
Abstract
Traumatic brain injury (TBI) is a major cause of mortality and morbidity with limited therapeutic options. Traumatic axonal injury (TAI) is an important component of TBI pathology. It is difficult to reproduce TAI in animal models of closed head injury, but in vitro stretch injury models reproduce clinical TAI pathology. Existing in vitro models employ primary rodent neurons or human cancer cell line cells in low throughput formats. This in vitro neuronal stretch injury model employs human induced pluripotent stem cell-derived neurons (hiPSCNs) in a 96 well format. Silicone membranes were attached to 96 well plate tops to create stretchable, culture substrates. A custom-built device was designed and validated to apply repeatable, biofidelic strains and strain rates to these plates. A high content approach was used to measure injury in a hypothesis-free manner. These measurements are shown to provide a sensitive, dose-dependent, multi-modal description of the response to mechanical insult. hiPSCNs transition from healthy to injured phenotype at approximately 35% Lagrangian strain. Continued development of this model may create novel opportunities for drug discovery and exploration of the role of human genotype in TAI pathology.
Similar content being viewed by others


Introduction
Traumatic brain injury (TBI) remains a major public health challenge, causing 52,000 deaths and 275,000 hospitalizations annually in the United States1. In addition, more than 1 million patients are diagnosed in the U.S. annually with concussion, which promotes neurodegeneration in some individuals2. TBI consists of several distinct neurological disorders, including edema, traumatic axonal injury (TAI), contusion and multiple types of hematoma. The study of these diverse disorders as a single disease entity is increasingly seen as a barrier to innovation since each has different pathology3. Therefore, this study focuses specifically on TAI. The human brain is soft and heavy. It stretches and shears under its own weight when the head is violently accelerated. This motion hyperextends and injures neurites, causing TAI4. TAI is difficult to produce in common lab animals because their brains are lighter than the human brain. More than 30 clinical trials have been conducted in TBI, and all have failed5. This may be because pre-clinical tools considered essential in other neurological conditions are unavailable in TBI. Primate modeling in the field stopped decades ago in response to animal welfare controversies and there is no widely accepted, biofidelic, high throughput model. This report presents an in vitro model of TAI employing human induced pluripotent stem cell-derived neurons (hiPSCNs) in a 96 well format with the goal of improving the predictive power and efficiency of pre-clinical drug discovery efforts.
hiPSCNs offer unique insight into the pathology of human disease by enabling human in vitro models with cells such as cardiomyocytes and neurons that are difficult to ethically obtain by biopsy from human subjects. Batch-to-batch variability in cells produced in academic labs has historically presented significant challenges, but the recent availability of hiPSCNs from commercial vendors with little batch-to-batch variability6 has broadened access to this technology and improved reproducibility across labs. iCell neurons from Cellular Dynamics are a mixed population of GABAergic and glutamatergic hiPSCNs expressing multiple ligand gated and voltage gated ion channels7. They exhibit spontaneous electrical activity after 7 days in culture8,9,10,11 that is comparable to the activity observed with primary rat neuron cultures12 or rat organotypic slice cultures10 Due to the long life cycle of the human, full functional maturation as evidenced by synchronous, bursting activity requires up to 90 days in vitro13,14. iCell neurons resemble primary rat neurons both in their susceptibility to α-synuclein stress and response to therapy15. They also reproduce important human pathologies that cannot be reproduced in rodent cells without genetic modification, such as intercellular tau transmission16. hiPSCNs offer unique insight into the genetics of human disease through the creation of patient-specific cells and isogenic cell lines that isolate the influence of a single genetic change. These opportunities are particularly attractive in the study of TBI, which is influenced by genotype in profound and poorly understood ways17,18,19 (reviewed in ref. 20). hiPSCNs have enabled important contributions to the study of neurological disorders21,22,23, but their application to neurotrauma has yet to be demonstrated.
An in vitro stretch injury model requires a repeatable, controllable, mechanical insult. Previous models have employed either uniaxial24,25,26 or biaxial27,28,29 loading modes. Biaxial loading induces channel-independent calcium influx via mechanoporation of the cell membrane, which is a widely accepted injury mechanism30,31,32. Uniaxial loading isolates calcium influx due to activation of stretch-sensitive ion channels33. Biaxial loading therefore more completely reproduces injury pathology. Also, biaxial stretch loads each cell equally, regardless of orientation, which supports cell-by-cell measures of pathology in high content analysis.
Neurites are highly viscoelastic34 so their response to stretch depends strongly on the rate of stretch28,35. Therefore, realistic strain rates are required to induce realistic phenotypes. Head impact in humans generates a pulse of stress that lasts <20 ms36. Air pressure-driven systems can achieve loading ramps in this domain, but engineering challenges associated with the compressibility of air frustrate efforts to expand these systems beyond a 6 well format37. Electromagnetic voice coils, which have to date been used primarily to load organotypic slice culture preparations38,39,40, achieve appropriate pulse durations without relying on air pressure and were therefore chosen as the driving element in this model. Biofidelic stretch injury reproduces the clinically observed process of secondary axotomy24,41,42. This process begins with a stretch insult that does not transect the neurite immediately but induces swellings that enlarge and eventually pinch off, transecting the neurite hours or days after the initial insult.
In this study, a custom-built device driven by an electromagnetic voice coil was used to apply a stretch insult to hiPSCNs in a 96 well format. Fluorescent images of intact cells in culture were analyzed to determine cell viability along with several measures of altered neurite morphology. A range of strains were applied to determine the response of these indicators to mechanical insult.
Results
In quality control testing of the flatness of 10 plates, 7 had no failed wells, 2 had a single failed well and one had 4 failed wells. These results are similar to those observed with commercial plates. In all cases, the failed wells were in the edge rows/columns which were not used for cell culture in experiments (see Methods).
The injury device generated a rapid, highly repeatable, stage displacement history (see Fig. 1A and Table 1). The post array sits in an aluminum block on the injury device that accommodates set screws for optimal alignment with the stage. When these set screws were omitted, a modest misalignment was introduced that created systematic variation in the membrane strain across the plate, with the highest strains on the right side (see Fig. 1B). This configuration was used for dose response experiments and the associated systematic variation was advantageous because it eliminated gaps in the spectrum of strains that would otherwise emerge between different levels of stage displacement (see Fig. 1C). The strain was negligible for the lowest amplitude of displacement tested but rose in an approximately linear manner at higher levels of displacement (see Fig. 1C). The strains in the x and y direction were indistinguishable, indicating that the strain field was equibiaxial28. Since low variation across the plate is desirable for applications other than dose response studies, membrane strain was also characterized with optimal alignment. This eliminated systematic variation across the plate (see Fig. 1D). The remaining random variation across well locations created a distribution with a mean of 0.45 and a standard deviation of 0.051 (see Fig. 1E). The average standard deviation of strains at a given well location over multiple tests on different plates was 0.065.

(A) Stage displacement histories over 10 pulses at a range of target amplitudes. (B) Average strain in each well in misaligned configuration with displacement of 2.9 mm (n = 5 measurements per well, average standard error per well = 0.056). Note that C4, D4, E4, F4, C9, D9, E9 and F9 are uninjured control wells. (C) The Lagrangian strain in the membrane increased with increasing stage displacement (n = 200–260 wells over 10 plates, bars = standard deviation). (D) Average strain in each well in optimally aligned configuration with displacement of 3.3 mm (n = 5 measurements per well, average standard error per well = 0.029). (E) Distribution of strains in optimally aligned configuration (circle = average strain in a single well location, square = average strain across all well locations, error bars = 1 standard deviation).
There is no evidence of astrocyte contamination in iCell neuron cultures (see Fig. 2). Fixed iCells were immunostained with microtubule-associated protein 2 (MAP2), glial fibrillary acidic protein (GFAP), and the nuclear marker Hoechst 33342. 818 MAP2-positive cells were detected across 6 images but no GFAP-positive cells were detected. Separate cultures of rat astrocytes were stained and imaged simultaneously using an identical protocol to control for errors in GFAP labeling (see Fig. 2B).

(A) Representative image of uninjured iCell neurons stained with MAP2, GFAP, and Hoechst 33342 demonstrating the absence of astrocytes. (B) Rat astrocyte culture stained and imaged with an identical protocol as a positive control.
The injury phenotype increased with increasing strain (see Fig. 3). At the time of injury, the iCell neurons were well-attached and established extensive neurite networks on the silicone-bottomed plates. 4 hours after injury, there was negligible evidence of injury at or below the 17% strain level. A clear injury phenotype emerged at the 38% strain level and rapidly approached saturation at higher strains. The injury phenotype had three components: cell death, shortening of neurites, and changes in neurite shape. Control neurites were thick and uniform (see Fig. 3B) while injured neurites were thin with round beads distributed along their length (see Fig. 3C).

(A) Representative images of neurons stained with calcein AM (green) and Hoechst 333342 (blue) 4 hours after injury at various levels of strain. As strain increases, the neurite network becomes less extensive and the number of calcein-AM negative nuclei increases, indicating cell death. (B) In the control condition, neurites have a large, constant thickness. (C) In injured neurons, neurites are shorter and thinner with beads (see white arrows) distributed along their length.
Neurite length and cell viability declined with increasing strain and are distributed in a sigmoidal fashion with respect to strain (see Fig. 4A,B). In the low strain domain (<0.2), there is no obvious increase in injury with increasing strain. In the intermediate strain domain (0.2 < strain < 0.4), injury increases sharply with increasing strain and in the high strain domain (>0.4), the level of injury saturates and becomes strain insensitive again.

(A) Mean neurite length per cell declines with increasing strain. (B) Cell viability declines with increasing strain. (C) Processes/cell declines with increasing strain. (D) Branches/cell declines with increasing strain. (E) Viable cells/image declines with increasing strain. (F) Dead cells/image increases with increasing strain. Injury metrics are plotting against well-specific strain. For phenotype measurements, n = 160 wells over 5 plates. For strain measurements, n = 800 wells over 50 plates (i.e. each point represents the average of 5 measurements (average standard error = 0.056). In both cases, the fit line represents a generalized logistic regression of the data (see Methods). Estimates for the coefficients of each fit along with confidence intervals and R2 values are presented in Table 2.
Cell viability and several measures of neurite morphology were calculated in each well (see Table 2). A generalized logistic function was fit to these datasets to quantify the variation of phenotype with strain (see Methods). The best fit parameters for this function are presented in Table 2. Et, the transition strain, was relatively consistent across the 9 different injury metrics computed, ranging between 0.305 and 0.361.
iCell Neurons stained positively for synaptophysin. (see Fig. 5). Injury with 2mm stage displacement did not significantly alter the synaptophysin density in cells overall. However, when the somatic and neuritic compartments were considered in isolation, injury was found to significantly increase somatic synaptophysin density. Injury caused a downward trend in neuritic synaptophysin density, but this trend was not statistically significant.Synaptophysin labeling of synapses revealed the conventional punctate organization (see Fig. 5A). However, it was not appropriate to count synaptophysin-positive puncta per unit length of neurite in this system because the injury induced a beaded morphology in neurites that caused uniform stain distribution to appear punctate. Therefore, synapse distribution was quantified by measuring the ratio of synaptophysin positive cell area to total cell area. Stretch injury did not alter this ratio (Fig. 5B). To further investigate this finding, the cell domain was split into somatic and neuritic domains and the ratio of synaptophysin positive area to total area was computed for each domain. There was a downward trend in the synaptophysin positive area of the neurites, although this trend was not statistically significant (Fig. 5C). By contrast, there was a statistically significant increase in the synaptophysin positive area of the soma (Fig. 5D, t-test with significance criterion Bonferroni corrected to p < 0.05/3).

(A) Representative image of uninjured iCell neurons stained with MAP2, Synaptophysin, and Hoechst 33342. Synaptophysin staining was punctate and distributed across soma and along neurites. (B) Injury did not change the ratio of synaptophysin positive cell area to total cell area (n = 12). (C) Injury did not significantly change the ratio of synaptophysin positive neurite area to total neurite area, although there was a modest downward trend (n = 12). (D) Injury significantly increased the ratio of synaptophysin positive soma area to total soma area (n = 12, *=t-test with significance criterion Bonferroni corrected to p < 0.05/3, bars = standard error).
Discussion
This model applied a repeatable, mechanical insult to hiPSCNs cultured in a 96 well format and induced a dose dependent, injury phenotype reproducing important aspects of clinical TAI neuropathology. The injury phenotype in some ways resembled that previously reported for in vitro stretch injury of rodent neurons and slice cultures. For example, multiple injury metrics transitioned from uninjured to severely injured levels at approximately 30–35% strain (see Table 2). These results agree with previous reports of biaxial in vitro models employing rat pup, organotypic, hippocampal, slice cultures, which found that a severe injury phenotype emerged either at 35% strain40 or between 20% and 50%38. 30% biaxial strain permeabilized embryonic rat neurons in vitro29. On the other hand, there are potentially significant differences between the response of hiPSCNs to mechanical insult and that previously reported for rodent neurons. For example, a study of embryonic rat neurons reported elevated calcium signaling at both 30% and 50% strain but no change in cell death at either strain level33. This study found that cell viability fell when strain reached 36% (see Table 2). This distinction may be attributable to differences in study design, but may also indicate human-specific pathology. This model is the first in vitro stretch injury model to employ hiPSCNs. This is significant because it allows patient-specific and isogenic cell lines (tools that have generated unique insights into other neurological disorders)22,23 to be applied to the study of neurotrauma. Also, it is the first 96 well model applying stretch at rates relevant to neuronal stretch injury. Realistic loading rates are essential because neuronal stretch injury is highly sensitive to loading rate34. Finally, it is the first model of stretch injury to quantify neurite morphology as an injury metric (this approach has only previously been used in the study of soluble neurotoxins43).
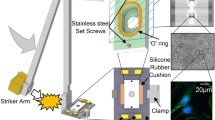
In this study, the silicone cell culture membrane was indented with a rigid post to induce strain. This approach has important advantages over applying air pressure to induce biaxial strain44. Cylindrical indentation of the membrane creates a spatially homogeneous, equibiaxial, strain field. This assertion is supported by theory45,46 and by the fact that the average Lagrangian strain in the X and Y directions were very similar at all levels of stage displacement (see Fig. 1B). This property of the model means that every cell in culture over the head of the post sees the same mechanical insult, regardless of its position or orientation. In biaxial air-driven systems, the circumferential strain peaks at the center and declines to zero at the edge so the strain environment varies continuously across the well bottom47. Also, the conventional geometry of a 96 well plate was retained because inducing deformation without air pressure eliminated the need for gaskets or other special features on the plate bottom (see Fig. 6B). These plates can therefore be manipulated and imaged using existing machinery for high throughput experiments. The relationship between stage displacement and strain in the silicone membrane was linear across all the displacement values tested except for the lowest value, which exhibited almost no strain. This may indicate imperfect zeroing at the start of the displacement pulse or it may indicate a stiction interaction between the silicone and the posts in which deformation begins only after a finite level of tension has been established.

(A) The injury device consists of a stage positioned above an array of Teflon-coated, aluminum posts and driven vertically by an electromagnetic voice coil. (B) The silicone-bottomed plate consists of a commercially-distributed plate top covalently bonded to a sheet of silicone. Since no sandwich construction or air tight gaskets are employed, the standard geometry of the 96 well plate is preserved. (C) Schematic depiction of the injury process. The plate and post array are shown in cross section. Initially, the plate is positioned so that the silicone membrane touches the posts. To induce injury, the plate is lowered, stretching the membrane over the rims of the posts. Posts can be omitted from the post array to create unstretched, control wells.
Morphological biomarkers of injury were employed instead of conventional molecular biomarkers such as amyloid precursor protein. This approach has previously been used to study neurotoxicity48,49 but this is its first application in a neuronal stretch injury model. This high content approach renders the experiment independent of any particular hypothesis about the molecular mechanism of injury and is therefore preferred in drug discovery studies50. Morphological metrics integrate the effects of multiple biochemical processes into a single outcome, which is particularly useful in a multi-modal disorder such as TAI. Degeneration of neurites in vivo disrupts neural networks and is therefore directly relevant to neurological deficits. The R2 value for the fit of neurite length/cell against strain was higher than that for cell viability, suggesting that the former is the more sensitive injury metric (see Table 2).
Mechanical injury modestly perturbed synapse distribution. The reason for an increase in somatic synaptic density with injury is unclear but it is worth noting that injury may exert a selective effect i.e. cells with higher somatic synaptic density may be more likely to survive mechanical insult.
This study is subject to several important limitations. As an in vitro model, it omits key features of the in situ condition, including the presence of other cell types and the three dimensional environment. Therefore, this model functions as a first step towards increasingly biofidelic human in vitro models incorporating these and other features of the in situ condition. Brain organoid technology is advancing rapidly51, and in vitro mechanical injury of neurons in three dimensional culture has been demonstrated52 (although extension of quantitative, automated, high content imaging to three dimensions is non-trivial). The model does not currently include any of the numerous other stressors that accompany mechanical stress in clinical TBI, such as oxidative stress, inflammation, and excitotoxicity. However, this can also be viewed as an advantage since it allows the experimenter to control these processes directly and reconstruct them using soluble agents53. Also, hiPSCNs are immature at the short culture time points employed in this study, with a gene expression profile typical of a neonate7. However, functional maturation can be achieved by prolonged (~90 days) culture13 and co-culture with astrocytes14. Prior neuronal stretch injury models have employed embryonic rodent neurons25,29,33, murine cell lines26,27 or human cancer cell lines24,54, so the feasibility of in vitro stretch injury to adult neurons from any source has yet to be demonstrated. Also, it is worth noting that approximately 1,850 children <1 year of age are admitted to U.S. hospitals with abusive head trauma every year so neonatal neurotrauma is an urgent and understudied clinical challenge55. While the 96 well format makes efficient use of expensive hiPSCNs, true high throughput drug screening will require greater consistency and this is the focus of ongoing development.
In summary, this paper presents a novel, human, in vitro model of TBI in a 96 well format. The model applies a realistic strain and strain rate to human cells to maximize biofidelity. The biomechanical insult is a spatially homogeneous, equibiaxial strain, and the outcome measures are derived from high content analysis of cell viability and morphology. The model induced a dose response relationship between mechanical insult and injury across several different injury metrics that is both internally consistent and consistent with previous findings in other in vitro models.
Methods
Device Description
The plate stretching device consists of a stage mounted on linear bearings above an array of Teflon-coated aluminum posts, each of which can be removed to create uninjured wells as controls (see Fig. 6). The stage is driven vertically by a LA43-67-000A voice coil actuator (BEI Kimco) with a 15 mm stroke and a peak theoretical acceleration of 2758 ms−2. Vertical displacement of the stage is monitored by a T1031-30A optical encoder with 0.1 μm resolution (Renishaw). The system is controlled and monitored using a rack-mounted computer. This computer hosts a cRIO 9024 Real Time PowerPC Controller that sits in a cRIO 9113 chassis and runs a real-time operating system (National Instruments). The cRIO 9024 runs the proportional - integral - derivative control loop that receives displacement information from the optical encoder and regulates the current supplied to the voice coil by a Xenus XTL servo drive (Copley Controls). The force generated by the voice coil is proportional to the current, closing the feedback loop that controls stage displacement. The control loop was tuned using automated routines supplied by the respective manufacturers. A dedicated LabVIEW code specifies the duration and magnitude of the displacement pulse. A separate code is used to record the displacement history at 5000 Hz via an NI 9411 digital input module mounted in the NI 9113 chassis (National Instruments).
Biomechanical Characterization
The relationship between the stage displacement and the strain in the well bottom was characterized using high speed video imaging of cell-free plates undergoing stretch. 60 wells, from position B2 to position G11, were characterized in this way (note that rows A and H and columns 1 and 12 were not used in experiments to eliminate possible confounds due to edge effects on cellular responses). A 1.5 mm diameter dot was stamped in permanent marker ink in the center of each well using a custom-fabricated, 3D printed stamp. Plates were positioned in contact with the post array as for cell injury experiments and stretched with 0.98, 1.94, 2.87, 3.82 and 4.67 mm amplitude stage displacements. High speed video of the well bottom was recorded at 1000 frames per second and 1280 × 1024 pixel resolution from 30 wells (6 rows and 5 columns) during each stretch experiment using a Fastcam Mini UX50 high-speed camera (Photron) and a flat dome light (CCS Inc). 10 plates were stretched at each peak stage displacement level and half of the plate was imaged each time so that 5 independent measurements could be made in each well at each level of peak stage displacement. Images of the well bottoms before onset of stretch and at peak stretch were selected for analysis. The height and width of the stamped spot in each well was measured manually using a Matlab script custom written for the purpose to quantify the deformation of the well bottom. Shear deformation was neglected in our analysis (see below) because it has repeatedly been shown to be negligible in previous investigations of this type of membrane strain45,46,56. Also, direct measurements of axial strain in perpendicular directions were indistinguishable (see Fig. 4), supporting the assumption of negligible shear strain.
The Lagrangian strains in the x and y directions, Exx and Eyy, in the plane of the well bottom were defined using equation (1)57.


where u is the change in length in the subscripted direction.
Shear deformation was neglected in this analysis, leading to the following simplified expressions:


Since neurons are randomly oriented in culture in this model, Exx and Eyy are equally important so the Lagrangian strain, E, for a given well was defined as the average of the two.
Plate Fabrication
Silicone membranes (Specialty Manufacturing Inc.) were covalently bonded to 96 well plate tops (see Fig. 6B) using a protocol adapted from Sukara et al.58. Briefly, 96 well plate tops were activated for 60 seconds using a plasma cleaner then submerged in 1.5% (3-Aminopropyl) triethoxysilane for 20 minutes. Plates were rinsed twice in deionized water and dried with pressurized air. Silicone membranes were cut into 7.5 × 11 cm rectangles and activated for 60 seconds in plasma cleaner. Plate bottoms and silicone membranes were aligned and clamped together for one hour using custom-built clamps. They were cured for a further 24 hours before use. Complete plates were sterilized in 70% ethanol for 15 minutes then rinsed in sterile water for 5 minutes before cell culture.
Cell Culture
Neurons derived from human iPSCs were supplied by Cellular Dynamics International under the brand name iCell neurons. Neurons were seeded at 33,750 cells/cm2 on to silicone-bottomed plates and maintained in proprietary cell culture media supplied by the vendor. Plates were treated with 0.01% poly-L-ornithine prior to seeding. Neurons were seeded in growth media containing 3.3 μg/ml laminin. For immunofluorescence, neuron density was increased to 67,500 cells/cm2 and laminin concentration was increased to 50 μg/ml to minimize losses during the repeated washing required by the immunofluorescence protocol. Plates were incubated at room temperature for 20 minutes after seeding to promote attachment and then maintained at 37 °C, 5% CO2.
Injury
Neurons were injured 48 hours after seeding. The zero position at which the plate touches the post array was established at the start of the experiment using a cell-free plate and a camera positioned above the post array to directly observe contact between the posts and the silicone membrane. Posts were lubricated with corn oil prior to each injury. To create stretch injury, a plate was clamped into the stage and lowered to the zero position. The plate was rapidly displaced down on to the post array and back to its initial position to stretch the membrane. After injury, plates were returned to the incubator. Posts were omitted at positions C4, D4, E4, F4, C9, D9, E9 and F9 to create uninjured control wells.
Live Cell Image Acquisition
Neurons were incubated for 5 minutes with 1 μg/ml Hoechst 33342 (Sigma, St. Louis, MO) and 2 μg/ml Calcein AM at room temperature 4 hours after injury and imaged immediately. Imaging was performed with a 10x magnification, 0.3 NA Plan Fluor lens on an Eclipse TE2000-U microscope (Nikon). The microscope was maintained at 37 °C during imaging using a Plexiglas enclosure fed by a thermally regulated air supply. All images were acquired near the center of the well in the domain above the rigid cylinder at the time of injury.
Immunofluorescent Imaging
Cells were fixed for 15 minutes in 4% formaldehyde in D-PBS at room temperature. Cells were washed with D-PBS and received 0.3% Triton-X (Sigma) in D-PBS for 5 minutes at room temperature. Cells were washed with D-PBS and incubated for 1 hour with 5% donkey serum in D-PBS at room temperature. Cells were labeled with 10 μg/ml MAP2 (abcam11267) and 1/1000 diluted GFAP (abcam7260) or 1/1000 Anti-Synaptophysin 1 (Synaptic Systems) primary antibodies in D-PBS with 5% donkey serum overnight at 4 °C. Cells were washed with D-PBS and received 10 μg/ml Alexa Flour 594 (Thermo Fisher) and 10 μg/ml Alexa Flour 488 (Thermo Fisher) secondary antibodies in D-PBS with 5% donkey serum for 1 hour at room temperature. Cells were washed with D-PBS and stained with 3 ng/ml Hoechst 33342 (Sigma) in D-PBS with 5% donkey serum for 10 minutes at room temperature and imaged immediately.
All immunofluorescent imaging employed a 20x, 0.75 NA Plan Apo lens on a Nikon Eclipse Ti microscope. Hoechst MAP2 GFAP labeled images were illuminated with a widefield epifluorescent X-Cite 120 LED lamp and captured using an Andor Zyla scMOS camera to create a 2048 × 2048 pixel image with a 0.33 μm pixel size. Hoechst was excited via a 361–389 nm bandpass filter and emission was collected through a 435–485 nm bandpass filter. Alexa 488 was excited via a 485–505 nm bandpass filter and emission was collected through a 525–555 nm bandpass filter. Alexa 594 was excited via a 540–580 nm bandpass filter and emission was collected through a 605–665 nm bandpass filter. Hoechst MAP2 synaptophysin images were captured using a Nikon C2 confocal microscope to maximize resolution of sub-cellular structure. Hoechst was excited with a 405 nm laser and emission was collected via a 428–463 nm bandpass filter. Alexa 488 was excited with a 488 nm laser and emission was collected via a 500–550 nm bandpass filter. Alexa 594 was excited with a 640 nm laser and emission was collected via a 575–625 nm long pass filter (these unconventional settings were used because they yielded better signal-to-background ratio in practice than conventional red channel settings). Confocal images were 2048 × 2048 pixels with 0.2 μm Nyquist sampled pixel size.
Image Analysis
Live cell images were preprocessed with CellProfiler software59. A 3 × 3 median filter was applied to the raw images and uneven, background illumination was corrected. MetaXpress software (Molecular Devices) was used to segment individual cells and compute outcome measures including viable cells/image, dead cells/image, neurite length, process number, and branch number (see Fig. 7).

(A) Neurons injured with 38% strain and imaged 4 hours post injury with calcein AM (green) and Hoechst 33342 staining (blue) (B) Automated segmentation of the image into cell bodies and neurites. Note that beads on neurites are rejected as cell bodies based on their size and the absence of Hoechst-positive nuclei. Extracellular nuclei are rejected as cell bodies based on the absence of calcein AM staining.
Immunofluorescent images of cells labeled with synaptophysin, MAP2, and Hoechst were analyzed in Cell Profiler. A 2 × 2 median filter was applied to all channels and the MAP2 channel was thresholded using the Background method59 to create a binary image of the area occupied by cells. A morphological erosion operation was applied to eliminate neurites from this image and create a binary image of the soma. The soma image was dilated by 3 pixels and subtracted from the cell image to create a binary image of the neurites. Each of these three domains: cell, soma, and neurites, was applied separately as a mask to the synaptophysin channel. The pixels within the mask were automatically thresholded using the maximum correlation thresholding method with 1 pixel Gaussian smoothing60 to identify the synaptophysin positive pixels within each domain. The ratio of synaptophysin positive area to total area in each domain was used to quantify the density of synaptophysin.
Statistical Methods
A generalized logistic function of the form

was fit to the injury metric data. In equation (5), y is the injury metric, E is the average of Exx and Eyy as defined in the Methods, y0 is the zero strain value of the metric, yf is the minimum value which the metric asymptotically approaches at high strain, k is a rate constant governing the transition between the initial and minimum asymptotes, and Et is the transition strain (i.e. the strain value at which the metric is half way between its initial and minimal values). Equation (5) was fit iteratively to the experimental data with the Levenberg – Marquardt algorithm using SPSS v.22 statistics software (IBM) to determine parameter estimates, confidence intervals, and R2 values. Initial values of parameters were estimated by visual inspection of scatter plots.
For comparison of synaptophysin density measures, a t-test was used. Since three comparisons were made, the conventional p < 0.05 threshold for significance was Bonferroni corrected to p < 0.05/3.
Additional Information
How to cite this article: Sherman, S. et al. Stretch Injury of Human Induced Pluripotent Stem Cell Derived Neurons in a 96 Well Format. Sci. Rep. 6, 34097; doi: 10.1038/srep34097 (2016).
References
Faul, M., Xu, L., Wald, M. M. & Coronado, V. ‘Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006’. Date of access: 01/09/2016 (2010).
Stern, R. A. et al. Long-term consequences of repetitive brain trauma: chronic traumatic encephalopathy. PM & R: the journal of injury, function, and rehabilitation 3, S460–S467, 10.1016/j.pmrj.2011.08.008 (2011).
Saatman, K. E. et al. Classification of traumatic brain injury for targeted therapies. Journal of neurotrauma 25, 719–738, 10.1089/neu.2008.0586 (2008).
Holbourn, A. H. S. Mechanics of Head Injuries. The Lancet 243, 483 (1943).
Kabadi, S. V. & Faden, A. I. Neuroprotective strategies for traumatic brain injury: improving clinical translation. International journal of molecular sciences 15, 1216–1236, 10.3390/ijms15011216 (2014).
Morrison, G. et al. Evaluation of inter-batch differences in stem-cell derived neurons. Stem cell research 16, 140–148, 10.1016/j.scr.2015.12.025 (2016).
Dage, J. L. et al. Pharmacological characterisation of ligand- and voltage-gated ion channels expressed in human iPSC-derived forebrain neurons. Psychopharmacology (Berl) 231, 1105–1124, 10.1007/s00213-013-3384-2 (2014).
Haythornthwaite, A. et al. Characterizing human ion channels in induced pluripotent stem cell-derived neurons. Journal of biomolecular screening 17, 1264–1272, 10.1177/1087057112457821 (2012).
Meneghello, G. et al. Evaluation of established human iPSC-derived neurons to model neurodegenerative diseases. Neuroscience 301, 204–212, 10.1016/j.neuroscience.2015.05.071 (2015).
Berry, B. J. et al. Morphological and functional characterization of human induced pluripotent stem cell-derived neurons (iCell Neurons) in defined culture systems. Biotechnol Prog 31, 1613–1622, 10.1002/btpr.2160 (2015).
Becker, N. et al. Minimized cell usage for stem cell-derived and primary cells on an automated patch clamp system. J Pharmacol Toxicol Methods 68, 82–87, 10.1016/j.vascn.2013.03.009 (2013).
Tukker, A. M. et al. Is the time right for in vitro neurotoxicity testing using human iPSC-derived neurons? ALTEX, 10.14573/altex.1510091 (2016).
Amin, H. et al. Electrical Responses and Spontaneous Activity of Human iPS-Derived Neuronal Networks Characterized for 3-month Culture with 4096-Electrode Arrays. Front Neurosci 10, 121, 10.3389/fnins.2016.00121 (2016).
Odawara, A., Saitoh, Y., Alhebshi, A. H., Gotoh, M. & Suzuki, I. Long-term electrophysiological activity and pharmacological response of a human induced pluripotent stem cell-derived neuron and astrocyte co-culture. Biochemical and biophysical research communications 443, 1176–1181, 10.1016/j.bbrc.2013.12.142 (2014).
Alhebshi, A. H., Odawara, A., Gotoh, M. & Suzuki, I. Thymoquinone protects cultured hippocampal and human induced pluripotent stem cells-derived neurons against alpha-synuclein-induced synapse damage. Neuroscience letters 570, 126–131, 10.1016/j.neulet.2013.09.049 (2014).
Usenovic, M. et al. Internalized Tau Oligomers Cause Neurodegeneration by Inducing Accumulation of Pathogenic Tau in Human Neurons Derived from Induced Pluripotent Stem Cells. The Journal of neuroscience: the official journal of the Society for Neuroscience 35, 14234–14250, 10.1523/JNEUROSCI.1523-15.2015 (2015).
Zhou, W. et al. Meta-analysis of APOE4 allele and outcome after traumatic brain injury. Journal of neurotrauma 25, 279–290, 10.1089/neu.2007.0489 (2008).
Failla, M. D., Conley, Y. P. & Wagner, A. K. Brain-Derived Neurotrophic Factor (BDNF) in Traumatic Brain Injury-Related Mortality: Interrelationships Between Genetics and Acute Systemic and Central Nervous System BDNF Profiles. Neurorehabilitation and neural repair 30, 83–93, 10.1177/1545968315586465 (2016).
Markos, S. M. et al. Genetic Variation in the Vesicular Monoamine Transporter: Preliminary Associations With Cognitive Outcomes After Severe Traumatic Brain Injury. The Journal of head trauma rehabilitation, 10.1097/HTR.0000000000000224 (2016).
McAllister, T. W. Genetic factors in traumatic brain injury. Handbook of clinical neurology 128, 723–739, 10.1016/B978-0-444-63521-1.00045-5 (2015).
Israel, M. A. et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482, 216–220, 10.1038/nature10821 (2012).
Marchetto, M. C. et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 143, 527–539, 10.1016/j.cell.2010.10.016 (2010).
Brennand, K. J. et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 473, 221–225, 10.1038/nature09915 (2011).
Smith, D. H., Wolf, J. A., Lusardi, T. A., Lee, V. M. & Meaney, D. F. High tolerance and delayed elastic response of cultured axons to dynamic stretch injury. The Journal of neuroscience: the official journal of the Society for Neuroscience 19, 4263–4269 (1999).
Lusardi, T. A., Wolf, J. A., Putt, M. E., Smith, D. H. & Meaney, D. F. Effect of acute calcium influx after mechanical stretch injury in vitro on the viability of hippocampal neurons. Journal of neurotrauma 21, 61–72, 10.1089/089771504772695959 (2004).
von Reyn, C. R., Mott, R. E., Siman, R., Smith, D. H. & Meaney, D. F. Mechanisms of calpain mediated proteolysis of voltage gated sodium channel alpha-subunits following in vitro dynamic stretch injury. Journal of neurochemistry 121, 793–805, 10.1111/j.1471-4159.2012.07735.x (2012).
Cargill, R. S. 2nd & Thibault, L. E. Acute alterations in [Ca2+]i in NG108-15 cells subjected to high strain rate deformation and chemical hypoxia: an in vitro model for neural trauma. Journal of neurotrauma 13, 395–407, 10.1089/neu.1996.13.395 (1996).
Geddes, D. M. & Cargill, R. S. An in Vitro Model of Neural Trauma: Device Characterization and Calcium Response to Mechanical Stretch. Journal of biomechanical engineering 123, 247, 10.1115/1.1374201 (2001).
Geddes, D. M., Cargill, R. S. 2nd & LaPlaca, M. C. Mechanical stretch to neurons results in a strain rate and magnitude-dependent increase in plasma membrane permeability. Journal of neurotrauma 20, 1039–1049, 10.1089/089771503770195885 (2003).
Singleton, R. H. & Povlishock, J. T. Identification and characterization of heterogeneous neuronal injury and death in regions of diffuse brain injury: evidence for multiple independent injury phenotypes. The Journal of neuroscience: the official journal of the Society for Neuroscience 24, 3543–3553, 10.1523/JNEUROSCI.5048-03.2004 (2004).
Pettus, E. H., Christman, C. W., Giebel, M. L. & Povlishock, J. T. Traumatically induced altered membrane permeability: its relationship to traumatically induced reactive axonal change. Journal of neurotrauma 11, 507–522, 10.1089/neu.1994.11.507 (1994).
Pettus, E. H. & Povlishock, J. T. Characterization of a distinct set of intra-axonal ultrastructural changes associated with traumatically induced alteration in axolemmal permeability. Brain research 722, 1–11 (1996).
Geddes-Klein, D. M., Schiffman, K. B. & Meaney, D. F. Mechanisms and consequences of neuronal stretch injury in vitro differ with the model of trauma. Journal of neurotrauma 23, 193–204, 10.1089/neu.2006.23.193 (2006).
Ahmadzadeh, H., Smith, D. H. & Shenoy, V. B. Viscoelasticity of tau proteins leads to strain rate-dependent breaking of microtubules during axonal stretch injury: predictions from a mathematical model. Biophysical journal 106, 1123–1133, 10.1016/j.bpj.2014.01.024 (2014).
Morrison, B. 3rd et al. A tissue level tolerance criterion for living brain developed with an in vitro model of traumatic mechanical loading. Stapp Car Crash J 47, 93–105 (2003).
Mao, H. et al. Development of a finite element human head model partially validated with thirty five experimental cases. Journal of biomechanical engineering 135, 111002, 10.1115/1.4025101 (2013).
Magou, G. C. et al. Engineering a high throughput axon injury system. Journal of neurotrauma 28, 2203–2218, 10.1089/neu.2010.1596 (2011).
Di Pietro, V. et al. S100B and Glial Fibrillary Acidic Protein as Indexes to Monitor Damage Severity in an In Vitro Model of Traumatic Brain Injury. Neurochemical research 40, 991–999, 10.1007/s11064-015-1554-9 (2015).
Cater, H. L. et al. Stretch-induced injury in organotypic hippocampal slice cultures reproduces in vivo post-traumatic neurodegeneration: role of glutamate receptors and voltage-dependent calcium channels. Journal of neurochemistry 101, 434–447, 10.1111/j.1471-4159.2006.04379.x (2007).
Morrison, B., Cater, H. L., Benham, C. D. & Sundstrom, L. E. An in vitro model of traumatic brain injury utilising two-dimensional stretch of organotypic hippocampal slice cultures. Journal of neuroscience methods 150, 192–201, 10.1016/j.jneumeth.2005.06.014 (2006).
Buki, A. & Povlishock, J. T. All roads lead to disconnection? –Traumatic axonal injury revisited. Acta neurochirurgica 148, 181–193, discussion 193–184, 10.1007/s00701-005-0674-4 (2006).
Maxwell, W. L., Povlishock, J. T. & Graham, D. L. A mechanistic analysis of nondisruptive axonal injury: a review. Journal of neurotrauma 14, 419–440 (1997).
Ryan, K. R. et al. Neurite outgrowth in human induced pluripotent stem cell-derived neurons as a high-throughput screen for developmental neurotoxicity or neurotoxicity. Neurotoxicology 53, 271–281, 10.1016/j.neuro.2016.02.003 (2016).
Ursekar, C. P. et al. Design and construction of an equibiaxial cell stretching system that is improved for biochemical analysis. PloS one 9, e90665, 10.1371/journal.pone.0090665 (2014).
Hung, C. T. & Williams, J. L. A method for inducing equi-biaxial and uniform strains in elastomeric membranes used as cell substrates. Journal of biomechanics 27, 227–232 (1994).
Schaffer, J. L. et al. Device for the application of a dynamic biaxially uniform and isotropic strain to a flexible cell culture membrane. Journal of orthopaedic research: official publication of the Orthopaedic Research Society 12, 709–719, 10.1002/jor.1100120514 (1994).
Williams, J. L., Chen, J. H. & Belloli, D. M. Strain fields on cell stressing devices employing clamped circular elastic diaphragms as substrates. Journal of biomechanical engineering 114, 377–384 (1992).
Sirenko, O., Hesley, J., Rusyn, I. & Cromwell, E. F. High-content high-throughput assays for characterizing the viability and morphology of human iPSC-derived neuronal cultures. Assay and drug development technologies 12, 536–547, 10.1089/adt.2014.592 (2014).
Wheeler, H. E., Wing, C., Delaney, S. M., Komatsu, M. & Dolan, M. E. Modeling chemotherapeutic neurotoxicity with human induced pluripotent stem cell-derived neuronal cells. PloS one 10, e0118020, 10.1371/journal.pone.0118020 (2015).
Lang, P., Yeow, K., Nichols, A. & Scheer, A. Cellular imaging in drug discovery. Nature reviews. Drug discovery 5, 343–356, 10.1038/nrd2008 (2006).
Lancaster, M. A. & Knoblich, J. A. Organogenesis in a dish: modeling development and disease using organoid technologies. Science 345, 1247125, 10.1126/science.1247125 (2014).
LaPlaca, M. C., Cullen, D. K., McLoughlin, J. J. & Cargill, R. S. High rate shear strain of three-dimensional neural cell cultures: a new in vitro traumatic brain injury model. Journal of biomechanics 38, 1093–1105, 10.1016/j.jbiomech.2004.05.032 (2005).
Morrison, B., Elkin, B. S., Dolle, J. P. & Yarmush, M. L. In vitro models of traumatic brain injury. Annual review of biomedical engineering 13, 91–126, 10.1146/annurev-bioeng-071910-124706 (2011).
Wolf, J. A., Stys, P. K., Lusardi, T., Meaney, D. & Smith, D. H. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. The Journal of neuroscience: the official journal of the Society for Neuroscience 21, 1923–1930 (2001).
Peterson, C., Xu, L., Florence, C. & Parks, S. E. Annual Cost of U.S. Hospital Visits for Pediatric Abusive Head Trauma. Child Maltreat 20, 162–169, 10.1177/1077559515583549 (2015).
Lee, A. A. et al. An equibiaxial strain system for cultured cells. The American journal of physiology 271, C1400–C1408 (1996).
Lai, W. M., R. D. & Krempl, E. Introduction to Continuum Mechanics. 134 (Butterworth-Heinemann, 1993).
Sunkara, V. et al. Simple room temperature bonding of thermoplastics and poly(dimethylsiloxane). Lab on a chip 11, 962–965, 10.1039/c0lc00272k (2011).
Carpenter, A. E. et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome biology 7, R100, 10.1186/gb-2006-7-10-r100 (2006).
Padmanabhan, K., Eddy, W. F. & Crowley, J. C. A novel algorithm for optimal image thresholding of biological data. Journal of neuroscience methods 193, 380–384, 10.1016/j.jneumeth.2010.08.031 (2010).
Acknowledgements
The authors would like to thank William Cohn and Richard De Maria for excellent technical assistance. They would also like to thank Barclay Morrison III, Eileen Dolan and Claudia Wing for helpful conversations, Vimal Patel, PhD, Medical and Scientific writer at NorthShore Neurological Institute, for copy editing this manuscript, and Sam Bettis and Sarah Fazal of the Cellular Screening Center at the University of Chicago for helpful conversations about data analysis.
Author information
Authors and Affiliations
Contributions
S.A.S. performed experiments and assisted with experimental design, figure preparation and manuscript preparation, J.K.P. performed experiments, analyzed data and assisted with experimental design, J.T.C. assisted with design and fabrication of the custom-built injury device, F.S.C. performed experiments and assisted with experimental design, S.R.O. assisted with design and fabrication of the custom-built injury device, J.D.F. designed the experiments, analyzed data and prepared the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Sherman, S., Phillips, J., Costa, J. et al. Stretch Injury of Human Induced Pluripotent Stem Cell Derived Neurons in a 96 Well Format. Sci Rep 6, 34097 (2016). https://doi.org/10.1038/srep34097
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep34097
This article is cited by
-
The interrelationships between neuronal viability, synaptic integrity, microglial responses, and amyloid-beta formation in an in vitro neurotrauma model
Scientific Reports (2022)
-
The neurodynamic treatment induces biological changes in sensory and motor neurons in vitro
Scientific Reports (2021)
-
Stretch-Induced Deformation as a Model to Study Dopaminergic Dysfunction in Traumatic Brain Injury
Neurochemical Research (2019)
-
Characterization of Biaxial Stretch as an In Vitro Model of Traumatic Brain Injury to the Blood-Brain Barrier
Molecular Neurobiology (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
