
Abstract
Decreased expression and activity of CaV1.2 calcium channels has been reported in pressure overload-induced cardiac hypertrophy and heart failure. However, the underlying mechanisms remain unknown. Here we identified in rodents a splice variant of CaV1.2 channel, named CaV1.2e21+22, that contained the pair of mutually exclusive exons 21 and 22. This variant was highly expressed in neonatal hearts. The abundance of this variant was gradually increased by 12.5-folds within 14 days of transverse aortic banding that induced cardiac hypertrophy in adult mouse hearts and was also elevated in left ventricles from patients with dilated cardiomyopathy. Although this variant did not conduct Ca2+ ions, it reduced the cell-surface expression of wild-type CaV1.2 channels and consequently decreased the whole-cell Ca2+ influx via the CaV1.2 channels. In addition, the CaV1.2e21+22 variant interacted with CaVβ subunits significantly more than wild-type CaV1.2 channels, and competition of CaVβ subunits by CaV1.2e21+22 consequently enhanced ubiquitination and subsequent proteasomal degradation of the wild-type CaV1.2 channels. Our findings show that the resurgence of a specific neonatal splice variant of CaV1.2 channels in adult heart under stress may contribute to heart failure.
Similar content being viewed by others

Introduction
Cardiac excitation-contraction coupling is mainly initiated by Ca2+ influx through L-type voltage gated CaV1.2 channels in cardiomyocytes via Ca2+-induced Ca2+ release mechanisms1. The CaV1.2 channel comprises a pore-forming α1 subunit and auxiliary α2δ and β subunits2. The accessory subunits modulate the channel biophysical properties and are involved in the anchorage, trafficking and post-translational modification of the pore-forming α1 subunit3. In particular, the CaVβ subunit was recently reported to promote the trafficking of CaV1.2 channels to the plasma membrane by inhibiting the proteasomal degradation of the channels4. Genetic deletion of either the pore-forming α1 subunit or CaVβ subunit led to embryonic death with cardiac defects5,6.
In cardiac hypertrophy and heart failure, linkage to alteration in Ca2+ influx via Cav1.2 channels has been controversial7,8. Clinical trials using Ca2+ channel blockers for heart failure have been disappointing with either no beneficial effects or a worse outcome of reduced ejection fraction9,10,11. Nevertheless, in human failing cardiomyocytes the density of CaV1.2 channels was decreased compared to normal cardiomyocytes12. In line with these findings, decreased CaV1.2 channel activity was recently reported to induce cardiac hypertrophy and heart failure in genetically modified mice8. More importantly, the hypertrophied cardiomyocytes induced by pressure overload showed drastic decrease in CaV1.2 channel density and activity due to reduced expression of the CaV1.2 channels. The mechanisms, however, by which the density and activity of CaV1.2 channels were reduced is unknown.
The pore-forming α1 subunit undergoes extensive alternative splicing that potentially generates multiple functionally diversified CaV1.2 variants in human13 and rodent hearts14. Alternative splicing could be developmentally regulated14,15 and involved in myocardial infarction16 and heart failure17. In human diseases, alternative splicing of α1 subunit has been reported in failing human ventricular cardiomyocytes and atherosclerotic human arteries17,18. Ectopic expression of some alternative splicing variants modulated the expression and activity of the CaV1.2 channels5,14. In the present study, we identified a CaV1.2 splice variant containing the mutually exclusive exons 21 and 22 (e21 + 22), named CaV1.2e21+22 channel, which was highly expressed in neonatal and hypertrophied adult hearts. As the newly identified channel variant does not conduct Ca2+ ions, we hypothesized that it may account for the reduced expression and activity of CaV1.2 channels in hypertrophied cardiomyocytes induced by pressure overload14.
Results
Differential expression of alternatively spliced isoforms of CaV1.2 channels in neonatal versus adult rat hearts
Mutually exclusive exons 21 and 22 encode the IIIS2 transmembrane segment and part of the linker region between IIIS1 and IIIS2. Restriction enzyme AvrII digests within exon 22 only, but not exon 21 (Fig. 1A). RT-PCR across exons 19 to 25 produced a fragment of 640 bp in length. Control cDNA containing exon 22 only was completely digested by Avr II. Under similar conditions, however, only a portion of the RT-PCR products from both neonatal and adult hearts were digested, suggesting the presence of a mixture of PCR products expressing exon 21 and exon 22 in four possible combinations of e21, e22, e(21 + 22) and ∆e(21 + 22) (Fig. 1B). The predicted PCR product sizes are 640 bp for e21 or e22, 700 bp for e(21 + 22) and 580 bp for ∆e(21 + 22) (Fig. 1C). The results were confirmed by sequencing the PCR products. Inclusion of both exons will generate a channel with one additional transmembrane segment and may result in a drastic change in the topology of the channel. In this study, we focused on the splice variant including both exons e(21 + 22): CaV1.2e21+22 channels. Transcript-scanning demonstrated that the abundance of CaV1.2e21+22 channels in rat neonatal heart (14.3%) was 2.5 times higher than that in adult heart (5.5%, P = 0.0124, Fig. 1D).

Inclusion level of exons 21+22 in CaV1.2 channels in neonatal hearts is higher than that in adult hearts.
(A) Exons 21 and 22 are mutually exclusive exons. RT-PCR across exons 19–25 could generate a fragment of 640 bp. Digestion with AvrII could produce two smaller fragments of 275 bp and 365 bp. Besides, aberrant exclusion or inclusion of both exons 21 and 22 would generate two fragments of 580 bp and 700 bp, respectively. (B) mRNA of CaV1.2 channels from both neonatal (NH) and adult (AH) hearts showed partial digestion by AvrII. Complete digestion was observed in control DNA containing exon 22. (C) Colony screening of a neonatal heart identified bands of three size classes: 580 bp, 640 bp and 700 bp. (D) Summary of aberrant splicing rate in rat neonatal hearts (NH, n = 5) and adult hearts (AH, n = 5). Data were shown as mean ± SEM. *p < 0.05.
Increased abundance of CaV1.2e21+22 channels in hypertrophied heart
Alteration in the expression of developmentally regulated CaV1.2 splice variants has been implicated in cardiac hypertrophy19 and heart failure17. To examine whether the alternative splicing isoform CaV1.2e21+22 in neonatal heart reemerges in the hypertrophic adult heart, we performed transverse aortic constriction (TAC) surgery on the mice as done previously8 to generate pressure-overload induced cardiac hypertrophy that gradually develops and reaches a peak on day 14 after TAC surgery20. As expected, left ventricular (LV) weight to body weight, measured in isolated ventricles, increased significantly after two weeks of TAC (Fig. 2A). Thickening of left ventricular anterior and posterior walls at end diastole (d) or end systole (s) was overt via echocardiography (Fig. 2B,C and Table 1). Cardiac hypertrophy was also evidenced by the gradual increase in Myh7 and decrease in Myh6 at mRNA level (Supplementary Fig. S1). Furthermore, the heart rate of those mice was significantly increased while cardiac function was depressed as indicated by the reduction in ejection fraction and fraction shortening (Table 1).

Abundance of cardiac CaV1.2e21+22 channels is increased in mice in response to TAC surgery.
(A) Increased ratio of left ventricle to body weight in TAC mice. (B) Representative M-mode echocardiography images of mouse hearts before and 14 days after TAC surgery indicating progression of cardiac hypertrophy. (C) Increased LVAWd and LVPWd in TAC mice. (D) Representative gel photos for transcript screening of exons 21 + 22 inclusion level. Each lane represents a single colony expressing exons 21 + 22 or exon 21/22. (E) Inclusion level of exons 21 + 22 increased from 0.52% to 6.49% with the development of cardiac hypertrophy induced by pressure overload within 14 days (n = 6). (F–H) Expression levels of total CaV1.2 channels and CaVβ2 subunits in left ventricles (n = 8). (I,J) Ubiquitination level of cardiac CaV1.2 channels in left ventricles (n = 8). Data were shown as mean ± SEM. *p < 0.05, #p < 0.01. 1-way ANOVA was performed for multiple comparisons in panel E.
In the hypertrophied heart, we first examined the expression level of CaV1.2 channels. Consistent with Goonasekera’s report8, total protein level of α1 subunit of CaV1.2 channels was reduced by 40% in mouse left ventricles 14 days after TAC surgery (p < 0.05, Fig. 2F,G). In addition, protein level of CaVβ2 subunit was reduced by 45% (p = 0.01, Fig. 2F,H). Compared to baseline, the ubiquitination of Cav1.2 channels was clearly enhanced in the hypertrophic ventricles (p < 0.05, Fig. 2I,J) indicating the involvement of proteasomal degradation. Alternative splicing of Cav1.2 channels or reemergence of fetal splicing isoforms has been suggested in stressed and failing hearts17,19, we therefore hypothesized that the neonatal isoform CaV1.2e21+22 may reemerge in the adult heart in pressure overload induced hypertrophy and subsequently disturb the expression level of normal CaV1.2 channels. To test this hypothesis, transcript-screening was performed in isolated left ventricles subjected to TAC. As shown in Fig. 2D,E, the abundance of the neonatal isoform CaV1.2e21+22 gradually increased by approximately 12.5-folds in the left ventricles, from 0.52% to 6.49% of total CaV1.2 channels, in 14 days of chronic pressure overload. In human hearts, the abundance of exons 21 + 22 was also significantly higher in the left ventricles from patients with dilated cardiomyopathy (DCM) than that from healthy donors (by 2.8 folds), but no elevation of exons 21 + 22 inclusion was observed in heart tissue from patients with ischemic cardiomyopathy (ICM, supplementary Fig. S2 and Table S1).
Functional characterization of CaV1.2e21+22 channels
To understand the pathological significance of CaV1.2e21+22 in hypertrophied heart, we characterized this isoform in vitro by heterologous expression in HEK 293 cells that do not have endogenous CaV1.2 channels. Compared to the robust ICa recorded from wild-type HA-tagged rat CaV1.2e22 channels (−18.8 ± 3.6 pA/pF at 0 mV), no currents were detected from CaV1.2e21+22 channels (Fig. 3A). Cellular localization of CaV1.2e21+22 channels was examined by expression of α1 subunit with or without β2a subunit in HEK 293 cells followed by surface protein biotinylation. Consistent with a previous report4, co-expression of β2a subunit increased the surface expression level of wild-type HA-CaV1.2e22 channels by 3.2-fold and the total expression level by 1.8-fold (Fig. 3B,C). However, CaV1.2e21+22 channels were nearly undetectable at the cell surface regardless of the expression of β2a subunit. Instead, the channel proteins were retained intracellularly (Fig. 3B,C). Of note, the total protein level of CaV1.2e21+22 channels was much lower than that of wild-type channels, indicating that intracellular degradation may have occurred. Together, these data suggest that β2a subunit failed to facilitate trafficking of CaV1.2e21+22 channels to the cell surface.

Characterization of exons 21 + 22-containing CaV1.2e21+22 channels.
(A) CaV1.2e21+22 channels were co-expressed with β2a and α2δ subunits in HEK293 cells. Whole cell patch-clamp recordings were performed on the cells expressing wild-type (n = 7) or CaV1.2e21+22 (n = 8) channels. (B,C) Detection and quantification of surface and total HA-CaV1.2e22 or CaV1.2e21+22 channels in the presence or absence of β2a subunit in transfected HEK293 cells (n = 4). Surface channels were biotinylated as indicated in the Methods. (D,E) Detection and quantification of β2a subunits bound to HA-CaV1.2e22 or CaV1.2e21+22 channels. β2a subunits were co-transfected with channels at different molar ratios of 0, 1/4, 1/2, 1/1, 2/1 or 4/1 (β2a/CaV1.2) in HEK293 cells treated with MG132 (2.5 μM, n = 4). (F,G) Detection and quantification of the ubiquitination levels of HA-CaV1.2e22 and CaV1.2-e21+22 channels in transfected HEK293 cells treated with MG132 (n = 3). e22, HA-CaV1.2e22 channels. e21 + 22, CaV1.2e21+22 channels. Data were shown as mean ± SEM, ns, non-significant, *p < 0.05, #p < 0.01.
To examine whether the intracellular retention of CaV1.2e21+22 channels was caused by attenuated binding to β2a subunit, CaV1.2e21+22 channels were co-transfected with β2a subunit at different molar ratios of 0, 1/4, 1/2, 1/1, 2/1 or 4/1 (β2a/CaV1.2) in HEK 293 cells in the presence of a proteasomal inhibitor, MG132 (2.5 μM). In contrast to our initial hypothesis, more β2a subunits were co-immunoprecipitated by CaV1.2e21+22 than the wild-type HA-CaV1.2e22 channels at the molar ratios of 1/4 (p = 0.0002), 1/2 (p = 0.009), and 1/1 (p = 0.048) (Fig. 3D,E). Furthermore, the ubiquitination level of CaV1.2e21+22 channels did not show significant difference in the presence or absence of β2a subunit (p = 0.65). This is in contrast to ubiquitination of wild-type channels which was drastically reduced upon co-expression with β2a subunit (p = 0.007, Fig. 3F,G). Taken together, the non-functional CaV1.2e21+22 channels showed stronger interactions with β2a subunit and its vulnerability to ubiquitination is not ameliorated by β2a subunit.
CaVβ subunits did not enhance total expression of CaV1.2e21+22 channels
To further confirm the roles of CaVβ subunits in regulation of CaV1.2e21+22 channel function, confocal microscopy was performed to image and evaluate the total expression of CaV1.2e21+22 channels with or without β2a subunits. As previously reported4, compared to cells that did not express β2a subunits, β2a subunit-expressing cells showed increased expression level of total HA-CaV1.2e22 channels with or without MG132 treatment (Fig. 4A), which is consistent with the finding that ubiquitination of HA-CaV1.2e22 channels was significantly reduced in the presence of β2a subunits under MG132 treatment (Fig. 3F,G). However, in line with the results as shown in Fig. 3B,C, the total expression of CaV1.2e21+22 channels was not markedly increased in β2a subunit-expressing cells with or without MG132 treatment (Fig. 4B), which also further supported that β2a subunits did not significantly prevent the ubiquitination of CaV1.2e21+22 channels (Fig. 3F,G).

CaVβ subunits do not enhance the total expression of CaV1.2e21+22 channels.
HA-CaV1.2e22 or CaV1.2e21+22 were co-transfected with α2δ and β2a subunit in HEK293 cells with or without MG132 treatment. β2a subunit was cloned in pIRES2-EGFP (as an indicator of β2a subunit expression). Immunostaining of total CaV1.2 channels and confocal imaging were performed after 48 h transfection. (A) β2a subunit-expressing cells showed up-regulation of total HA-CaV1.2e22 channels with or without MG132 treatment, compared to cells not expressing β2a subunits. (B) Total CaV1.2e21+22 channels were not markedly altered in β2a subunit-expressing cells with or without MG132 treatment. Scale bar, 20 μm.
CaV1.2e21+22 channels down-regulated expression of CaV1.2e22 channels in a dominant-negative manner
Based on the observed stronger interaction between CaV1.2e21+22 channels and β2a subunit, we hypothesized that CaV1.2e21+22 channels may modulate the function of wild-type CaV1.2 channels by depleting or competing for free β2a subunits. To test this hypothesis, CaV1.2e21+22 channels were co-transfected with CaVβ-dependent HA-CaV1.2e22 channels or CaVβ-independent CaV3.1 channels in HEK 293 cells. As measured in external solution containing 1.8 mM Ca2+, the current density of CaV1.2 channels at 0 mV was lowered from −16.7 ± 2.5 pA/pF to −9.3 ± 1.0 pA/pF (p = 0.007, Fig. 5A). In contrast, the current density of CaV3.1 channels at −20 mV remained unchanged (−9.9 ± 1.2 pA/pF vs. −9.2 ± 1.4 pA/pF, p = 0.671, Fig. 5B). To further investigate whether this dominant-negative effect on CaV1.2 channels is due to reduction of free CaVβ subunits by CaV1.2e21+22 channels, the CaV1.2 I-II loop containing the AID domain that binds CaVβ subunit was substituted into CaVβ-independent CaV3.1 channel to generate a chimeric CaV3.1GCGGG channel. Compared to CaV3.1 channels, the chimeric channels displayed a dramatic increase in current amplitude and a 40 mV leftward shift in the I–V relationship (Fig. 5B,C) as previously reported21. Upon co-expression with CaV1.2e21+22 channels, the current density of CaV3.1GCGGG channels at −60 mV was significantly reduced from −128.4 ± 22.2 pA/pF to −51.6 ± 16.8 pA/pF (Fig. 5C).

CaV1.2e21+22 channels produce dominant-negative effects on L-type CaV1.2 channels, but not on T-type CaV3.1 channels.
CaV1.2e21+22 channels were co-transfected at a ratio of 1:1 with HA-CaV1.2e22, CaV3.1 or chimeric CaV3.1GCGGG channels containing CaV1.2 I-II loop in HEK293 cells with or without MG132 treatment. As control, CaV1.2e21+22 channels were replaced with pcDNA3 vectors for co-transfection. I-V curves were obtained in an external solution containing 1.8 mM Ca2+. For western blot assays, cells were biotinylated for surface proteins 36 hrs after transfection and then lysed for analysis. (A–C) Effects of CaV1.2e21+22 channels on the current density of wild-type CaV1.2e22 channels (Vector, n = 19; e21 + 22, n = 18), CaV3.1 channels (Vector, n = 9; e21 + 22, n = 11) or the chimeric CaV3.1-GCGGG channels (Vector, n = 10; e21 + 22, n = 9). *p < 0.05, #p < 0.01. (D–I) Effects of CaV1.2e21+22 channels on the surface and total expression levels of HA-CaV1.2e22 channels (D,E), CaV3.1 channels (F,G) or chimeric CaV3.1GCGGG channels (H,I, n = 3). Transferrin receptor (TfR) was used as surface protein loading control. e22, wild-type HA-CaV1.2e22 channel. e21 + 22, aberrant CaV1.2e21+22 channel. Data were shown as mean ± SEM, ns, non-significant, *p < 0.05, #p < 0.01, 1-way ANOVA with post hoc Bonferroni’s test was performed for multiple comparisons.
To further delineate the mechanisms underlying the differential regulation of current density by CaV1.2e21+22 channels, the total and surface expression levels of all the three calcium channels were examined in HEK 293 cells co-transfected with β2a subunit. As expected, CaV1.2e21+22 channels did not affect the surface and total expression levels of CaV3.1 channels (Fig. 5F,G). However, co-expression of CaV1.2e21+22 channels significantly reduced the surface (upper panel, Fig. 5D,E) and total (lower panel, Fig. 5D,E) levels of CaV1.2 channels and the chimeric CaV3.1GCGGG channels (Fig. 5H,I). The reduction in expression levels of CaVβ-dependent channels was prevented by MG132 treatment indicating proteasomal degradation of those channels co-expressed with the CaV1.2e21+22 isoform (Fig. 5D,E).
CaV1.2e21+22 channels enhanced ubiquitination of CaVβ-binding calcium channels
As most proteasomal degradation involves ubiquitin, the ubiquitination levels of all the three calcium channels were evaluated in HEK 293 cells co-transfected with CaV1.2e21+22 channels. As shown by western blot analyses, the relative intensity of ubiquitinated CaV1.2 channels (Ub-CaV1.2) to total CaV1.2 channels was greatly enhanced by the presence of CaV1.2e21+22 channels (p = 0.015, Fig. 6A,B), and the increase in ubiquitination of CaV1.2 channels was augmented by MG132 treatment (p = 0.007, Fig. 6A,B). In contrast, the ubiquitination of CaV3.1 channels was not affected by the presence of CaV1.2e21+22 channels or MG132 treatment (Fig. 6C,D). While introduction of the CaVβ-binding domain into this CaVβ-independent channel markedly increased its ubiquitination by CaV1.2e21+22 channels (Fig. 6E,F). These results suggested that the augmentation of ubiquitination of calcium channels by CaV1.2e21+22 channels is attributed to the CaVβ-binding domain.

CaV1.2e21+22 channels enhance ubiquitination of L-type CaV1.2 channels, but not T-type CaV3.1 channels.
CaV1.2e21+22 channels were co-transfected at a ratio of 1:1 with HA-CaV1.2e22, CaV3.1 or the chimeric CaV3.1GCGGG channels in HEK293 cells with or without MG132 treatment. Anti-HA and anti-CaV3.1 were used to pull down the protein complexes from cell lysates for ubiquitnation analysis of HA-CaV1.2e22 (A,B), wild-type CaV3.1 channels (C,D) or chimeric CaV3.1GCGGG channels (E,F) with or without MG132 treatment (n = 3). e22, HA-CaV1.2e22 channel. e21 + 22, CaV1.2e21+22 channel. Ub-CaV1.2, ubiquitinated CaV1.2 channels. Data were shown as mean ± SEM, ns, non-significant, *p < 0.05, #p < 0.01, 1-way ANOVA with post hoc Bonferroni’s test was performed for multiple comparisons.
CaV1.2e21+22 channels competed for CaVβ subunits with CaV1.2 channels
To substantiate the notion that the reduced expression level and increased ubiquitination of CaV1.2 channels by CaV1.2e21+22 channels were due to competition with CaV1.2 channels for available CaVβ subunits, CaV1.2e21+22 channels were co-transfected with CaV1.2, CaV3.1 or the chimeric CaV3.1GCGGG channels at a molar ratio of 0, 1/4, 1/2 or 1 in HEK 293 cells treated with MG132. As indicated by Western blot, the relative intensity of β2a subunit to CaV1.2 channels was gradually reduced with increase of CaV1.2e21+22 channels (Fig. 7A,B). While no β2a subunit was co-immunoprecipitated with wild-type CaV3.1 channels (Fig. 7C) the relative intensity of β2a subunit to chimeric CaV3.1GCGGG channels was clearly attenuated by CaV1.2e21+22 channels in a dose-dependent manner (Fig. 7D,E).

CaV1.2e21+22 channels compete for β2a subunit with L-type CaV1.2 channels, but not with T-type CaV3.1 channels, in a dose-dependent manner.
CaV1.2e21+22 channels were co-transfected at indicated ratios with HA-CaV1.2e22, CaV3.1 or chimeric CaV3.1GCGGG channels in HEK 293 cells treated with MG132. Anti-HA and anti-CaV3.1 were used for co-immunoprecipitation. Effects of CaV1.2e21+22 channels on the binding of β2a subunits to HA-CaV1.2e22 (A,B), CaV3.1 channels (C) or chimeric CaV3.1GCGGG channels (E) were analyzed by Western blotting (n = 3). e22, wild-type HA-CaV1.2e22 channel. e21 + 22, aberrant CaV1.2e21+22 channel. Data were shown as mean ± SEM, ns, non-significant, *p < 0.05, #p < 0.01, 1-way ANOVA with post hoc Bonferroni’s test was performed for multiple comparisons.
Discussion
This study identified a novel alternatively spliced isoform of CaV1.2 channels, CaV1.2e21+22. The expression of CaV1.2e21+22 diminishes during postnatal cardiac maturation and re-emerges in pressure-overload induced cardiac hypertrophy. This fetal-like alternative splicing pattern of CaV1.2 channels in the hypertrophied heart is in agreement with a recent report that alternative splicing events in response to TAC displayed reciprocal expression changes during postnatal cardiac development versus heart failure22. Despite its physiological significance during cardiac maturation, the role of the re-emergence of CaV1.2e21+22 in response to cardiac stress is unknown.
Mutations in CaV1.2 channels are associated with multiple heart diseases including Timothy syndrome that is characterized by a long QT interval and ventricular arrhythmia due to sustained activation of CaV1.2 channels23,24 and Brugada syndrome that is notable for a short QT interval and sudden cardiac death due to inactivation of CaV1.2 channels25. While the role of CaV1.2 channels in electrical heart diseases is well known, its role in mechanical or structural heart diseases remains less understood. In failing human or animal hearts, the density and currents of CaV1.2 channels were reportedly reduced26 or unchanged27. In causal studies with genetic modified animals, the conclusion is so far controversial. Increase in Ca2+ influx through CaV1.2 channels by cardiac specific over-expression of β2a subunit7 or α1C subunit28 in mice was reported to induce cardiac hypertrophy and cardiomyopathy. Unexpectedly, decrease in Ca2+ influx through CaV1.2 channels in α1C+/− mice resulted in a similar phenotype8. The disparities might be attributed to activation of calcineurin activation or neurohumoral effects8. Alternatively, it might be partly explained by the existence of two distinct subsets of the channels24,29,30: One subset assembled in the T-tubules for calcium-induced calcium release with ryanodine receptors for excitation-contraction coupling31,32, and the other subset (~50% in mice)33 enriched in caveolae to activate the transcription factor NFAT (nuclear factor of activated T cells) for cardiac hypertrophy. In line with this notion, we and Goonasekera et al. found that the protein level (Fig. 2) and activity of CaV1.2 channels were reduced in pressure overload-induced hypertrophic hearts23. Detailed analysis showed that the density and current of CaV1.2 channels both significantly declined in the isolated cardiomyocytes from those failing hearts. Although it is hard to prove which subset of channels decreased, according to their distinct functions, one may suspect that the caveolae-localized channels were affected the most in such a scenario.
One crucial question following Goonasekera’s study is how the channel expression and activity was reduced in response to pressure overload. As alternative splicing occurs frequently at 19 out of 55 exons that constitute the CaV1.2 gene, Cacna1c, in rodent heart and artery34,35, and some alternatively spliced isoforms were suggested to dominant-negatively suppress expression and channel conductivity of calcium channels14,36, we therefore hypothesized that the fetal splice variant CaV1.2e21+22 may reemerge in adult heart in response to cardiac stress, based on the recent findings by Gao and colleagues22, and disrupt the expression and activity of CaV1.2 channels. In agreement with Goonasekera’s findings, the total expression of CaV1.2 channels and CaVβ2 subunits were significantly reduced in left ventricles in response to TAC surgery (Fig. 2F–H). More importantly, the abundance of the CaV1.2e21+22 splice variant in mouse left ventricles was gradually increased up to 12.5 folds within 14 days after TAC (Fig. 2), and also elevated in left ventricles of DCM patients (Supplementary Fig. S2). Aberrant splicing of CaV1.2 channels was reported, though in very few studies, in cardiovascular diseases. For example, the abundance of exon 31- and exon 32-containing CaV1.2 isoforms significantly changed in end stage failing human hearts17 and smooth muscle CaV1.2 channel including exon 21 was completely replaced by a single isoform containing alternative exon 22 in human atherosclerosis18. However, the patho-physiological significance of these splicing events is unknown.
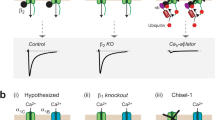
In the present study, we demonstrated that CaV1.2-e21+22 splice variant was retained intracellularly and it did not conduct Ca2+ (Fig. 3B,C). Strikingly, stronger binding to CaVβ subunit did not increase the accumulation and trafficking of CaV1.2e21+22 channels to cell surface, which appears contradictory to the conceptual model proposed by the Colecraft’s group21. In that model, it was proposed that following CaVβ interaction, a conformational rearrangement of the C-terminus attenuates the strength of ER retention signals within the C-terminus relative to the export signals found within the I-II loop. The net result would be enhanced trafficking to plasma membrane21. However, the C-terminus of rabbit CaV2.1 channels was shown to specifically interact with CaVβ4 subunit; meanwhile it also displayed a lower binding to CaVβ2 subunit37. In addition, Qin et al. also proposed that CaVβ2a inhibition of the inhibitory effect of Gβγ (G protein βγ dimers) on R-type CaV2.3 channel activity could be explained by the competitive displacement of Gβγ from its C-terminal binding site by the CaVβ2a subunit38. Based on these findings, we speculate that the presence of two transmembrane segments, exons 21 and 22, in CaV1.2e21+22 channel may induce conformational changes in the C-terminus, which may lead to a stronger binding to CaVβ subunit and prevent the CaVβ-dependent conformational rearrangement of the C-terminus as proposed in wild-type CaV1.2 channel. As a result, CaV1.2e21+22 channels fail to be transported to cell membrane and are trapped in ER to be degraded as misfolded proteins.
CaV1.2e21+22 channels suppressed expression of CaV1.2 channels via a dominant-negative mechanism. It has been shown that the misfolded calcium channels could drive wild-type channels toward proteasomal degradation, leading to a significant dominant-negative effect14,36. For example, the truncated variants of P/Q-type CaV2.1 channels could drive wild-type CaV2.1 channels into ER-associated degradation system by directly binding to domain I-II region36, which required an intact N-terminus39. However, our data did not support a direct interaction between CaV1.2e21+22 channels and wild-type CaV1.2 channels. Furthermore, the obvious dominant-negative effect of CaV1.2-e21+22 channels on the chimeric CaV3.1GCGGG channels (Fig. 5) excluded a major role of the N-terminus suggested for CaV2.1 channels39. Therefore the dominant-negative effect of CaV1.2e21+22 channels is likely attributed to a disparate mechanism. As CaV1.2e21+22 channels showed significantly stronger binding to CaVβ subunits than that of the wild type channels, this aberrant isoform may act as a CaVβ subunit trap by competing for free CaVβ subunits. Dose-dependent inhibition of the interaction between CaVβ subunits and CaV1.2 or CaV3.1GCGGG channels by CaV1.2e21+22 channels further supported this hypothesis. The competition for CaVβ subunits by CaV1.2e21+22 channels may result in a shortage of free CaVβ subunits for CaV1.2 channels and eventually lead to impaired membrane targeting, elevated ubiquitination and thereby increased degradation of CaV1.2 channels. Accordingly, the enhanced ubiquitination and diminished expression8 of CaV1.2 channels in the hypertrophied mouse heart induced by pressure overload are presumably caused by the reemergence of CaV1.2e21+22 channels under stress (Fig. 2E). However, it is noteworthy that there are two major isoforms for CaVβ subunits (β2 and β3 subunit), quantitatively in the order of β2b > β3 > β2a in the heart40. Thus whether the competition for CaVβ subunits by CaV1.2e21+22 channels is CaVβ isoform-dependent in cardiomyocytes will warrant further study.
Altogether, we may not anticipate that overexpression of CaV1.2e21+22 channels in the heart will induce cardiac hypertrophy, rather CaV1.2e21+22 channels could dominant-negatively disturb particularly the caveolae-localized CaV1.2 channels and activate the calcineurin/NFAT in response to hypertrophic stresses. Nevertheless, it is tempting to speculate that, in the patients suffering aortic stenosis or severe hypertension, CaV1.2e21+22 channels may reemerge in the heart and disturb the expression and activity of CaV1.2 channels, in particular of those channels localized in the caveolae, and consequently lead to cardiac hypertrophy.
In conclusion, we have identified and functionally characterized a naturally occurring fetal splice variant of CaV1.2 channels (CaV1.2e21+22 channels) that reemerged in adult mouse heart under stress and consequently disturbed the expression and activity of CaV1.2 channels. In addition, we demonstrated that this splice isoform augmented the ubiquitination of CaV1.2 channels for proteasomal degradation, impaired membrane targeting of the channels and reduced the channel expression and activity by competing for available CaVβ subunits. These data may provide a new insight of the dynamics of CaV1.2 channels at molecular level in the setting of cardiac hypertrophy.
Methods
Study approval
All human heart samples obtained from Cardiovascular Research Institute, National University of Singapore (NUS), were de-identified and pre-existing. All the experiments on the human heart tissues were performed in accordance with guidelines and protocols approved by the NUS Institutional Review Board (Reference code: 12-405). One normal human heart total RNA was purchased from Clontech (636532). All animal experiments were performed in accordance with guidelines and protocols approved by the Institutional Animal Care and Use Committee of National University of Singapore.
Induction of cardiac hypertrophy in mice
C57BL/6 mice were purchased from Jackson Laboratory and maintained at the Comparative Medicine Animal Vivarium at National University of Singapore. Experiments were carried out on adult male C57BL mice (10–12 weeks). Mice were anesthetized with a cocktail of 0.5 mg/kg Domitor, 5 mg/kg Dormicum and 0.05 mg/kg Fentanyl via intra-peritoneal injection, intubated and ventilated with a rodent ventilator (Harvard Apparatus). Transverse aortic constriction (TAC) was performed as previously described41. Briefly, the transverse aortic arch was exposed by a median sternotomy and bonded against a blunt 27-gauge needle with a 7-0 suture followed by prompt removal of the needle. Sham operated mice underwent the same procedure without aortic binding. Left ventricles were isolated for qPCR analysis of Myh6 and Myh7, transcript screening of exon 21 + 22 or biochemical analysis of of CaV1.2 channels and CaVβ2 subunits. Echocardiography was performed with Vevo 2100 from Visualsonics.
Transcript screening
As previously described14, total RNA was isolated using Trizol method total RNA was isolated using Trizol method from neonatal rat hearts, or left ventricles of adult rats, mice or patients with DCM or ICM. Then first strand cDNA was synthesized with Superscript II and oligo(dT)18 primers. PCR products (Primers for screening in rat heart: sense primer 5′-ACACTGCAGGTGAAGAGGATG-3′ and antisense primer 5′-TTTCCCTTGAAGAGCTGGACC-3′. For mouse heart: sense primer 5′-GAGCTGCACCTTAAGGAAAAGG-3′ and antisense primer 5′-GGATGCCAAAGGAGATGAGG-3′. For human heart: sense primer 5′-CCACCGCATTGTCAATGACAC-3′ and antisense primer 5′-CACGATGTTCCCGATGGTC-3′) were cloned into the pGEM-T Easy vector. Following transformation, each transformant was picked and grown in a single well in a 96-well plate. Colony PCR was performed with the same set of primers and conditions to identify the component of exons in each colony. 192 colonies were selected for each sample and at least 5 clones from each cDNAs group were sequenced to verify the exon specific PCR results.
DNA constructs
β2a and α2δ subunits have been described previously42. Chimeric CaV3.1GCGGG channel21 was kindly provided by A/Prof Henry Colecraft from Columbia University, and rat HA-CaV1.2e22 channel (wild type CaV1.2 channel)43 was from Prof Emmanuel Bourinet from Institut de Génomique Fonctionnelle. The cloning of rat CaV1.2e21+22 channel was achieved by inserting a PCR fragment containing exon 21 + 22 into the wild-type channel using NotI and KpnI sites.
Cell culture and transfection
HEK293 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Gibco) containing 10% fetal bovine serum (Gibco) and 1% penicillin–streptomycin and maintained at 37 °C in a humidified atmosphere containing 95% air and 5% CO2. For co-immunoprecipitation experiments, if not specified, HEK293 cells were cultured in 6-well plates, and were transiently transfected with different calcium channels, β2a subunit, α2δ subunit using calcium phosphate methods. In some experiments, cells were treated with a proteasomal inhibitor MG132 (2.5 μM) for 16 hrs at 24 hrs after transfection in order to prevent the proteasomal degradation of calcium channels. For whole cell patch-clamp recordings, HEK293 cells cultured on the coverslips coated with poly-D-lysine in 35 mm dishes were transiently transfected with different calcium channels at a molar ratio of 1:1 unless otherwise stated.
Co-immunoprecipitation
Co-immunoprecipitation was performed as described previously with modification. In brief, proteins harvested from transfected HEK293 cells were incubated with primary antibodies overnight at 4 °C with gentle rotation, followed by incubation with 20 μl of protein A/G agarose (Pierce) for another 1 h at 4 °C. The beads were washed 3 times using cold PBS and then denatured in 2X SDS sample loading buffer by boiling at 95 °C for 10 min. Proteins were then used for western blot analysis.
Surface protein biotinylation
To determine the level of CaV1.2e21+22 channels localized on the cell surface, CaV1.2e21+22 channels were biotinylated using an EZ-LinkTM Sulfo-NHS-Biotinylation Kit (Pierce) as previously described with modifications44. Briefly, cells were incubated with 0.25 mg/ml Biotin for 1h at 4 °C. Unbound biotin was removed by incubation with quenching buffer for 20 min and washed by PBS buffer. After measurement of protein concentration with Bradford assay, cell lysates were incubated with NeutrAvidin (Pierce) overnight to pull down the biotinylated surface proteins. The precipitates were boiled in 2X sample loading buffer to elute Avidin-bound for SDS-PAGE analysis. GAPDH was used as a cytoplasmic marker to assess whether the surface biotinylated fractions include cytoplasmic channels (Data were not included).
Ubiquitination assay
CaV1.2e21+22 channels were transiently transfected with HA-CaV1.2e22, wild-type CaV3.1 or chimeric CaV3.1GCGGG channels in HEK 293 cells. Twenty-four hours after transfection, cells were treated with MG132 (2.5 μM) overnight and then lysed in PBS buffer containing 1% SDS and 1 mM EDTA. Cell lysates were boiled for 5 min at 95 °C, votexed for 10 sec, and then boiled for another 3 min at 95 °C. Ubiquitinated substrates in the supernatant were immunoprecipitated with anti-CaV3.1 or anti-HA, washed 3 times with cold PBS buffer, and resolved by 8% SDS-PAGE gel.
Western blot
Cells were harvested using lysis buffer (50 mM Tris, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, pH 7.4) containing protease inhibitor cocktails (Roche) at 48 hrs after transfection. After measurement of protein concentration, cell lysates were separated by 8% SDS-PAGE for 50 min at 150 V and transferred onto PVDF membrane at 30 V overnight at 4 °C. Subsequently, the membrane was blocked with 5% non-fat milk for 1h at room temperature and then incubated overnight at 4 °C with primary antibodies: rabbit anti-CaV1.2 (1:1000, ACC-003, Alomone), anti-CaV3.1 (1:1000, ACC-021), rabbit anti-β2 (1:1000, ACC-105), mouse anti-ubiquitin (1:1000, 13–1600, Invitrogen), rabbit anti-HA (1:1000, 71–5500), mouse anti-TfR (1:1000, 13–6800), mouse anti-β-actin (1:5000, A1978, Sigma) or mouse anti-GAPDH (1:2000, G8795). The membrane was washed three times with TBS-T buffer and then incubated with corresponding HRP-conjugated secondary antibodies (1:5000) for 1h at room temperature. After washing, proteins were detected using West Pico or Femto Chemiluminescent Substrate (Pierce). The blots were analyzed with ImageJ software (NIH).
Confocal imaging
HA-CaV1.2e22 or CaV1.2e21+22, α2δ and β2a subunit in pIRES2-EGFP as an indicator of β2a subunit expression were co-transfected in HEK293 cells cultured in 35 mm dish using calcium phosphate method. As described previously4, 48 h after transfection, cells were passaged to 2 wells with coated coverslips in 12-well plate, followed by 10 μM MG132 treatment for 2 h with cells in one of the wells. After that, cells were washed by cold PBS containing 5% FBS and fixed in 4% paraformaldehyde for 15 min. Following permeablization using 0.2% Tween-20/PBS for 15 min and blocking by 10% FBS/PBS for 20 min, cells were stained with rabbit anti-CaV1.2 (1:100, Alomone) at 37 °C for 60 min. Alexa Fluor 594-conjugated goat α-rabbit IgG antibody (Molecular Probes, 1:300) was used as secondary antibody to incubate with cells for 60 min in room temperature.
Cells were imaged using a Zeiss LSM-510 Meta confocal microscope with a 63 × 1.4NA oil immersion lens in the inverted position. EGFP was visualized by excitation with an argon laser (488 nm) and emission detected using a long-pass 530-nm filter. AF-594 antibody was visualized by excitation with a HeNe laser (543 nm) and emission detected using a 585–615 nm bandpass filter. Image acquisition was performed with identical gain, contrast, laser excitation, pinhole aperture and laser scanning speed for 3 rounds of cultures.
Electrophysiological recordings
As previously described, patch-clamp recordings were performed at 24–72 hrs after transfection using an Axopatch 200B amplifier (Molecular Device). The external solution contained 144 mM TEA-MeSO3, 10 mM HEPES, 1.8 mM CaCl2 (pH 7.4 adjusted with CsOH and osmolarity 300–310 mOsm with glucose). The internal solution contained 138 mM Cs-MeSO3, 5 mM CsCl, 0.5 mM EGTA, 10 mM HEPES, 1 mM MgCl2, 2 mg/ml Mg-ATP (pH 7.3 adjusted with CsOH and osmolarity 300–310 mOsm with glucose). To determine the whole cell current-voltage (I-V) relationships, currents were recorded by holding the cell at −90 mV (or −100 mV for CaV3.1GCGGG channel) before stepping to various potentials from −90 to 60 mV (or −100 to 40 mV for CaV3.1GCGGG channel) over 900 ms. The I-V curve was fitted with the equation: ICa = Gmax(V − Erev)/(1 + exp((V − V1/2)/k), where Gmax is the maximum conductance; Erev is the reversal potential; V1/2 is the half-activation potential; and k is the slope.
Statistics
All results were presented as mean ± SEM. Student’s t test was performed to compare two independent groups and one-way ANOVA followed by Bonferroni post hoc test was used for multiple group comparisons. P < 0.05 was considered significant.
Additional Information
How to cite this article: Hu, Z. et al. Aberrant Splicing Promotes Proteasomal Degradation of L-type CaV1.2 Calcium Channels by Competitive Binding for CaVβ Subunits in Cardiac Hypertrophy. Sci. Rep. 6, 35247; doi: 10.1038/srep35247 (2016).
References
Hofmann, F., Flockerzi, V., Kahl, S. & Wegener, J. W. L-type CaV1.2 calcium channels: from in vitro findings to in vivo function. Physiol Rev 94, 303–326, doi: 10.1152/physrev.00016.2013 (2014).
Dolphin, A. C. Calcium channel auxiliary alpha2delta and beta subunits: trafficking and one step beyond. Nat Rev Neurosci 13, 542–555, doi: 10.1038/nrn3311(2012).
Catterall, W. A. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol 3, a003947, doi: 10.1101/cshperspect.a003947 (2011).
Altier, C. et al. The Cavbeta subunit prevents RFP2-mediated ubiquitination and proteasomal degradation of L-type channels. Nat Neurosci 14, 173–180, doi: 10.1038/nn.2712 (2011).
Weissgerber, P. et al. Reduced cardiac L-type Ca2+ current in Ca(V)beta2-/- embryos impairs cardiac development and contraction with secondary defects in vascular maturation. Circ Res 99, 749–757, doi: 10.1161/01.RES.0000243978.15182.c1 (2006).
Seisenberger, C. et al. Functional embryonic cardiomyocytes after disruption of the L-type alpha1C (Cav1.2) calcium channel gene in the mouse. J Biol Chem 275, 39193–39199, doi: 10.1074/jbc.M006467200 (2000).
Chen, X. et al. Calcium influx through Cav1.2 is a proximal signal for pathological cardiomyocyte hypertrophy. J Mol Cell Cardiol 50, 460–470, doi: 10.1016/j.yjmcc.2010.11.012 (2011).
Goonasekera, S. A. et al. Decreased cardiac L-type Ca(2)(+) channel activity induces hypertrophy and heart failure in mice. J Clin Invest 122, 280–290, doi: 10.1172/JCI58227 (2012).
Patel, K. et al. Calcium channel blockers and outcomes in older patients with heart failure and preserved ejection fraction. Circ Heart Fail 7, 945–952, doi: 10.1161/CIRCHEARTFAILURE.114.001301 (2014).
de Vries, R. J., van Veldhuisen, D. J. & Dunselman, P. H. Efficacy and safety of calcium channel blockers in heart failure: focus on recent trials with second-generation dihydropyridines. Am Heart J 139, 185–194 (2000).
Packer, M. et al. Effect of amlodipine on the survival of patients with severe chronic heart failure due to a nonischemic cardiomyopathy: results of the PRAISE-2 study (prospective randomized amlodipine survival evaluation 2). JACC Heart Fail 1, 308–314, doi: 10.1016/j.jchf.2013.04.004 (2013).
Chen, X. et al. L-type Ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ Res 91, 517–524 (2002).
Tang, Z. Z. et al. Transcript scanning reveals novel and extensive splice variations in human l-type voltage-gated calcium channel, Cav1.2 alpha1 subunit. J Biol Chem 279, 44335–44343, doi: 10.1074/jbc.M407023200 (2004).
Liao, P. et al. Alternative splicing generates a novel truncated Cav1.2 channel in neonatal rat heart. J Biol Chem 290, 9262–9272, doi: 10.1074/jbc.M114.594911 (2015).
Tang, Z. Z., Zheng, S., Nikolic, J. & Black, D. L. Developmental control of CaV1.2 L-type calcium channel splicing by Fox proteins. Mol Cell Biol 29, 4757–4765, doi: 10.1128/MCB.00608-09 (2009).
Liao, P. et al. Molecular alteration of Ca(v)1.2 calcium channel in chronic myocardial infarction. Pflugers Arch 458, 701–711, doi: 10.1007/s00424-009-0652-4 (2009).
Yang, Y. et al. L-type Ca2+ channel alpha 1c subunit isoform switching in failing human ventricular myocardium. J Mol Cell Cardiol 32, 973–984, doi: 10.1006/jmcc.2000.1138 (2000).
Tiwari, S., Zhang, Y., Heller, J., Abernethy, D. R. & Soldatov, N. M. Atherosclerosis-related molecular alteration of the human CaV1.2 calcium channel alpha1C subunit. Proc Natl Acad Sci USA 103, 17024–17029, doi: 10.1073/pnas.0606539103 (2006).
Gidh-Jain, M., Huang, B., Jain, P., Battula, V. & el-Sherif, N. Reemergence of the fetal pattern of L-type calcium channel gene expression in non infarcted myocardium during left ventricular remodeling. Biochem Biophys Res Commun 216, 892–897 (1995).
Sano, M. et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 446, 444–448, doi: 10.1038/nature05602 (2007).
Fang, K. & Colecraft, H. M. Mechanism of auxiliary beta-subunit-mediated membrane targeting of L-type (Ca(V)1.2) channels. J Physiol 589, 4437–4455, doi: 10.1113/jphysiol.2011.214247 (2011).
Gao, C. et al. RBFox1-mediated RNA splicing regulates cardiac hypertrophy and heart failure. J Clin Invest 126, 195–206, doi: 10.1172/JCI84015 (2016).
Splawski, I. et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31, doi: 10.1016/j.cell.2004.09.011 (2004).
Dixon, R. E., Cheng, E. P., Mercado, J. L. & Santana, L. F. L-type Ca2+ channel function during Timothy syndrome. Trends Cardiovasc Med 22, 72–76, doi: 10.1016/j.tcm.2012.06.015 (2012).
Antzelevitch, C. et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 115, 442–449, doi: 10.1161/CIRCULATIONAHA.106.668392 (2007).
Ming, Z., Nordin, C., Siri, F. & Aronson, R. S. Reduced calcium current density in single myocytes isolated from hypertrophied failing guinea pig hearts. J Mol Cell Cardiol 26, 1133–1143, doi: 10.1006/jmcc.1994.113 (1994).
Mewes, T. & Ravens, U. L-type calcium currents of human myocytes from ventricle of non-failing and failing hearts and from atrium. J Mol Cell Cardiol 26, 1307–1320, doi: 10.1006/jmcc.1994.1149 (1994).
Wang, S. et al. Dilated cardiomyopathy with increased SR Ca2+ loading preceded by a hypercontractile state and diastolic failure in the alpha(1C)TG mouse. PLoS One 4, e4133, doi: 10.1371/journal.pone.0004133 (2009).
Glukhov, A. V. et al. Direct Evidence for Microdomain-Specific Localization and Remodeling of Functional L-Type Calcium Channels in Rat and Human Atrial Myocytes. Circulation 132, 2372–2384, doi: 10.1161/CIRCULATIONAHA.115.018131 (2015).
Makarewich, C. A. et al. A caveolae-targeted L-type Ca(2)+ channel antagonist inhibits hypertrophic signaling without reducing cardiac contractility. Circ Res 110, 669–674, doi: 10.1161/CIRCRESAHA.111.264028 (2012).
Horiuchi-Hirose, M. et al. Decrease in the density of t-tubular L-type Ca2+ channel currents in failing ventricular myocytes. Am J Physiol Heart Circ Physiol 300, H978–H988, doi: 10.1152/ajpheart.00508.2010 (2011).
Kashihara, T. et al. beta(2)-Adrenergic and M(2)-muscarinic receptors decrease basal t-tubular L-type Ca2+ channel activity and suppress ventricular contractility in heart failure. Eur J Pharmacol 724, 122–131, doi: 10.1016/j.ejphar.2013.12.037 (2014).
Nichols, C. B. et al. Sympathetic stimulation of adult cardiomyocytes requires association of AKAP5 with a subpopulation of L-type calcium channels. Circ Res 107, 747–756, doi: 10.1161/CIRCRESAHA.109.216127 (2010).
Liao, P., Zhang, H. Y. & Soong, T. W. Alternative splicing of voltage-gated calcium channels: from molecular biology to disease. Pflugers Arch 458, 481–487, doi: 10.1007/s00424-009-0635-5 (2009).
Cheng, X. et al. Alternative splicing of Cav1.2 channel exons in smooth muscle cells of resistance-size arteries generates currents with unique electrophysiological properties. Am J Physiol Heart Circ Physiol 297, H680–H688, doi: 10.1152/ajpheart.00109.2009 (2009).
Mezghrani, A. et al. A destructive interaction mechanism accounts for dominant-negative effects of misfolded mutants of voltage-gated calcium channels. J Neurosci 28, 4501–4511, doi: 10.1523/JNEUROSCI.2844-07.2008 (2008).
Walker, D., Bichet, D., Campbell, K. P. & De Waard, M. A beta 4 isoform-specific interaction site in the carboxyl-terminal region of the voltage-dependent Ca2+ channel alpha 1A subunit. J Biol Chem 273, 2361–2367 (1998).
Qin, N., Platano, D., Olcese, R., Stefani, E. & Birnbaumer, L. Direct interaction of gbetagamma with a C-terminal gbetagamma-binding domain of the Ca2+ channel alpha1 subunit is responsible for channel inhibition by G protein-coupled receptors. Proc Natl Acad Sci USA 94, 8866–8871 (1997).
Page, K. M. et al. N terminus is key to the dominant negative suppression of Ca(V)2 calcium channels: implications for episodic ataxia type 2. J Biol Chem 285, 835–844, doi: 10.1074/jbc.M109.065045 (2010).
Hullin, R. et al. Cardiac L-type calcium channel beta-subunits expressed in human heart have differential effects on single channel characteristics. J Biol Chem 278, 21623–21630, doi: 10.1074/jbc.M211164200 (2003).
Rockman, H. A. et al. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc Natl Acad Sci USA 88, 8277–8281 (1991).
Wang, J. et al. Splice variant specific modulation of CaV1.2 calcium channel by galectin-1 regulates arterial constriction. Circ Res 109, 1250–1258, doi: 10.1161/CIRCRESAHA.111.248849 (2011).
Altier, C. et al. Trafficking of L-type calcium channels mediated by the postsynaptic scaffolding protein AKAP79. J Biol Chem 277, 33598–33603, doi: 10.1074/jbc.M202476200 (2002).
Bannister, J. P. et al. Ca(V)1.2 channel N-terminal splice variants modulate functional surface expression in resistance size artery smooth muscle cells. J Biol Chem 286, 15058–15066, doi: 10.1074/jbc.M110.182816 (2011).
Acknowledgements
This work was supported by the National Medical Research Council of Singapore (NMRC/CBRG/0020/2012 to T.W.S.) and the National University Health Systems (NUHSRO/2014/086/AF-Partner/02 to T.W.S.), and the National Medical Research Council CS-IRG grant (to D.P.V.d.K.) and the ATTRaCT SPF grant to D.P.V.d.K.), and the President’s Graduate Fellowship from National University of Singapore to Z.Y.H. We are grateful to Prof. Emmanuel Bourinet from Institut de Génomique Fonctionnelle for construct of rat HA-CaV1.2e22 channel. We thank Mr Xiaoyuan Wang and Ms Suet Yen Chong from Department of Surgery for assistance of animal experiments and qPCR experiments, and Dr Esther Koh Geok Liang from Advanced Imaging Laboratory in Center for Life Science for technical help.
Author information
Authors and Affiliations
Contributions
T.W.S. conceived and supervised this project. Z.H. performed most of the biochemical assays. J.-W.W. generated the TAC mouse models, evaluated the cardiac function of TAC mice and harvested the left ventricles. D.Y. and Z.H. conducted the path-clamp recordings. Z.H. and J.-W.W. wrote the initial draft of the manuscript. T.W.S., P.L., H.M.C., D.P.V.d.K., J.L.S. and R.F. edited the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Hu, Z., Wang, JW., Yu, D. et al. Aberrant Splicing Promotes Proteasomal Degradation of L-type CaV1.2 Calcium Channels by Competitive Binding for CaVβ Subunits in Cardiac Hypertrophy. Sci Rep 6, 35247 (2016). https://doi.org/10.1038/srep35247
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep35247
This article is cited by
-
Development and characterization of a monoclonal antibody blocking human TRPM4 channel
Scientific Reports (2021)
-
TRPM4-specific blocking antibody attenuates reperfusion injury in a rat model of stroke
Pflügers Archiv - European Journal of Physiology (2019)
-
Leukocytic Toll-Like Receptor 2 Deficiency Preserves Cardiac Function And Reduces Fibrosis In Sustained Pressure Overload
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
