodegenerative diseases. Cell Mol Immunol 2, 113–122 (2005).
Condello, C., Yuan, P., Schain, A. & Grutzendler, J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Abeta42 hotspots around plaques. Nat Commun 6, 6176 (2015).
Wilkinson, B. L. & Landreth, G. E. The microglial NADPH oxidase complex as a source of oxidative stress in Alzheimer’s disease. J Neuroinflammation 3, 30 (2006).
Simonyi, A. et al. Targeting NADPH oxidase and phospholipases A2 in Alzheimer’s disease. Molecular neurobiology 41, 73–86 (2010).
Goldsbury, C., Whiteman, I. T., Jeong, E. V. & Lim, Y. A. Oxidative stress increases levels of endogenous amyloid-beta peptides secreted from primary chick brain neurons. Aging Cell 7, 771–775 (2008).
Janelsins, M. C. et al. Early correlation of microglial activation with enhanced tumor necrosis factor-alpha and monocyte chemoattractant protein-1 expression specifically within the entorhinal cortex of triple transgenic Alzheimer’s disease mice. J Neuroinflammation 2, 23 (2005).
Sommer, G. et al. Amyloid precursor protein expression is induced by tumor necrosis factor alpha in 3T3-L1 adipocytes. J Cell Biochem 108, 1418–1422 (2009).
Milton, N. G. Role of hydrogen peroxide in the aetiology of Alzheimer’s disease: implications for treatment. Drugs Aging 21, 81–100 (2004).
Pu, K. et al. Semiconducting polymer nanoparticles as photoacoustic molecular imaging probes in living mice. Nat Nanotechnol 9, 233–239 (2014).
Pu, K., Shuhendler, A. J. & Rao, J. Semiconducting polymer nanoprobe for in vivo imaging of reactive oxygen and nitrogen species. Angewandte Chemie 52, 10325–10329 (2013).
Wu, W. et al. Bioluminescent probe for hydrogen peroxide imaging in vitro and in vivo. Analytical chemistry 86, 9800–9806 (2014).
Van de Bittner, G. C., Dubikovskaya, E. A., Bertozzi, C. R. & Chang, C. J. In vivo imaging of hydrogen peroxide production in a murine tumor model with a chemoselective bioluminescent reporter. Proceedings of the National Academy of Sciences of the United States of America 107, 21316–21321 (2010).
Srikun, D., Miller, E. W., Domaille, D. W. & Chang, C. J. An ICT-based approach to ratiometric fluorescence imaging of hydrogen peroxide produced in living cells. Journal of the American Chemical Society 130, 4596–4597 (2008).
Dickinson, B. C. & Chang, C. J. A targetable fluorescent probe for imaging hydrogen peroxide in the mitochondria of living cells. Journal of the American Chemical Society 130, 9638–9639 (2008).
Abo, M. et al. Development of a highly sensitive fluorescence probe for hydrogen peroxide. Journal of the American Chemical Society 133, 10629–10637 (2011).
Ran, C. et al. Design, synthesis, and testing of difluoroboron-derivatized curcumins as near-infrared probes for in vivo detection of amyloid-beta deposits. J Am Chem Soc 131, 15257–15261 (2009).
Ran, C. & Moore, A. Spectral unmixing imaging of wavelength-responsive fluorescent probes: an application for the real-time report of amyloid Beta species in Alzheimer’s disease. Molecular imaging and biology: MIB: the official publication of the Academy of Molecular Imaging 14, 293–300 (2012).
Zhang, X. et al. Design and synthesis of curcumin analogues for in vivo fluorescence imaging and inhibiting copper-induced cross-linking of amyloid Beta species in Alzheimer’s disease. Journal of the American Chemical Society 135, 16397–16409 (2013).
Zhang, X. et al. Near-infrared fluorescence molecular imaging of amyloid beta species and monitoring therapy in animal models of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America 112, 9734–9739 (2015).
Hsiao, K. et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274, 99–102 (1996).
Garcia-Alloza, M. et al. Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiology of disease 24, 516–524 (2006).
Giandomenico, A. R., Cerniglia, G. E., Biaglow, J. E., Stevens, C. W. & Koch, C. J. The importance of sodium pyruvate in assessing damage produced by hydrogen peroxide. Free Radic Biol Med 23, 426–434 (1997).
Zhou, D., Qian, J., Chang, H., Xi, B. & Sun, R. P. Pyruvate administered to newborn rats with insulin-induced hypoglycemic brain injury reduces neuronal death and cognitive impairment. Eur J Pediatr 171, 103–109 (2012).
Acknowledgements
This work was supported by NIH K25AG036760 and R21AG050158 awards (C.R.). We thank Pamela Pantazopoulos, B.S. for proofreading this manuscript. We also thank China Scholarship Council of Ministry of Education of China for supporting (J.Y. and J.Y.).
Author information
Authors and Affiliations
Contributions
J.Y., Y.X. and C.R. designed the study; J.Y., J.Y. and C.R. performed experiments; J.Y., Y.X. and C.R. wrote the manuscript; S.L. and A.M. provided suggestions for the experiments and critical comments on the manuscript; C.R. supervised the study.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yang, J., Yang, J., Liang, S. et al. Imaging hydrogen peroxide in Alzheimer’s disease via cascade signal amplification. Sci Rep 6, 35613 (2016). https://doi.org/10.1038/srep35613
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep35613
This article is cited by
-
COA-Cl Evokes Protective Responses Against H2O2-and 6-OHDA-Induced Toxic Injury in PC12 Cells
Neurotoxicity Research (2022)
-
Jatamansinol from Nardostachys jatamansi Ameliorates Tau-Induced Neurotoxicity in Drosophila Alzheimer’s Disease Model
Molecular Neurobiology (2022)
-
Similarity of therapeutic networks induced by a multi-component herbal remedy, Ukgansan, in neurovascular unit cells
Scientific Reports (2020)
-
Computational modeling of curcumin-based fluorescent probe molecules
Theoretical Chemistry Accounts (2019)
-
Two-photon excited fluorescent silica nanoparticles loaded with iron(II) as a probe for determination and imaging of hydrogen peroxide in living cells
Microchimica Acta (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
