
Abstract
Hybrid seed production in rice relies on cytoplasmic male sterility (CMS) induced by specific mitochondrial proteins, whose deleterious effects are suppressed by nuclear Restorer of Fertility (RF) genes. The majority of RF proteins belong to a specific clade of the RNA-binding pentatricopeptide repeat protein family. We have characterised ‘restorer-of-fertility-like’ (RFL) sequences from 13 Oryza genomes and the Brachypodium distachyon genome. The majority of the RFL sequences are found in genomic clusters located at two or three chromosomal loci with only a minor proportion being present as isolated genes. The RFL genomic cluster located on Oryza chromosome 10, the location of almost all known active rice RF genes, shows extreme variation in structure and gene content between species. We show evidence for homologous recombination events as an efficient mechanism for generating the huge repertoire of RNA sequence recognition motifs within RFL proteins and a major driver of RFL sequence evolution. The RFL sequences identified here will improve our understanding of the molecular basis of CMS and fertility restoration in plants and will accelerate the development of new breeding strategies.
Similar content being viewed by others


Introduction
Proper functioning of a plant cell depends on coordinated expression of genes encoded in all three genomes (nuclear, mitochondrial, plastid). Despite this functional interdependence, inheritance and evolution of the nuclear and organellar genomes are quite different. Whereas nuclear genes are regularly re-shuffled during meiosis, the organellar genes are typically strictly uniparentally inherited1. Organellar genes and the nuclear genes encoding organellar proteins co-evolve via compensatory mutations1,2. The tight epistasis between nuclear and organellar genotypes can be the source of genetic incompatibilities after a new nuclear genome has been introduced into a cytoplasmic background1. Mitochondrial-nuclear genome incompatibilities caused by mitochondrially-encoded traits can lead to plant sterility and in this way can prevent interspecific crosses by creating barriers between the nucleus and foreign mitochondria1,2,3. Cytoplasmic male sterility (CMS) is one of the best investigated examples of mitochondrial-nuclear genome incompatibility as it has a direct application in production of F1 hybrids in crops4,5.
In breeding hybrids, CMS is used as a way of controlling self-pollination of autogamous plants4,5. The mitochondrial genomes of CMS plants encode proteins that induce the abortion of pollen development and thus male sterility4,6. The majority of investigated CMS-associated ORFs feature chimeric structures composed partly of conserved mitochondrial gene sequences (often the 5′ region containing transcription and translation starts) and partly of unique sequences6,7,8. How CMS-associated ORFs lead to the abortion of pollen development is largely unknown4,9. Deficiencies in mitochondrial energy production, toxicity of the CMS-causing proteins and premature programmed cell death of tapetal cells have been proposed as possible scenarios explaining this phenomenon4,6,10. In nature and in F1 hybrid breeding programs, expression of these mitochondrial CMS-inducing ORFs is controlled by restorer of fertility (RF) proteins. Nuclearly encoded RF proteins are imported into mitochondria where they block the expression of CMS-inducing ORFs8. The exact mechanism by which they achieve this is not known, but it is presumed that RF proteins bind directly to the CMS-inducing transcript, preventing translation or inducing RNA cleavage4,11. Recent studies indicate that additional proteins may be required for proper function of RF proteins in rice12,13 and restorer-of-fertility-like (RFL) proteins in Arabidopsis thaliana14,15.
RFL proteins form a distinct group of pentatricopeptide repeat (PPR) proteins11,16, a huge family of sequence-specific organellar RNA-binding proteins that participate in a wide range of post-transcriptional processes leading to the maturation of organellar transcripts17,18. PPR proteins can be divided into P and PLS subfamilies17,18,19. PLS-class proteins are predominantly involved in RNA editing, whereas P-class PPR proteins are involved in stabilisation of organellar transcripts and intron splicing17,18,19. RFL proteins belong to the P-class PPR subfamily and are characterised by the presence of tandem arrays of 15 to 20 PPR motifs each composed of 35 amino acid residues16. High substitution rates observed for particular amino acids within otherwise very conserved PPR motifs, indicating diversifying selection, prompted the conclusion that these residues might be directly involved in binding to RNA targets16. These discoveries provided the foundation for the development of a “PPR code” which allows the prediction of RNA targets of naturally occurring PPR proteins20,21,22 as well as the design of synthetic PPR proteins that can bind RNA molecules of interest23,24. Sequence specificity is ensured by distinct patterns of hydrogen bonding between each RNA base and the amino acid side chains at positions 5 and 35 in the aligned PPR motif 25.
In recent years, genes encoding RF proteins have been cloned from various plant species (reviewed in refs 5 and 11). The best studied cereal CMS/Rf systems are in the genus Oryza (Supplementary Table S1 and references therein). The Rf-1 locus located on chromosome 10 in rice has been isolated independently by several groups and shown to restore fertility in BT-type CMS (Supplementary Table S1)26,27,28. Rf-1 encodes a protein composed of 791 amino acids comprising 18 tandem PPR motifs26,27. Later it was discovered that in the elite restorer line Minghui63 (MH63), the locus on chromosome 10 encodes two Rf-1 genes, which were named Rf1a and Rf1b8. Rf1b orthologs in 6 restoring and 6 non-restoring lines differ by single amino acid substitutions8. RF1A was proposed to restore male fertility by blocking production of the suspected CMS-inducing protein ORF79 via endonucleolytic cleavage of the B-atp6/orf79 transcript. RF1B most likely also causes degradation of this dicistronic mRNA via an unknown mechanism8. RF1a has been demonstrated to be epistatic to RF1b8.
In CMS-WA rice, an interaction involving mitochondrially encoded, CMS-conferring Wild Abortive 352 (WA352) protein, nuclearly encoded COX11 protein and two RF genes has been described29. It was proposed that WA352 protein, produced exclusively in the tapetum of CMS-WA plants, interacts with COX11 and suppresses its function29. This suppression induces premature programmed tapetal cell death and leads to pollen abortion29. Two genes, Rf3 and Rf4, located on rice chromosomes 1 and 10 respectively, can restore CMS-WA30,31. PPR9-782-M and PPR782a, RF4 candidate proteins from the elite restorer line MH63 and cultivar IR24 respectively, are 86% identical to the RF1A restorer of CMS-BT rice and are encoded within the same chromosomal region32,33. Recently, two genes designated Rf5 and Rf6 were determined to restore fertility in Hong-Lian CMS rice12,13.
Three candidate RF genes and several additional RFL genes have been reported in sorghum34,35,36. The first to be identified was the Rf1 locus on chromosome 8, which, unlike all other RF proteins so far, encodes a PLS-class PPR protein36. The Rf2 gene located within a genomic cluster of RFL genes on chromosome 2 has been reported to restore fertility in the A1 cytoplasm34 and Rf5 located on chromosome 5 restores fertility in both A1 and A2 cytoplasms35. The mitochondrially-encoded, CMS-associated ORFs causing sterility in sorghum A1 and A2 cytoplasms have not been identified yet.
Apart from these examples from rice and sorghum, no RF genes encoding PPR proteins have been cloned and characterised from other cereal crops. Although CMS-based hybrid systems can be established without Rf sequence information, such knowledge will certainly accelerate marker-assisted selection and transfer of Rf alleles into elite breeding lines through traditional breeding. The obtained sequences could, however, also be directly introduced into desired lines by transgenic approaches. Intensive efforts are being made to identify restorer genes for msm1 and msm2 male-sterile cytoplasms in barley, and recently high-resolution genetic and physical mapping narrowed the region containing the Rfm1 locus in barley to the short arm of chromosome 6H37. Similarly, although several major restoring alleles in maize including Rf1 for CMS-Texas (CMS-T)38, Rf3 for CMS-USDA (CMS-S)39 and Rf4 for CMS-Charrua (CMS-C)40 have been mapped for decades, their sequences remain to be isolated.
With recent advances in sequencing technology, a whole plethora of fully or partially sequenced plant genomes and transcriptomes have become available. We took advantage of these large-scale data sets to systematically identify and characterise 158 RFL genes from 13 rice genomes and Brachypodium distachyon. We have compared several alternative methods for distinguishing RFL sequences from other P-class PPR proteins, resulting in a rapid but robust and effective pipeline. Subtle but characteristic features of PPR motifs in RFL proteins separate them from the remaining P-class PPR proteins in cereals. Only a few RFL genes are found as singlets with the vast majority organised into genomic clusters showing relatively low interspecific synteny. To explore mechanisms underlying the high sequence diversity we analysed an RFL cluster in nine rice species and were able to confirm recombination events as major factors driving evolution of this unusual subfamily of PPR proteins. We discuss the possible mode of action of RFL proteins and the implications for plant fertility. The catalogue of cereal RFL sequences gathered in this study will be a useful resource for experimental approaches and will help in identifying RFL sequences in newly mapped genetic regions predicted to contain a restorer-of-fertility gene.
Results
Bioinformatics pipeline for identifying RFL sequences in genomic sequence data
Thirteen Oryza genomes and the Brachypodium distachyon genome were obtained from public sequence depositories (Supplementary Table S2). Introns are extremely rare or absent within RFL coding sequences16, allowing accurate annotation of RFL sequences from six frame translations of genomic DNA. Predicted ORFs were screened for PPR motifs using hidden Markov models41. Out of 9.4 million potential ORFs in the 14 genomes, 8729 contained predicted PPR motifs (Supplementary Table S3). 1736 sequences encoding 10 or more P-class PPR motifs were retained for further analysis. The number of P-class PPR proteins composed of ten or more PPR motifs varied from 105 in Oryza glaberrima to 144 in Oryza longistaminata (Supplementary Table S3). RFL genes generally show higher sequence similarity to their intra-specific paralogs than to putatively orthologous RFL genes from other species16. This phenomenon means that they can be identified by phylogenetic methods16 or other sequence clustering approaches42. We applied four different methods to identify the RFL proteins within the P-class PPR protein sets: creation of orthologous sequence clusters using OrthoMCL43 or OrthoFinder44, sequence clustering with CD-Hit45 and phylogenetic analysis following multiple alignment (Fig. 1 and Supplementary Tables S3–S5).

Identification of RFL sequences in 13 Oryza genomes and the Brachypodium distachyon genome.
(a) Comparison of the assignment of the P-class PPR proteins into sequence clusters by OrthoFinder, OrthoMCL and CD-Hit. For OrthoMCL and OrthoFinder, P-class PPR sequences from all 14 genomes were analysed at once and the average number of RFL sequences per genome is shown. For CD-Hit analysis, each genome was analysed separately and only the representative outcome of clustering the sequences from O. sativa japonica is shown. Clusters composed of RFL sequences are highlighted in red. (b) Radial tree of 1736 P-class PPR-protein sequences from 13 rice genomes and the B. distachyon genome, as well as 36 reference RFL sequences16. Sequences were aligned with MAFFT v7.18766 and the tree constructed using FastTree 2.1.8 software67. The tree was visualised in Geneious 8.1.6 (www.geneious.com). The RFL clade is highlighted in red. (c) Comparison of the number of RFL sequences identified by the four different approaches illustrated by a Venn diagram. Out of 158 identified RFL sequences, 138 were identified by all four methods and additional 16 sequences were identified by all methods other than CD-Hit. Three sequences were not found by OrthoFinder and one sequence was identified only by OrthoMCL. (d) Contribution of RFL sequences to the total number of P-class PPRs in 13 Oryza genomes and the genome of B. distachyon. The total number of P-class PPR sequences per plant species is highlighted in light green and the number of RFL sequences in black, respectively.
All four methods tested in this study converged on a set of 138 RFL sequences (Fig. 1c), with another 16 found by all methods other than CD-Hit (Fig. 1c and Supplementary Table S6). In addition, three sequences were found by all methods other than OrthoFinder and one sequence was found only by OrthoMCL (Fig. 1c and Supplementary Table S6). The preferred method depends on the scale of the analysis and the time and computational resources available. The analysed Oryza genomes contained from 2 (O. glaberrima) to 18 (O. sativa indica) RFL genes of 10 or more PPR motifs (Fig. 1d and Supplementary Table S3).
Analysis of RFL clades in Oryza
To study the evolutionary relationships between the identified RFLs, the 147 Oryza RFL sequences were aligned with previously published RFs and RFLs (Supplementary Table S1) and a phylogeny constructed (Figs 1b and 2). The RFL sequences form a single monophyletic clade that stands out from other P-class clades by showing a much greater degree of recent divergence (Fig. 1b). Within the RFL clade, the sequences group into three subclades that correspond to genomic clusters located on chromosomes 4, 8 and 10, respectively (Fig. 2). The largest genomic cluster is on chromosome 10 (Fig. 2), which has been reported previously to contain active Rf-1a, Rf-1b and Rf4 genes in different rice accessions8,27,33,46. The proportion of RFL genes in genomic clusters ranges from zero in O. brachyantha to 93% in O. sativa japonica (Table 1).

Phylogenetic analysis of RFL sequences encoded in Oryza genomes.
Multiple sequence alignment of 147 RFL sequences identified in 13 rice genomes supplemented by 27 sequences of published RF and RFLs (Supplementary Table S1) was performed with MAFFT v7.187. The tree was generated with FastTree 2.1.5 and coloured in Geneious 8.1.6 (www.geneious.com). The RFL sequences located on chromosome 4 are highlighted in green, chromosome 8 in orange and chromosome 10 in blue, respectively. Published RFL sequences are highlighted in red and, apart from RF6, are all located on chromosome 10. Due to space limitations, not all tip labels are shown. The scale bar represents the number of substitutions per site.
Mechanisms contributing to the evolutionary plasticity of RFL genes in rice
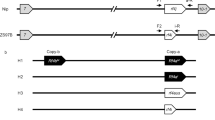
Various mechanisms including homologous recombination, gene conversion, duplication and selection have been proposed to contribute to the genome-wide diversity of RFL-gene loci in plants16,47,48. In order to investigate such phenomena within the largest rice RFL cluster on chromosome 10, the corresponding regions spanning ~500 kbp of nine Oryza species were compared (Fig. 3). Local pairwise alignments revealed that the colinearity of the genomic sequences tends to break at the sites of RFL loci (Supplementary Figure S1). The number of RFL genes in the cluster composed of 10 or more PPR motifs varied from 8 in O. sativa indica to zero in O. brachyantha (Fig. 3, Supplementary Table S7). Within the cluster, two regions carrying a variable number of RFL genes can be distinguished (Fig. 3). The first is located between two conserved genes encoding a KH-domain protein and a DNA-directed RNA polymerase (Fig. 3). These flanking genes identify this region as that carrying the Rf4 restorer gene in O. sativa japonica Zhonghua 1133 (Supplementary Figure S2). The second region is located between genes encoding an acetyltransferase and a serine/threonine-protein kinase (Fig. 3 and Supplementary Table S7). These flanking genes identify this region as that carrying the RF1a restorer8,26,28 (Supplementary Figure S2). Interestingly, O. meridionalis and O. barthii both contain an RFL gene with high sequence similarity to Rf-1A in O. sativa indica (Fig. 3). In O. nivara the Rf-region 2 seems to have been “broken” and subsequently translocated upstream in the cluster as indicated by the presence of two RFL sequences and the gene encoding the kinase protein (Fig. 3). Apart from the RFL genes located within the Rf-regions 1 and 2, several other genes are found nearby, including Rf-1B8 and another RFL gene located downstream in the cluster (Fig. 3). Both genes are present in all rice species carrying the A genome type (Fig. 3). In O. glumipatula and O. barthii a single RFL gene was found to be located upstream of the conserved gene encoding the alpha-galactosidase (Fig. 3 and Supplementary Table S7). It seems possible that an RFL gene located at this position in O. nivara was involved in a recombination event that has caused the partial translocation of Rf-region 2 (Fig. 3). The three RFL sequences found within the genomic cluster in O. punctata which carries the B genome type differ from the other RFLs found in the cluster. Two of the sequences form a distinct branch in the phylogenetic tree shown in Supplementary Figure S3, reflecting the evolutionary distance between A and B genome types in Oryza.

Structural analysis of the RFL cluster located on chromosome 10 in nine Oryza genomes.
Genomic regions spanning ~500 kbp of chromosome 10 from O. sativa indica (OsI), O. sativa japonica (OsJ), O. rufipogon (Or), O. nivara (On), O. glumipatula (Oglu), O. barthii (Oba), O. meridionalis (Om), O. punctata (Op), and O. brachyantha (Obr), were obtained from EnsemblPlants (http://plants.ensembl.org/index.html) and gene annotations were visualized in KONG Cloning Suite (www.kongcloning.com). Oryza genome types (AA, FF, BB) are shown in parentheses. Genes annotated in EnsemblPlants are shown as boxes, with conserved genes listed in Supplementary Table S7 highlighted in blue. RFL genes are shown as arrowheads, with red indicating ten or more PPR motifs and yellow less than ten. The direction of the arrowhead indicates the orientation of the RFL gene. Two regions described previously to contain active RF genes are indicated as Rf-region 133 and Rf-region 228, and are located between conserved genes b-c and d-e, respectively. The annotation of the RFL genes (Rf-1A-D) is based on the phylogenetic tree presented in Supplementary Figure S3. The Rf-1B gene shown in the figure has been annotated according to8 and is different from the Rf-1B gene named by others28,68. The dashed lines connect syntenic loci between species. The conserved genes encode the following proteins alpha-galactosidase (a), KH-domain-containing protein (b), DNA-directed RNA polymerase (c), acyltransferase (d), serine/threonine-protein kinase (e), glutamyl-tRNA reductase (f).
The results of the structural analysis of the RFL loci in all nine rice accessions and the high sequence similarity of the RFL genes (Fig. 3 and Supplementary Figure S3), suggest that the structural complexity of RFL clusters originates from gene duplications allowing for homologous recombination and unequal crossing over to take place.
Recombination analysis of RFL sequences located in the cluster on chromosome 10 in O. sativa indica with the Recombination Detection Program (RDP4)49 identified several potential recombination events (Supplementary Figure S4) and emphasises the chimeric structure of RFL genes. Such recombination events can lead to translocations and insertions of partial or whole RFL sequences within a cluster and by doing so will contribute to the overall sequence plasticity. Insertion of a partial RFL sequence within an already present RFL gene has the immediate consequence of altering RNA recognition by the gene product. In the longer term, duplicated RFL genes will diverge and slowly gain the ability to bind new mitochondrial RNA targets.
Duplication, deletion, insertion and transposition of PPR motifs in RFL genes
The modular structure of PPR proteins implies that not only duplications of whole genes but also motif duplications, deletions, insertions or transpositions by recombination can give rise to functional protein variants with altered target recognition48. To look for evidence of such events, PPR motifs from all RFL proteins encoded on chromosome 10 in O. rufipogon, O. sativa indica and O. sativa japonica were aligned and used to build a distance tree (Fig. 4a). These three species were chosen because they are the most important with respect to the development of CMS-based breeding systems in rice. The distance tree of O. rufipogon PPR motifs revealed duplications and insertions, a transposition, and also deletions of one or several PPR motifs (Fig. 4b). Similar results were seen for O. sativa indica and japonica (Supplementary Figures S5 and S6).

Analysis of the PPR motifs present in RFL proteins encoded within the cluster on chromosome 10 in O. rufipogon.
(a) Tree illustrating the phylogenetic relationships between PPR motifs extracted from protein sequences and numbered starting from the amino-terminus. In total, 69 PPR motifs from 5 RFL sequences have been aligned. (b) Schematic representation of RFL proteins identified in O. rufipogon. The PPR motifs are coloured and numbered according to the tree in (a). Putative duplication, deletions and insertions of PPR motifs have been indicated.
Characteristic features of RFL proteins
The high diversity and rapid evolution of RFL sequences might suggest relaxed selection on them, but by several criteria they appear to be closer to consensus PPR proteins than other, more highly conserved members of the family. RFL proteins tend to have more PPR motifs per protein (Fig. 5a), and these motifs are generally better matches to the PPR consensus (as judged by hmmsearch scores) than those in other P-class proteins (Fig. 5b). The differences are even more prominent when total protein hmmsearch scores are compared (Fig. 5c). The sequence logos obtained from the analysis of ~34000 P-class PPR motifs and ~3000 motifs extracted from RFL proteins are almost identical (Supplementary Figure S7). The frequency with which different base-recognising combinations occur within RFL PPR motifs is generally similar to that seen for other P-class PPR proteins, but there are some subtle but significant differences (Fig. 5d). The commonest purine-recognising combinations in P-class PPR proteins are generally TD (G) and TN (A), but in RFL proteins they are SD (G) and SN (A). GD and GN are also unusually common in RFL proteins, whereas the C-recognising combinations NS and NT are unusually rare (Fig. 5d). The overall effect is that whereas P-class PPR proteins have a strong predicted bias toward pyrimidines, this is much less pronounced in RFL proteins (Fig. 5e).

Characterisation of RFL proteins from Oryza and B. distachyon.
(a) Number of PPR motifs in the identified P-class and RFL proteins. (b) Distribution of PPR motif scores for P-class and RFL proteins. The scores are domain scores per motif from hmmsearch using the P motif to search41. (c) Distribution of total protein scores (full sequence scores from hmmsearch using the P motif to search). (d) Frequency of amino acid combinations at positions 5 and 35 of PPR motifs in RFL and P-class PPR proteins. The 25 most frequent combinations are shown, sorted by abundance in P-class PPRs. Asterisks illustrate the combinations that are significantly more frequent in RFLs compared to P-class PPRs (p < 0.01) as listed in Supplementary Table S8. (e) Proportion of predicted recognised RNA bases (A, C, G, U) for P-class and RFL proteins. For the calculations, the set of combinations that add up to at least 50% of all combinations was used (Supplementary Table S9). (f) Combinations of amino acid residues at positions 5 and 35 of PPR motifs and the RNA sequence predicted to be recognised by these combinations20 for the Rf-1C orthologues26 located within the Rf-region 2 of the RFL cluster on chromosome 10.
High levels of sequence divergence coupled with strong conservation of overall structure are consistent with positive (diversifying) selection on specific residues within the sequence. Positive selection on the base-recognising residues within RFL sequences has been shown before16 and would be expected to lead to exceptionally high diversity at these positions. This is illustrated by a comparison of amino acid residues at positions 5 and 35 of PPR motifs extracted from Rf-1C orthologues26 in 7 rice species (Fig. 5f). High variation in these amino acid residues, supplemented by the insertion/deletion of whole PPR motifs originating from homologous recombination and unequal crossover, results in stark changes in the RNA sequence predicted to be recognized by a particular PPR protein (Fig. 5f).
Discussion
RFL proteins share many characteristics with another protein family that is greatly expanded in plants, the family of disease resistance (R-) proteins typically composed of a nucleotide-binding site (NBS) and leucine-rich repeats (LRR)50. These shared characteristics include long arrays of tandem repeats within each protein and chromosomal clusters of related genes that evolve in a manner that is strikingly different to the genes around them. The rapid evolution of NBS-LRR proteins is thought to be driven by selection for pathogen recognition in an evolutionary “arms-race”50. The similarly rapid evolution of RFL genes has been proposed to be driven by an analogous “arms-race” with the plant’s own mitochondrial genome3,16. RFL proteins induce alterations in processing of their mitochondrial target RNAs, leading in some cases to complete suppression of the transcript, or at least the protein encoded by the transcript26,27,28,32,33. In this way they can prevent the expression of mitochondrial ORFs that otherwise lead to CMS or other forms of nuclear-mitochondrial incompatibility. The “arms-race” ensues because mitochondrial mutations causing CMS can be selected for under some circumstances (as they favour transmission of the mutated mitochondrial genome via the seed), but once CMS is frequent in a population, nuclear RF genes are strongly favoured in their turn. Hence the selection pressures on R genes and RFL genes are similar, probably explaining their similar evolutionary behaviour.
The high diversity in RFL genes is focused on the residues that contribute the most to RNA binding specificity16. Only one or two changes at these residues are sufficient to alter binding specificity from one target sequence to another in other PPR proteins20 and this is presumably true for RFL proteins too. In this case, we can confidently predict that in many cases the diverse sequences present at the same locus in different Oryza species (e.g. Fig. 5f) will have different functions because they can bind different RNA targets. These RNA targets are potentially predictable using the ‘code’ proposed to describe RNA sequence recognition by PPR proteins20,21,22. The identification of the exact binding sites of RF proteins will greatly accelerate the characterisation of the molecular mechanisms involved in fertility restoration. The mode of action of RF proteins remains largely unknown, but they are frequently implicated in endonucleolytic cleavage of their target RNAs8. As RF proteins are composed almost entirely of PPR motifs with no additional C- or N-terminal domains likely to possess endonuclease activity, it seems likely that additional proteins must be involved14,15.
The data on the Oryza RFL clusters are consistent with recombinational processes driving most of the sequence evolution, rather than simple point mutations. The high-level of intra-cluster similarity, contrasted with the low level of inter-cluster and inter-species similarity, strongly suggests that there is sequence exchange between genes within each cluster. The most likely explanation is unequal pairing at meiosis followed by gene conversion or unequal crossing over, leading to the duplications, insertions and translocations of single PPR motifs or whole genes that we observed. A plausible depiction of some of these processes is shown in Fig. 6. The outcome is a continual flux of new RFL variants into the gene pool that can potentially act on any new mitochondrial sequences that appear in the equally recombinogenic mitochondrial genome. It is evident that in addition to creating functional variants, these processes will also randomly create many non-functional variants with truncations, deletions or other defects. Indeed, the Oryza RFL clusters analysed here are littered with presumably non-functional RFL fragments seen in the six-frame translations (some of these are shown in Fig. 3), hence the cut-off at 10 PPR motifs that we used. These fragments may however indirectly serve a purpose as inducers or generators of small non-coding RNAs as well as reservoirs of structural variation, enhancing the diversity of novel RFL variants.

Mechanisms contributing to the evolutionary plasticity of RFL genes in Oryza.
(a) Tandem duplication. (b) Recombination via equal crossing over (c) Recombination via unequal crossing over causing change in gene number (d) Recombination via unequal crossing over, causing change in gene length. Arrows represent RFL sequences, with differences in shading representing divergent sequences.
A genome-wide analysis of the RNA-dependent RNA POLYMERASE6/DICER-LIKE4 pathway in Arabidopsis showed that several RFL transcripts are targeted by trans-acting short interfering RNAs (ta-siRNAs)51. Ta-siRNAs are produced by a mechanism that yields 21-nucleotide, phased siRNAs from TAS transcripts that are initially processed by miRNA-guided cleavage51. Similarly, rice osa-miR1425 targets RF1 mRNAs52. Out of five predicted gene targets of miR1425 four encode RFL proteins, including Os08g01650, Os10g35436, Os10g35640, Os10g3524052. Plant R genes have been reported to be a frequent target of miRNAs53, especially those forming chromosomal clusters50. In addition, as seen for RFL genes in Arabidopsis, miR2109/miR2118/miR1507 in Medicago, miR482/miR2118 in tomato, and miR6019/miR6020 in tobacco have been shown to guide the cleavage of transcripts of NBS-LRRs, and to trigger secondary phased siRNA production by RNA-dependent RNA polymerase54,55,56. The functional consequences of this potential regulation by gene-silencing pathways are still unclear.
In the past, the primary interest in RF genes has been their role in breeding hybrid crops, with several of them being widely used in agriculture even before their identity and mode of action were known. RF genes have played an important role in the development of hybrid rice varieties. The availability of suitable restorer lines was the major factor that contributed to the success of the “three-line” (CMS, maintainer and restorer line) system based on indica WA-CMS in hybrid rice production57. CMS-WA remains the most widely used single source of sterile cytoplasm for commercial hybrid seed production since 1970, not only because its pollen sterility is complete, but also because the fertility of hybrids between CMS-WA and restorer lines is readily recovered57. In recent years several RF genes have been cloned in rice (Supplementary Table S1) and with the increasing availability of sequenced genomes from other cereals, more RF sequences are forthcoming. Our analysis revealed about a dozen putatively functional RFL genes per rice species on average (Supplementary Table S3). In O. brachyantha and O. glaberrima, however, many fewer were found. In comparison to all other Oryza species analysed here, O. brachyantha has the smallest genome size (Supplementary Table S3) and far fewer gene models have been predicted in its genome sequence than in other rice species, suggesting an amplification of many gene families in domesticated rice58. In particular, disease resistance-related gene families (e.g. the NBS-LRR family) are highly overrepresented in O. sativa58. Whereas O. brachyantha branches early in the Oryza lineage59, O. glaberrima was domesticated from the wild progenitor O. barthii, an A genome species, and is related to other A genome rice species including O. sativa japonica and indica60. Recently, using O. glaberrima as a cytoplasm donor, a new indica CMS, designated African rice (AF)-CMS, was developed and corresponding introgression lines carrying major restorer genes from O. glaberrima were created61. The presence of active Rf loci in O. glaberrima conflicts with the low number of RFL genes found in this study. We suspect that the two RFL genes found in the available genome sequence (Table 1) probably do not represent an exhaustive catalogue of the RFL genes in the O. glaberrima genome.
In this study, we have analysed a total of 147 Oryza RFL sequences from 13 genomes. This knowledge will expedite marker-assisted selection and transfer of Rf alleles into elite breeding lines through traditional breeding. The identified sequences could, however, also be directly introduced into desired lines by transgenic approaches. As their fascinating properties and unusual evolutionary behaviour become better understood, other potential applications of RFL proteins besides fertility restoration are coming to the fore. PPR proteins are being investigated for their potential as custom-made RNA processing tools24,62, and of all the natural PPR proteins, RFL proteins are perhaps the best suited for biotechnological manipulation given their recurrent selection for diversity in binding site selection.
Methods
Genomic sequence data used in the study
The compressed fasta files containing the genomic sequence data of 13 rice genomes were either downloaded from EnsemblPlants (http://plants.ensembl.org/index.html)63 or the Rice Annotation Project Database (http://rapdb.dna.affrc.go.jp/gb1/index.html)64. The genome sequence of B. distachyon was downloaded from Phytozome (http://phytozome.jgi.doe.gov/pz/portal.html). A detailed list of the names of downloaded files, genome versions and release dates can be found in Supplementary Table S2.
Bioinformatics pipeline for identifying RFL genes in genomic sequence data
The DNA sequences were screened for open reading frames (ORFs) in six-frame translations with the getorf program of the EMBOSS 6.6.0 package65. Predicted ORFs longer than 92 codons were screened for the presence of P- and PLS-class PPR motifs using hmmsearch from the HMMER 3.1b package (hmmer.org) and hidden Markov models defined by hmmbuild41. The post-processing of hmmsearch results was carried out according to rules described previously41. Sequences containing 10 or more P-class PPR motifs were retained for further analysis, as a previous study has shown that RFL genes are primarily comprised of tandem arrays of 15 to 20 PPR motifs16.
OrthoMCL, OrthoFinder and CD-Hit analysis
Sequence clustering methods were used to identify candidate RF sequences. One approach employed the OrthoMCL algorithm43 via the OrthoMCL-DB website (http://www.orthomcl.org/orthomcl/). In a second approach, OrthoFinder from https://github.com/davidemms/OrthoFinder44 was used to cluster P-class PPR proteins from each data set. The resulting output files were screened for groups containing reference RFLs16. A third approach used CD-Hit version 4.6.4 from https://github.com/weizhongli/cdhit/releases45. Our preliminary CD-Hit analyses on the whole pool of 1736 P-class PPR proteins showed that, even with the lowest identity threshold of 40%, CD-Hit failed to group RFLs from multiple species into a single cluster. Separately analysing the data sets from each species overcame that problem. CD-Hit was run with the settings -c 0.4 -n 2 -d 200 -G0 -aS 0.1.
Phylogenetic analysis
For identification of RFL sequences based on phylogeny, P-class PPR proteins were aligned with a parallel version of MAFFT v7.18766. The resulting alignment file was used for tree generation with FastTree 2.1.867. The resulting tree was analysed in Geneious 8.1.6 (http://www.geneious.com/). The RFL clade was identified by the location of reference RFLs16.
Synteny analysis
For the analysis of genomic fragments carrying the RFL cluster on rice chromosome 10, the regions spanning ~500 Mbp were obtained from EnsemblPlants (http://plants.ensembl.org) and the gene annotations were visualized in KONG Cloning Suite (www.kongcloning.com). The RFL genes were manually annotated.
Additional Information
How to cite this article: Melonek, J. et al. Evolutionary plasticity of restorer-of-fertility-like proteins in rice. Sci. Rep. 6, 35152; doi: 10.1038/srep35152 (2016).
References
Birky, C. W. The inheritance of genes in mitochondria and chloroplasts: Laws, mechanisms, and models. Annu Rev Genet 35, 125–148, 10.1146/annurev.genet.35.102401.090231 (2001).
Greiner, S. & Bock, R. Tuning a menage a trois: Co-evolution and co-adaptation of nuclear and organellar genomes in plants. Bioessays 35, 354–365, 10.1002/bies.201200137 (2013).
Touzet, P. & Budar, F. Unveiling the molecular arms race between two conflicting genomes in cytoplasmic male sterility? Trends Plant Sci 9, 568–570, 10.1016/j.tplants.2004.10.001 (2004).
Chase, C. D. Cytoplasmic male sterility: a window to the world of plant mitochondrial-nuclear interactions. Trends Genet 23, 81–90, 10.1016/j.tig.2006.12.004 (2007).
Chen, L. & Liu, Y. G. Male sterility and fertility restoration in crops. Annu Rev Plant Biol 65, 579–606, 10.1146/annurev-arplant-050213-040119 (2014).
Hanson, M. R. & Bentolila, S. Interactions of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell 16, S154–169, 10.1105/tpc.015966 (2004).
Akagi, H., Sakamoto, M., Shinjyo, C., Shimada, H. & Fujimura, T. A unique sequence located downstream from the rice mitochondrial atp6 may cause male-sterility. Curr Genet 25, 52–58, 10.1007/Bf00712968 (1994).
Wang, Z. H. et al. Cytoplasmic male sterility of rice with boro II cytoplasm is caused by a cytotoxic peptide and is restored by two related PPR motif genes via distinct modes of mRNA silencing. Plant Cell 18, 676–687, 10.1105/tpc.105.038240 (2006).
Guo, J. X. & Liu, Y. G. The genetic and molecular basis of cytoplasmic male sterility and fertility restoration in rice. Chin Sci Bull 54, 2404–2409, 10.1007/s11434-009-0322-0 (2009).
Schnable, P. S. & Wise, R. P. The molecular basis of cytoplasmic male sterility and fertility restoration. Trends Plant Sci 3, 175–180, 10.1016/S1360-1385(98)01235-7 (1998).
Dahan, J. & Mireau, H. The Rf and Rf-like PPR in higher plants, a fast-evolving subclass of PPR genes. RNA Biol 10, 1469–1476, 10.4161/rna.25568 (2013).
Hu, J. et al. The rice pentatricopeptide repeat protein RF5 restores fertility in Hong-Lian cytoplasmic male-sterile lines via a complex with the glycine-rich protein GRP162. Plant Cell 24, 109–122, 10.1105/tpc.111.093211 (2012).
Huang, W. et al. Pentatricopeptide-repeat family protein RF6 functions with hexokinase 6 to rescue rice cytoplasmic male sterility. Proc Natl Acad Sci USA 112, 14984–14989, 10.1073/pnas.1511748112 (2015).
Fujii, S. et al. The Restorer-of-fertility-like 2 pentatricopeptide repeat protein and RNase P are required for the processing of mitochondrial orf291 RNA in Arabidopsis. Plant J, 10.1111/tpj.13185 (2016).
Stoll, B. & Binder, S. Two NYN domain containing putative nucleases are involved in transcript maturation in Arabidopsis mitochondria. Plant J, 10.1111/tpj.13111 (2015).
Fujii, S., Bond, C. S. & Small, I. D. Selection patterns on restorer-like genes reveal a conflict between nuclear and mitochondrial genomes throughout angiosperm evolution. Proc Natl Acad Sci USA 108, 1723–1728, 10.1073/pnas.1007667108 (2011).
Barkan, A. & Small, I. Pentatricopeptide repeat proteins in plants. Annu Rev Plant Biol 65, 415–442, 10.1146/annurev-arplant-050213-040159 (2014).
Schmitz-Linneweber, C. & Small, I. Pentatricopeptide repeat proteins: a socket set for organelle gene expression. Trends Plant Sci 13, 663–670, 10.1016/j.tplants.2008.10.001 (2008).
Lurin, C. et al. Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell 16, 2089–2103, 10.1105/tpc.104.022236 (2004).
Barkan, A. et al. A combinatorial amino acid code for RNA recognition by pentatricopeptide repeat proteins. PLoS Genet 8, 10.1371/journal.pgen.1002910 (2012).
Nakamura, T., Yagi, Y. & Kobayashi, K. Mechanistic insight into pentatricopeptide repeat proteins as sequence-specific RNA-binding proteins for organellar RNAs in plants. Plant Cell Physiol 53, 1171–1179, 10.1093/pcp/pcs069 (2012).
Yagi, Y., Hayashi, S., Kobayashi, K., Hirayama, T. & Nakamura, T. Elucidation of the RNA recognition code for pentatricopeptide repeat proteins involved in organelle RNA editing in plants. PLoS One 8, 10.1371/journal.pone.0057286 (2013).
Shen, C. C. et al. Specific RNA recognition by designer pentatricopeptide repeat protein. Mol Plant 8, 667–670, 10.1016/j.molp.2015.01.001 (2015).
Yagi, Y., Nakamura, T. & Small, I. The potential for manipulating RNA with pentatricopeptide repeat proteins. Plant J 78, 772–782, 10.1111/tpj.12377 (2014).
Shen, C. et al. Structural basis for specific single-stranded RNA recognition by designer pentatricopeptide repeat proteins. Nat Commun 7, 11285, 10.1038/ncomms11285 (2016).
Akagi, H. et al. Positional cloning of the rice Rf-1 gene, a restorer of BT-type cytoplasmic male sterility that encodes a mitochondria-targeting PPR protein. Theor Appl Genet 108, 1449–1457, 10.1007/s00122-004-1591-2 (2004).
Kazama, T. & Toriyama, K. A pentatricopeptide repeat-containing gene that promotes the processing of aberrant atp6 RNA of cytoplasmic male-sterile rice. FEBS Lett 544, 99–102, 10.1016/S0014-5793(03)00480-0 (2003).
Komori, T. et al. Map-based cloning of a fertility restorer gene, Rf-1, in rice (Oryza sativa L.). Plant J 37, 315–325, 10.1111/j.1365-313X.2004.01961.x (2004).
Luo, D. P. et al. A detrimental mitochondrial-nuclear interaction causes cytoplasmic male sterility in rice. Nat Genet 45, 573–U157, 10.1038/ng.2570 (2013).
Ahmadikhah, A. & Karlov, G. I. Molecular mapping of the fertility-restoration gene Rf4 for WA-cytoplasmic male sterility in rice. Plant Breed 125, 363–367, 10.1111/j.1439-0523.2006.01246.x (2006).
Lu, Y., Virmani, S. S., Zhang, G., Bharaj, T. S. & Huang, N. Mapping of the Rf-3 nuclear fertility-restoring gene for WA cytoplasmic male sterility in rice using RAPD and RFLP markers. Theor Appl Genet 94, 27–33, 10.1007/s001220050377 (1997).
Kazama, T. & Toriyama, K. A fertility restorer gene, Rf4, widely used for hybrid rice breeding encodes a pentatricopeptide repeat protein. Rice (N Y) 7, 10.1186/s12284-014-0028-z (2014).
Tang, H. W. et al. The rice restorer Rf4 for wild-abortive cytoplasmic male sterility encodes a mitochondrial-localized PPR protein that functions in reduction of WA352 transcripts. Mol Plant 7, 1497–1500, 10.1093/mp/ssu047 (2014).
Jordan, D. R., Mace, E. S., Henzell, R. G., Klein, P. E. & Klein, R. R. Molecular mapping and candidate gene identification of the Rf2 gene for pollen fertility restoration in sorghum [Sorghum bicolor (L.) Moench]. Theor Appl Genet 120, 1279–1287, 10.1007/s00122-009-1255-3 (2010).
Jordan, D. R. et al. Mapping and characterization of Rf 5: a new gene conditioning pollen fertility restoration in A1 and A2 cytoplasm in sorghum (Sorghum bicolor (L.) Moench). Theor Appl Genet 123, 383–396, 10.1007/s00122-011-1591-y (2011).
Klein, R. R. et al. Fertility restorer locus Rf1 [corrected] of sorghum (Sorghum bicolor L.) encodes a pentatricopeptide repeat protein not present in the colinear region of rice chromosome 12. Theor Appl Genet 111, 994–1012, 10.1007/s00122-005-2011-y (2005).
Ui, H. et al. High-resolution genetic mapping and physical map construction for the fertility restorer Rfm1 locus in barley. Theor Appl Genet 128, 283–290, 10.1007/s00122-014-2428-2 (2015).
Duvick, D. N., Snyder, R. J. & Anderson, E. G. The chromosomal location of Rfl, a restorer gene for cytoplasmic pollen sterile maize. Genetics 46, 1245–1252 (1961).
Kamps, T. L. & Chase, C. D. RFLP mapping of the maize gametophytic restorer-of-fertility locus (rf3) and aberrant pollen transmission of the nonrestoring rf3 allele. Theor Appl Genet 95, 525–531, DOI 10.1007/s001220050593 (1997).
Sisco, P. H. Duplications complicate genetic-mapping of Rf4, a restorer gene for cms-C cytoplasmic male-sterility in corn. Crop Sci 31, 1263–1266 (1991).
Cheng, S. F. et al. Redefining the structural motifs that determine RNA binding and RNA editing by pentatricopeptide repeat proteins in land plants. Plant J 85, 532–547, 10.1111/tpj.13121 (2016).
Sykes, T. et al. In-silico identification of candidate genes for fertility restoration in cytoplasmic male sterile perennial ryegrass (Lolium perenne L.). Genome Biol Evol, 10.1093/gbe/evw047 (2016).
Li, L., Stoeckert, C. J. Jr. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13, 2178–2189, 10.1101/gr.1224503 (2003).
Emms, D. M. & Kelly, S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol 16, 157, 10.1186/s13059-015-0721-2 (2015).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659, 10.1093/bioinformatics/btl158 (2006).
Kazama, T., Nakamura, T., Watanabe, M., Sugita, M. & Toriyama, K. Suppression mechanism of mitochondrial ORF79 accumulation by Rf1 protein in BT-type cytoplasmic male sterile rice. Plant J 55, 619–628, 10.1111/j.1365-313X.2008.03529.x (2008).
Geddy, R. & Brown, G. G. Genes encoding pentatricopeptide repeat (PPR) proteins are not conserved in location in plant genomes and may be subject to diversifying selection. BMC Genomics 8, 130, 10.1186/1471-2164-8-130 (2007).
Mora, J. R. H., Rivals, E., Mireau, H. & Budar, F. Sequence analysis of two alleles reveals that intra-and intergenic recombination played a role in the evolution of the radish fertility restorer (Rfo). BMC Plant Biol 10, 10.1186/1471-2229-10-35 (2010).
Martin, D. P., Murrell, B., Golden, M., Khoosal, A. & Muhire, B. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol 1, vev003, 10.1093/ve/vev003 (2015).
Zhang, R., Murat, F., Pont, C., Langin, T. & Salse, J. Paleo-evolutionary plasticity of plant disease resistance genes. BMC Genomics 15, 187, 10.1186/1471-2164-15-187 (2014).
Howell, M. D. et al. Genome-wide analysis of the RNA-DEPENDENT RNA POLYMERASE6/DICER-LIKE4 pathway in Arabidopsis reveals dependency on miRNA- and tasiRNA-directed targeting. Plant Cell 19, 926–942, 10.1105/tpc.107.050062 (2007).
Lu, C. et al. Genome-wide analysis for discovery of rice microRNAs reveals natural antisense microRNAs (nat-miRNAs). Proc Natl Acad Sci USA 105, 4951–4956, 10.1073/pnas.0708743105 (2008).
Fahlgren, N. et al. High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS One 2, e219, 10.1371/journal.pone.0000219 (2007).
Li, F. et al. MicroRNA regulation of plant innate immune receptors. Proc Natl Acad Sci USA 109, 1790–1795, 10.1073/pnas.1118282109 (2012).
Shivaprasad, P. V. et al. A microRNA superfamily regulates nucleotide binding site-leucine-rich repeats and other mRNAs. Plant Cell 24, 859–874, 10.1105/tpc.111.095380 (2012).
Zhai, J. X. et al. MicroRNAs as master regulators of the plant NB-LRR defense gene family via the production of phased, trans-acting siRNAs. Genes Dev 25, 2540–2553, 10.1101/gad.177527.111 (2011).
Huang, J. Z., E, Z. G., Zhang, H. L. & Shu, Q. Y. Workable male sterility systems for hybrid rice: Genetics, biochemistry, molecular biology, and utilization. Rice (N Y) 7, 13, 10.1186/s12284-014-0013-6 (2014).
Chen, J. F. et al. Whole-genome sequencing of Oryza brachyantha reveals mechanisms underlying Oryza genome evolution. Nat Commun 4, 10.1038/ncomms2596 (2013).
Zou, X. H. et al. Analysis of 142 genes resolves the rapid diversification of the rice genus. Genome Biol 9, 10.1186/gb-2008-9-3-r49 (2008).
Wambugu, P. W., Brozynska, M., Furtado, A., Waters, D. L. & Henry, R. J. Relationships of wild and domesticated rices (Oryza AA genome species) based upon whole chloroplast genome sequences. Sci Rep 5, 10.1038/srep13957 (2015).
Huang, F. et al. Genetically characterizing a new indica cytoplasmic male sterility with Oryza glaberrima cytoplasm for its potential use in hybrid rice production. Crop Sci 53, 132–140, 10.2135/cropsci2012.07.0444 (2013).
Wei, H. & Wang, Z. Engineering RNA-binding proteins with diverse activities. Wiley Interdiscip Rev RNA 6, 597–613, 10.1002/wrna.1296 (2015).
Kersey, P. J. et al. Ensembl Genomes 2013: scaling up access to genome-wide data. Nucleic Acids Res 42, D546–D552, 10.1093/nar/gkt979 (2014).
Sakai, H. et al. Rice Annotation Project Database (RAP-DB): an integrative and interactive database for rice genomics. Plant Cell Physiol 54, e6, 10.1093/pcp/pcs183 (2013).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet 16, 276–277, 10.1016/S0168-9525(00)02024-2 (2000).
Katoh, K. & Toh, H. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 26, 1899–1900, 10.1093/bioinformatics/btq224 (2010).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490, 10.1371/journal.pone.0009490 (2010).
Kato, H. et al. Structural diversity and evolution of the Rf-1 locus in the genus Oryza. Heredity (Edinb) 99, 516–524, 10.1038/sj.hdy.6801026 (2007).
Acknowledgements
I.S. and J.M. benefitted from support by the Australian Research Council Centre of Excellence in Plant Energy Biology (CE140100008) and Groupe Limagrain. The stay of J.D.S. at The University of Western Australia was funded by the European Social Fund and the state budget of the Czech Republic through the Operational Program Education for Competitiveness.
Author information
Authors and Affiliations
Contributions
I.S. and J.M. conceived the study. I.S., J.M. and J.D.S. developed the bioinformatics pipeline used in the study. J.M. and I.S. analysed the data. I.S. and J.M. wrote the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
This work has been partially funded by Groupe Limagrain.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Melonek, J., Stone, J. & Small, I. Evolutionary plasticity of restorer-of-fertility-like proteins in rice. Sci Rep 6, 35152 (2016). https://doi.org/10.1038/srep35152
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep35152
This article is cited by
-
The genetic basis of cytoplasmic male sterility and fertility restoration in wheat
Nature Communications (2021)
-
Map and sequence-based chromosome walking towards cloning of the male fertility restoration gene Rf5 linked to R11 in sunflower
Scientific Reports (2021)
-
Inheritance of alloplasmic fertility restoration in eggplant (Solanum melongena L.)
Genetic Resources and Crop Evolution (2021)
-
The molecular basis for allelic differences suggests Restorer-of-fertility 1 is a complex locus in sugar beet (Beta vulgaris L.)
BMC Plant Biology (2020)
-
Genome-wide identification of the restorer-of-fertility-like (RFL) gene family in Brassica napus and expression analysis in Shaan2A cytoplasmic male sterility
BMC Genomics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
