
Abstract
Triple negative breast cancer (TNBC) is the most aggressive breast cancer subtype with no targeted treatment available. Our previous study identified 38 TNBC-specific genes with altered expression comparing tumour to normal samples. This study aimed to establish whether DNA methylation contributed to these expression changes in the same cohort as well as disease progression from primary breast tumour to lymph node metastasis associated with changes in the epigenome. We obtained DNA from 23 primary TNBC samples, 12 matched lymph node metastases, and 11 matched normal adjacent tissues and assayed for differential methylation profiles using Illumina HumanMethylation450 BeadChips. The results were validated in an independent cohort of 70 primary TNBC samples. The expression of 16/38 TNBC-specific genes was associated with alteration in DNA methylation. Novel methylation changes between primary tumours and lymph node metastases, as well as those associated with survival were identified. Altered methylation of 18 genes associated with lymph node metastasis were identified and validated. This study reveals the important role DNA methylation plays in altered gene expression of TNBC-specific genes and lymph node metastases. The novel insights into progression of TNBC to secondary disease may provide potential prognostic indicators for this hard-to-treat breast cancer subtype.
Similar content being viewed by others

Introduction
Breast cancer is responsible for the highest cancer incidence in women worldwide and its rates are increasing. Out of all breast cancer subtypes, triple negative breast cancer (TNBC) is the most aggressive, accounting for somewhere between 10 and 20% of all diagnosed breast cancers1,2,3. TNBC is recognised for its more frequent and rapid progression to metastasis, high rate of BRCA1 mutations4, chromosome instability5 and is more commonly diagnosed in younger women (pre-menopausal) as well as women of African-American descent6. It lacks the expression of estrogen and progesterone receptors (ER and PR) as well as the human epidermal growth factor receptor 2 (HER2). Treatment which target these receptors are ineffective in TNBC and the aggressive nature of TNBC drives the urgent need for new treatment targets.
The development and progression of cancer is due to the accumulation of multiple genetic and epigenetic change. DNA methylation is the most well-studied epigenetic change, which culminates in altered gene expression7,8. DNA methylation is mediated by DNA-methyltransferase (DNMT), which leads to a conformational change of the nucleosomes, where histones are drawn tighter together thereby excluding transcription factors access such that genes cannot be transcribed and expressed. Generally, there is a global decrease of DNA methylation (hypomethylation) in cancer cells, which leads to increased genomic instability9. Nevertheless, an increase in DNA methylation (hypermethylation) has been identified at tumour suppressor genes in multiple cancers10.
DNA methylation loci are attractive candidates as biomarkers for TNBC as they are more stable than RNA or proteins and are readily detectable in tissue samples and blood11. In TNBC, the methylation pattern of a number of cancer-related genes has been analysed12. Additionally, methylation patterns have been used to differentiate breast cancer subtypes13. Sharma et al. discovered that the methylation of the BRCA1 promoter region is associated with worse overall survival and relapse-free survival in TNBC14. A recent study by Stirzaker et al. used whole genome DNA methylation analysis to identify a signature which divided TNBC into three prognostic subgroups and identified differentially methylated regions (DMRs) associated with overall survival15. However, there have been no studies to date that have addressed genome-wide methylation change during disease progression from the primary tumour to lymph node metastasis in TNBC.
In this study, we aimed to identify whether DNA methylation contributed to the altered expression of 38 genes we identified previously in TNBC16. By performing whole genome methylation analysis of 23 grade 3 primary invasive ductal carcinomas (IDC) and 11 matched normal adjacent tissues (NAT), we determined that 42% of our TNBC specific genes had significantly altered methylation. Furthermore, by comparing IDC to NAT and IDC to 12 matched lymph node metastases (LN), we identified a set of DNA methylation aberrations associated with the progression of TNBC from primary tumour to LN metastases. We validated the methylation changes of 18 genes associated with LN metastasis with a regional DNA methylation analysis in an independent cohort. Additionally, we identified nine methylation probes, that have significantly altered methylation in LN samples to be associated with survival in TNBC. This is the first whole genome methylation study in TNBC including matched IDC, LN, and NAT samples. We were able correlate the findings of this study with the gene expression results of our previous report using the same sample cohort. A number of previously identified genes show differential methylation suggesting the potential functional relevance of these changes.
Results
Methylation profiles are altered in TNBC
We performed 450 K DNA methylation BeadChip array analysis (Illumina) in two independent TNBC cohorts. The study cohort contained 23 TNBC primary IDCs and the validation cohort contained 70 TNBC primary IDCs. All primary tumour samples were compared to three pooled NAT samples and one singular NAT (11 samples in total).
By comparing the methylation of the IDC samples to the NAT samples we identified 44,005 differentially methylated probes in the study cohort (52.2% hypomethylated, 47.8% hypermethylated) and 45,263 probes in the validation cohort (40.2% hypomethylated, 59.8% hypermethylated). We validated 29,612 probes (=67.29%) in the independent validation cohort. Of the 29,612 validated probes, 41.4% were hypomethylated and 58.6% were hypermethylated. Within the validated probes there were 1,849 promoter-associated probes (52.8% hypomethylated, 47.2% hypermethylated) and 9,161 probes within enhancer regions (44.9% hypomethylated, 55.1% hypermethylated). In total, there were over 10,000 probes within enhancer or promoter regions that were significantly methylated. Performing a pathway enrichment analysis of these probes identified many important cancer pathways influenced by the respective genes. The pathways with the highest enrichment scores for hypomethylated genes were: Axon guidance, Rap1 signaling pathway, Platelet activation, Mucin type O-Glycan biosynthesis, and MAPK signaling pathway. The pathways with the highest enrichment scores for hypermethylated genes were: ECM-receptor interaction, Pathways in cancer, PI3K-Akt signaling pathway, focal adhesion, and signaling pathways regulating pluripotency of stem cells (Supplementary Table 1). We next identified differentially methylated regions (DMRs, a minimum of three significant consecutive probes). In the study cohort we identified 2,373 DMRs (10,082 probes) and in the validation cohort we identified 2,932 DMRs (12,938 probes), 72.62% (1,756 DMRs/7,523 probes) were common in both cohorts (Supplementary Table 2). The results of this analysis are shown in Fig. 1.

Summary of the DNA methylation comparing primary tumours (IDC) versus matched normal adjacent tissue (NAT) in the study cohort (blue) and the validation cohort (orange).
The top two Venn diagrams show hyper- and hypomethylation of single loci, and the bottom two Venn diagrams show hyper- and hypomethylation of differentially methylated regions (DMRs). The number of validated methylation probes is shown in the middle of each Venn diagram. Underneath the validated number of probes, the number of these probes that are located within promoter and enhancer regions is shown (top two Venn diagrams).
Genes differentially expressed in TNBC compared to NAT are associated with altered methylation patterns
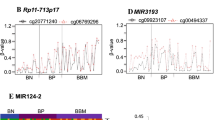
We have previously identified 66 genes to be differentially expressed in primary tumour samples compared to normal adjacent tissue in TNBC16. Therefore, in this study we aimed to identify the contribution of DNA methylation aberration to these gene expression changes. We determined that 26 of the 66 genes had significantly altered methylation of single loci (a total of 63 probes, 47.6% hypomethylated, 52.4% hypermethylated) in both the study and the validation cohorts (Supplementary Table 3). Of the significant probes there were nine within enhancers and four within promoter regions. Additionally, eight of the 66 genes had significantly altered regional methylation (40 probes, 30% hypomethylated, 70% hypermethylated) in both cohorts (Table 1). Of the eight genes with significantly altered regional methylation, one of these (EGR1) was significantly associated with overall survival in one probe (cg07336840). High DNA methylation of this probe was associated with better survival as shown in Fig. 2.

Survival analysis of the EGR1 probe cg07336840.
The y-axis shows the percent of survival of patients within the validation cohort. The x-axis shows the number of months of survival since diagnosis. The blue line represents patients with low DNA methylation of this probe, whereas the red line represents patients with high DNA methylation of this probe.
Relationship of methylation changes to altered expression of TNBC-specific genes
Previously we identified 38 genes that had altered expression in the TNBC subtype but not in other breast cancer subtypes, using two independent cohorts16. In the first cohort 28 TNBC-specific genes and in the second 14 TNBC specific genes were identified. There were four genes common to both cohorts (ANKRD30A, ANP32E, DSC2, and IL6ST). Here, we sought to investigate the DNA methylation changes of these 38 TNBC-specific genes.
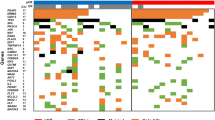
We found that 16 of the 38 TNBC-specific genes were associated with differentially methylated probes in the study and validation cohorts (41 probes) (Table 2 and Fig. 3). A set of five genes (ANKRD30B, COL14A1, IGF1, IL6ST, MEG3) exhibited regional methylation differences (28 probes) in both cohorts. Three of which showed very strong methylation changes in both cohorts (>20% methylation change), these were ANKRD30B (7 hyper-methylated probes), COL14A1 (6 hyper-methylated probes), and MEG3 (8 hyper-methylated probes). Of the four TNBC-specific genes that were common in both analyses in our previous study, there was one (IL6ST) that showed significantly altered methylation in 3 probes in both cohorts (Table 2). The methylation change for IL6ST can be classed as a DMR. However, no probes within these regions were significantly associated with survival (data not shown).

Unsupervised Hierarchical clustering of the DNA methylation of the significant 16/38 TNBC specific genes.
Primary tumour TNBC (IDC) samples are shown in red and matched normal adjacent tissue (NAT) samples are shown in blue in the sample tree on the left (y-axis). Genes are clustered along the x-axis. Hypomethylation is shown in blue, hypermethylation is shown in red and equivocal methylation is shown in grey.
Methylation changes associated with lymph node metastases
In our previous study16 we identified 83 genes that showed altered expression both in primary tumours with LN metastases and in their matched LN metastasis, but were unaltered in lymph node negative tumours. This led to the rationale that the expression of these genes may be affected by DNA methylation and is a contributing factor to tumour progression. To interrogate this, we performed 450 K DNA methylation arrays on 12 LN metastasis samples from the same cohort used in our previous study16 and compared the DNA methylation from these samples to that of the NAT samples. A total of 51,563 probes had significantly altered methylation when comparing LN to NAT samples (46.7% hypermethylated, 53.3% hypomethylated) of which, there were 2,350 significant DMRs (these DMRs contain 11,218 probes). Furthermore, 38 of the 83 LN metastasis-associated genes had significantly altered DNA methylation in 107 probes. Of these, 14 genes were present in DMRs (74 probes). The gene expression and DNA methylation changes of these 14 genes is shown in Supplementary Table 4.
It was not possible to validate the methylation changes in the validation cohort due to the lack of LN samples. However, we hypothesised that due to their altered expression in LN metastases, they would be associated with survival outcome. We performed survival analysis on the 70 tumour samples from our validation cohort using the methylation analysis of the 74 probes that comprised the 14 DMRs. Nine of the 74 probes were significantly associated with survival in the TNBC validation cohort (cg18108818, cg20464151, cg09933058, cg24173596, cg07336840, cg08500417, cg20066782, cg04028606, cg00185066) (Fig. 4). Eight of these probes (cg18108818, cg20464151, cg24173596, cg07336840, cg08500417, cg20066782, cg04028606, cg00185066) were associated with improved survival when they were highly methylated and one probe (cg09933058) was associated with worse survival when it was highly methylated.

Survival analysis of nine probes that show significant methylation changes comparing lymph node metastasis to matched normal adjacent tissue.
The x-axis shows the number of months of survival since diagnosis. The green line represents patients with low DNA methylation of this probe, whereas the red line represents patients with high DNA methylation of this probe.
Due to the lack of LN samples in the validation cohort, Methyl-Binding-Domain-Capture (MBDcap) sequencing in an independent cohort of 7 LN and 4 NAT samples was used to validate the direction of DNA methylation of the 38 genes that showed significantly altered methylation when comparing LN to NAT samples in the study cohort. The MBDcap sequencing data analysed 1 kb regions covering all 38 genes associated with LN metastasis (identified in our previous gene expression analyses) starting 2 kb upstream from the first transcription start site to the end of the gene. Of the 38 genes that showed significant altered methylation in 107 probes, 18 of these had significant regional DNA methylation changes in the same direction (hyper/hypomethylation) using MBDcap sequencing data. The results of this analysis can be seen in Table 3.
Tumour versus lymph node
We have previously reported that miRNA and gene expression patterns are highly similar in LN metastases and the primary tumour within our study cohort16,17. We next compared whether the methylation changes occurring in LN metastases were similar to those occurring in the primary tumour. The two comparisons of IDC vs NAT and LN vs NAT yielded an overlap of 88.75% (=39,057 probes) or 89.93% (2134 DMRs), indicating that IDC and matched LN metastases DNA methylation alterations are highly similar.
We next compared the DNA methylation of all IDC samples with the DNA methylation of all LN samples to determine methylation changes specifically associated with metastases that were not present in the primary tumour. This comparison revealed that 5,221 probes (58.1% hypomethylated, 41.9% hypermethylated) and 104 DMRs (=366 probes) showed significantly altered methylation. Over 2,000 of these significantly associated probes were located within enhancer or promoter regions. Pathway enrichment analysis revealed the following significant pathways for hypomethylated genes: Inflammatory mediator regulation of TRP channels, Fructose and mannose metabolism, Regulation of lipolysis in adipocytes, Estrogen signaling pathway, and Platelet activation. Pathway enrichment analysis revealed the following significant pathways for hypermethylated genes: Glycosaminoglycan biosynthesis - heparan sulfate/heparin, Axon guidance, Gastric acid secretion, GABAergic synapse, and Glycosaminoglycan biosynthesis - chondroitin sulfate/dermatan sulfate (Supplementary Table 5).
Discussion
We have previously identified gene expression changes in TNBC primary tumours compared to matched NAT as well as gene expression changes associated with the progression of TNBC from primary tumour to lymph node metastasis. In this study we performed whole genome DNA profiling to determine the contribution of DNA methylation to these gene expression changes as DNA methylation is known to gene silencing events. Here we used the same sample cohort to identify DNA methylation changes that were associated with the previously observed alterations in gene expression by: (1) comparing tumour and matched normal samples and; (2) that were associated with lymph node metastasis.
First, we aimed to identify a global methylation profile of TNBC primary tumour samples (IDC) compared to matched normal adjacent tissue (NAT) to provide information about tumour specific differences. We identified and validated global hypermethylation and hypomethylation that included single loci as well as differentially methylated regions (DMRs) (a minimum of three significant consecutive probes). Since DNA methylation contributes to gene expression changes18, the differences in the methylation profiles between these two tissue types were expected. There has only been one other whole genome DNA methylation study in TNBC15, which focused on the prognostic value of DNA methylation patterns. The study by Stirzaker et al. identified 308 hypermethylated genes by comparing IDC versus NAT samples using Methylation-Binding-Domain Capture sequencing data15. We identified 227 (73.7%) of these genes in our study cohort to be significantly hypermethylated. The global analysis of DNA methylation revealed a higher number of hypermethylated probes and DMRs (>17,000 probes, >1,300 DMRs) than hypomethylated probes and DMRs (>12,000 probes, 307 DMRs) comparing the two tissue types, which has also been reported by Stirzaker et al.15. Interestingly over 10,000 of these probes are located within enhancer or promoter regions. Activation/inactivation of enhancers can affect the transcription of the host gene so that they can act as alternative promoters19,20,21. Our study identified a number of pathways that were associated with altered methylation within enhancer/promoter regions, including the estrogen signalling pathway. In particular, DNA methylation has been associated with hormone receptor status of breast cancer patients22,23 and our current study suggests that DNA methylation may be involved in the downregulation of the estrogen receptor in TNBC patients, which was also shown in ref. 15.
Next, we focused more specifically on the genes identified in our previous study16. There we identified 66 genes to be differentially expressed in tumour versus normal samples. Our current study revealed that 26 of the 66 genes showed significantly altered DNA methylation, which was verified in two independent cohorts. The majority of these (19 genes = 73%) were negatively correlated, such that where gene expression was upregulated, DNA methylation was decreased and vice versa. However, there were genes whose DNA methylation profile was positively correlated with their gene expression (gene expression was upregulated when DNA methylation was increased, and vice versa). This does not mean that the DNA methylation of these genes does not contribute to their gene expression levels as described recently24. There Wan et al.24 hypothesised two mechanisms of DNA methylation-dependent gene regulation: (1) the dogma of gene repression due to DNA methylation; and (2) gene activation through DNA methylation. They found positively correlated gene-methylation relationships to be in more conserved regions and mainly in promoter regions, which suggests that these positive correlations have a regulatory role and do not just happen by chance. It may also indicate differential promoter usage as seen in ref. 25.
Our previous study found that EGR1 gene expression is downregulated comparing tumour versus normal. The methylation of EGR1 was found to be negatively correlated with its gene expression in our TNBC cohort, where there were five significant CpG methylation probes that were hypermethylated in both cohorts comparing these two tissue types. This gene has also been identified by other studies comparing IDC versus NAT in TNBC26. It is a zinc finger protein that acts as a transcriptional regulator functioning as a tumour suppressor by regulating other tumour suppressors including TGFβ1, PTEN, p53, and fibronectin27. We were able to correlate the DNA methylation of one of the significant probes within EGR1 (cg07336840) with overall survival (p < 0.05). High DNA methylation at this locus is significantly associated with better overall survival. Interestingly, the study by Stirzaker et al. identified a DMR located in the WT-1 gene to be associated with poorer overall survival in TNBC patients15. EGR1 and WT-1 are members of the same family (early growth response – zinc-finger family) but with mostly opposing functions, EGR1 activates the transcription of genes that WT-1 represses28,29.
We previously identified that the gene expression of 38 genes is specific to TNBC16. Here we investigated the DNA methylation levels of these genes to determine the contribution of DNA methylation to gene expression. We showed that half (16/38) of the TNBC specific genes showed significantly altered DNA methylation at 41 probes; and of these there are five genes classed as DMRs (28 probes). These five genes are ANKRD30B, COL14A1, IGF1, IL6ST, and MEG3. ANKRD30B has been shown to be expressed in breast, brain, and testicular tumours30 but it has not been studied in TNBC to date. Stirzaker et al.15 also identified significant hypermethylation of COL14A1 when comparing tumour versus normal tissue15. However, there is a need for functional analysis of COL14A1 in TNBC. IGF1 has been the focus of multiple TNBC studies and is known to regulate cell proliferation and survival, and has been suggested as a potential treatment target for TNBC31,32,33. We have previously identified IL6ST as a TNBC-specific gene and its gene expression to be associated with overall survival (increased gene expression → better survival)16. Our previous study showed MEG3 is associated with lymph node metastasis16. It is a long non-coding RNA that is known to be down-regulated in multiple cancers and to regulate cell proliferation through the p53-tumour suppressor pathway34,35.
Due to its aggressive nature and increased number of metastasis, TNBC patients have much poorer outcomes relative to other subtypes. Therefore, we aimed to identify differences during early cancer progression from the primary tumour site to lymph node metastasis (LN). Eighty three genes were previously shown to be associated with LN metastasis16. Here we revealed that the expression of 38 of these genes may be influenced by methylation changes at single DNA methylation loci. Of these there are 14 genes that were differentially methylated in DMRs (over 74 probes). The survival analysis on our validation cohort using these 74 probes (Fig. 4) showed that nine probes were significantly associated with survival. Seven probes associated with five genes (SPRY2, EGR1, GREB1, ITIH5, LRRC17) were associated with better survival having higher methylation, whereas one probe for AMIGO2 (cg09933058) shows better survival with low methylation. Interestingly, of the significant DMRs comparing IDC versus NAT only one probe showed significant association with survival (EGR1 (cg07336840)) which is also in a DMR comparing LN versus NAT. SPRY2 and AMIGO2 have not previously been studied in TNBC. However, SPRY2 is a known tumour suppressor that regulates the RAS-ERK pathway36,37. AMIGO2 has been shown to be differentially expressed in other cancers including gastric adenocarcinomas, and it is known to effect ploidy, chromosomal stability, cell adhesion/migration, and tumourigenicity38. It also controls cell survival and angiogenesis via Akt activation39. The DMR of LRRC17 is associated with survival in three probes (high methylation/better survival). Interestingly, this gene has been identified as a TNBC specific gene previously40. However, no functional analysis of LRRC17 in TNBC has been done. Promoter methylation of the tumour suppressor ITIH5 has been suggested as early breast cancer detection biomarker41. Finally, GREB1 is a key estrogen regulator42 and is expressed in hormone responsive breast cancers43 but not in TNBC16.
Due to the lack of LN samples in our validation cohort, we utilised Methylation-Binding-Domain Capture sequencing (MBDcap seq) data from 7 LN and 4 NAT samples to validate the direction of methylation change of the genes that are associated with LN metastasis. Differential methylation was validated for 18 of the 38 genes associated with LN metastasis. The MBDcap seq provided regional methylation analysis covering 2 kb upstream from the first transcription start site to the end of the gene of interest, these regions were broken up into 1 kb tiles. The majority of genes (12 of 14) show a negative correlation between gene expression and DNA methylation. However, there are four genes that show negative and positive correlation in different probes. These are TSHZ2 (known to be down-regulated in breast and prostate cancer44), ITIH5 (promoter methylation is an early breast cancer detection biomarker41), GREB1 (estrogen regulator42,43), MEG3 (long non-coding RNA known to be down-regulated in cancer34,35), and RELN (methylated and down-regulated in pancreatic cancer, where its expression has been associated with increased cell motility, invasiveness and colony-forming ability45 but has not been described in breast cancer). This could mean that some loci overpower others or potentially, that during tumour progression to lymph nodes, the methylation becomes tissue specific and changes. Further research into loci specific DNA methylation during cancer progression is urgently needed to explain these phenomena. Additionally, we reviewed the connection of the 18 validated genes to epithelial-mesenchymal transition (EMT) (a process which cells undergo to travel to distant sites, leading to distant cancerous disease). The majority of these genes (12/18) have a known connection to EMT, which we summarised in Table 4. These findings support the importance of these genes during cancer progression.
In conclusion, this is the first whole genome DNA methylation analysis in a TNBC cohort including matched lymph node metastases. Here we identified and validated a global DNA methylation profile, which we correlated with our previously published gene expression findings in tumour, matched lymph node and matched normal adjacent tissue. Our findings show that DNA methylation contributes to the deregulation of gene expression changes and is associated with overall survival.
Methods
Study design
The study cohort comprised a total of 23 grade three invasive ductal carcinomas (IDC), 12 matched lymph node metastasis (LN), and 11 matched normal adjacent tissues (NAT), from which DNA was isolated and screened using the Illumina HumanMethylation450 BeadChips to reveal DNA methylation changes across the genome. All samples in the study cohort were obtained as formalin-fixed, paraffin-embedded (FFPE) blocks from the archives of the Hunter Area Pathology Service, John Hunter Hospital, Newcastle, Australia. This cohort has been described previously17. A pathologist confirmed the triple negative phenotype, areas of NAT, invasive cancer and LN metastasis. As previously described, 1.5 mm punch biopsies were used to isolate areas of IDC, LN and NAT from these sections.
The validation cohort used in this study contained 70 IDC samples derived from the Australian Breast Cancer Tissue Bank (ABCTB) and the same 11 NAT samples used in the study cohort. All participants consented to the use of their tissue in this study. Details regarding this cohort are shown in Table 5.
To assess the direction of DNA methylation in LN metastasis samples, we utilised Methylation-Binding-Domain-Capture sequencing data from 7 LN samples and 4 NAT samples (Supplementary Table 6).
Ethics statement
All experiments were performed in accordance with approved guidelines and regulations.
This study, including all experimental protocols, was granted a waiver of consent in accordance with the National Statement on Ethical Conduct in Research Involving Humans.
This study, including all experimental protocols, complies with the Helsinki Declaration with ethical approval from the Hunter New England Human Research Ethics Committee (Approval number: 09/05/20/5.02).
Written informed consent was obtained from all patients included in this study.
DNA extraction
The Gentra Puregene Tissue Kit (Qiagen, Venlo, Limburg, Netherlands) was used to isolate DNA from FFPE tissue following the manufacturers’ instruction with few alterations. The protocol including the alterations was as follows.
All biopsy samples were placed in 1.5 ml tubes. Five hundred microliters of Xylene were added in a fume hood and incubated with constant gentle mixing at 55 °C. This was followed by centrifugation at 16,000 g for 3 minutes. The supernatant was discarded and the Xylene wash step repeated twice. Five hundred microliters of 100% ethanol was added and incubated for 5 mins under constant mixing at room temperature. For the cell lysis, 300 μl of Cell Lysis Solution was added to each tube and incubated for 10 mins at 70 °C. Twenty microliters Proteinase K (20 mg/ml) were added to each sample, mixed for 20 sec and incubated at 55 °C overnight. On day two, a further 10 μl Proteinase K was added to each sample, mixed and again incubated overnight at 55 °C. On day three, 5 freeze-thaw cycles were performed, 5 mins on dry ice and 5 mins at 95 °C. All samples were brought back to room temperature. Two microlitres of RNase A solution (4 mg/ml) was added to each sample, inverted 25 times and then incubated at 37 °C for one hour. This was followed by a protein precipitation step by adding 100 μl Protein Precipitation solution to the cell lysates, mixed, incubated on ice for 5 mins, followed by centrifugation for 5 mins at 4 °C at × 21,100 g (full speed). For DNA precipitation the supernatant was transferred into a new tube and 300 μl of 100% isopropanol were added. The solutions were mixed by inverting 30 times. Followed by full speed centrifugation at 4 °C for 15 mins. Supernatant was discarded, and 300 μl cold 70% ethanol were added. All samples were centrifuged for one minute at full speed, the supernatant discarded and all samples were subject to further centrifugation for a further minute at full speed. The DNA pellet was air-dried and dissolved in 20 μl of DNAse-free water with constant mixing for one hour. The DNA was stored at −20 °C until used.
DNA was quantitated using the Qubit dsDNA BR Assay Kit according to the manufacturer’s instructions (Life Technologies, Carlsbad, CA, United States of America).
Illumina Infinium HD FFPPE methylation arrays
The array results have been deposited in Gene Expression Omnibus (GEO) with Accession No. GSE78758.
The Infinium HD FFPE quality control (QC) Assay (Illumina, San Diega, CA, United States of America) was used to assess the integrity of the DNA used for methylation analysis. It was performed using a Real-time PCR assay according to the manufacturers’ instructions (Applied Biosystems 7500 Fast Real-Time PCR system). All samples were assayed in triplicates. Two microliters of genomic DNA (at 1 ng/μl) was used for each reaction. The threshold cycle (Ct) was calculated for each individual sample. Replicates where the Ct diverged by more than half a unit were excluded. The average Ct was calculated for each sample as well as the QC template reagent (Illumina). To calculate the delta Ct (ΔCt) the average Ct of the QC template reagent was subtracted from the average Ct of each sample. The ΔCt had to remain below 5 for the samples to pass the quality control test. Of the initial 37 IDC samples, 27 had enough DNA (>250 ng) for further analysis, and 23 of these passed the quality control (ΔCt < 5), all 12 LN samples passed the QC stage as well as the 3 NAT pooled samples and one of the single NAT samples passed the QC.
Bisulfite conversion was performed using the EZ-96 DNA Methylation Kit (Zymo Research, Irvine, CA, United States of America).
Next, FFPE Restoration was undertaken following the Infinium HD FFPE Restore Protocol (Illumina).
Infinium HD FFPE Methylation Assay (Illumina) with the hybridisation, washing and staining of the arrays as well as the scanning (iScan) of the HumanMethylation 450 K BeadChip arrays was performed using the manufacturers’ instructions.
Methylation array analysis
The data from all samples was imported in form of the idat files into Genomic Suite 6.6 (Partek, St Louis, Missouri, United States of America) and Illumina normalisation was performed. ANOVA analysis was performed to detect differentially methylated probes between groups (IDC versus NAT, LN versus NAT, and IDC versus LN). Significance was granted if p < 0.05 and the estimated difference between groups (Δβ) was <−0.1 or >0.1, signifying a methylation change of at least 10%.
These analyses were performed on single loci (=probes on the BeadChips) and on differentially methylated regions (DMR) – a minimum of three significant consecutive probes. The focus in this study was the 38 triple negative specific genes which we identified in our previous study16.
All samples of the validation cohort were treated and analysed in the same way. Due to the lack of LN samples in this cohort, only the IDC versus NAT comparisons were performed.
Pathway enrichment analysis was performed using Genomic Suite 6.6 (Partek). All significant probes were filtered to include probes that are within enhancer and/or promoter regions. These were then used for pathway enrichment analysis, which is a tool within Genomic Suite 6.6 (Partek). The enrichment score is the negative natural log of the enrichment p-value derived from the Fisher’s exact test of the pathway enrichment analysis.
MDB-Cap Sequencing
We compared our findings (comparing LN versus NAT) with MBDcap sequencing data provided by Dr. Clare Stirzaker and Prof. Susan Clark from the Garvan Institute Sydney. The methylation profiling was performed as previously described in ref. 15. This analysis provides regional DNA methylation information therefore a strict validation with our CpG-specific analysis was not possible. The regions that were analysed using the MBDcap data included our genes of interest (83 genes that are associated with LN metastasis, identified in ref. 16) starting 2 kb upstream of the first transcription start site (TSS) to the 3′ end of the genes. These regions are broken up into 1kb tiles. The methylation profiling was performed as described by Stirzaker, et al.15 where the same sample cohort had been used (excluding the LN samples) and methylation differences between LN and NAT samples were assessed using a Student’s t-test (using the R base package) (p < 0.05).
Additional Information
How to cite this article: Mathe, A. et al. DNA methylation profile of triple negative breast cancer-specific genes comparing lymph node positive patients to lymph node negative patients. Sci. Rep. 6, 33435; doi: 10.1038/srep33435 (2016).
References
Carotenuto, P. et al. Triple negative breast cancer: from molecular portrait to therapeutic intervention. Crit Rev Eukaryot Gene Expr 20, 17–34 (2010).
Chacon, R. D. & Costanzo, M. V. Triple-negative breast cancer. Breast Cancer Res 12 Suppl 2, S3 (2010).
Chen, J. Q. & Russo, J. E. Ralpha-negative and triple negative breast cancer: molecular features and potential therapeutic approaches. Biochim Biophys Acta 1796, 162–175 (2009).
Wong-Brown, M. W. et al. Prevalence of BRCA1 and BRCA2 germline mutations in patients with triple-negative breast cancer. Breast Cancer Res Treat 150, 71–80 (2015).
Szasz, A. M. et al. The CIN4 chromosomal instability qPCR classifier defines tumor aneuploidy and stratifies outcome in grade 2 breast cancer. PLos One 8, e56707 (2013).
Reis-Filho, J. S. & Tutt, A. N. Triple negative tumours: a critical review. Histopathology 52, 108–118 (2008).
Portela, A. & Esteller, M. Epigenetic modifications and human disease. Nat Biotechnol 28, 1057–1068 (2010).
Hirst, M. & Marra, M. A. Epigenetics and human disease. Int J Biochem Cell Biol 41, 136–146 (2009).
Feinberg, A. P. & Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 301, 89–92 (1983).
Esteller, M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene 21, 5427–5440 (2002).
How Kit, A., Nielsen, H. M. & Tost, J. DNA methylation based biomarkers: practical considerations and applications. Biochimie 94, 2314–2337 (2012).
Branham, M. T. et al. Methylation profile of triple-negative breast carcinomas. Oncogenesis 1, e17 (2012).
Feng, W. et al. Correlation between CpG methylation profiles and hormone receptor status in breast cancers. Breast Cancer Res 9, R57 (2007).
Sharma, P. et al. The prognostic value of promoter methylation in early stage triple negative breast cancer. J Cancer Ther Res 3, 1–11 (2014).
Stirzaker, C. et al. Methylome sequencing in triple-negative breast cancer reveals distinct methylation clusters with prognostic value. Nat Commun 6, 5899 (2015).
Mathe, A. et al. Novel genes associated with lymph node metastasis in triple negative breast cancer. Sci Rep 5, 15832 (2015).
Avery-Kiejda, K. A., Braye, S. G., Mathe, A., Forbes, J. F. & Scott, R. J. Decreased expression of key tumour suppressor microRNAs is associated with lymph node metastases in triple negative breast cancer. BMC Cancer 14, 51 (2014).
Mathe, A., Scott, R. J. & Avery-Kiejda, K. A. miRNAs and Other Epigenetic Changes as Biomarkers in Triple Negative Breast Cancer. Int J Mol Sci 16, 28347–28376 (2015).
Heintzman, N. D. et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112 (2009).
Kowalczyk, M. S. et al. Intragenic enhancers act as alternative promoters. Mol Cell 45, 447–458 (2012).
Stone, A. et al. DNA methylation of oestrogen-regulated enhancers defines endocrine sensitivity in breast cancer. Nat Commun 6, 7758 (2015).
Conway, K. et al. DNA methylation profiling in the Carolina Breast Cancer Study defines cancer subclasses differing in clinicopathologic characteristics and survival. Breast Cancer Res 16, 450 (2014).
Stefansson, O. A. et al. A DNA methylation-based definition of biologically distinct breast cancer subtypes. Mol Oncol 9, 555–568 (2015).
Wan, J. et al. Characterization of tissue-specific differential DNA methylation suggests distinct modes of positive and negative gene expression regulation. BMC Genomics 16, 49 (2015).
Bert, S. A. et al. Regional activation of the cancer genome by long-range epigenetic remodeling. Cancer Cell 23, 9–22 (2013).
Cascione, L. et al. Integrated microRNA and mRNA signatures associated with survival in triple negative breast cancer. PLos One 8, e55910 (2013).
Baron, V., Adamson, E. D., Calogero, A., Ragona, G. & Mercola, D. The transcription factor Egr1 is a direct regulator of multiple tumor suppressors including TGFbeta1, PTEN, p53, and fibronectin. Cancer Gene Ther 13, 115–124 (2006).
Dey, B. R. et al. Repression of the transforming growth factor-beta 1 gene by the Wilms’ tumor suppressor WT1 gene product. Mol Endocrinol 8, 595–602 (1994).
Sukhatme, V. P. et al. A zinc finger-encoding gene coregulated with c-fos during growth and differentiation, and after cellular depolarization. Cell 53, 37–43 (1988).
Jager, D. et al. Identification of a tissue-specific putative transcription factor in breast tissue by serological screening of a breast cancer library. Cancer Res 61, 2055–2061 (2001).
Chandran, S., Harmey, J. & Toomey, S. Inhibition of IGF signalling pathway in MDA-MB-231 triple negative breast cancer cells. BMC Proc 6, O14 (2012).
Davison, Z., de Blacquière, G. E., Westley, B. R. & May, F. E. B. Insulin-like Growth Factor-Dependent Proliferation and Survival of Triple-Negative Breast Cancer Cells: Implications for Therapy. Neoplasia 13, 504–515 (2011).
Lindner, R. et al. Molecular Phenotypes in Triple Negative Breast Cancer from African American Patients Suggest Targets for Therapy. PLoS One 8(2013).
Sun, M. et al. Downregulated long noncoding RNA MEG3 is associated with poor prognosis and promotes cell proliferation in gastric cancer. Tumour Biol 35, 1065–1073 (2014).
Wang, P., Ren, Z. & Sun, P. Overexpression of the long non-coding RNA MEG3 impairs in vitro glioma cell proliferation. J Cell Biochem 113, 1868–1874 (2012).
Kwak, H. J. et al. Downregulation of Spry2 by miR-21 triggers malignancy in human gliomas. Oncogene 30, 2433–2442 (2011).
Sharma, S. B. et al. MicroRNAs 206 and 21 cooperate to promote RAS-extracellular signal-regulated kinase signaling by suppressing the translation of RASA1 and SPRED1. Mol Cell Biol 34, 4143–4164 (2014).
Rabenau, K. E. et al. DEGA//AMIGO-2, a leucine-rich repeat family member, differentially expressed in human gastric adenocarcinoma: effects on ploidy, chromosomal stability, cell adhesion//migration and tumorigenicity. Oncogene 23, 5056–5067 (2004).
Park, H. et al. AMIGO2, a novel membrane anchor of PDK1, controls cell survival and angiogenesis via Akt activation. J Cell Biol 211, 619–637 (2015).
Zaka, M. U., Yonghong, P. & Sutton, C. W. Microarray big data integrated analysis to identify robust diagnostic signature for triple negative breast cancer. in Computational Intelligence in Big Data (CIBD), 2014 IEEE Symposium on 1–7 (2014).
Kloten, V. et al. Promoter hypermethylation of the tumor-suppressor genes ITIH5, DKK3, and RASSF1A as novel biomarkers for blood-based breast cancer screening. Breast Cancer Res 15, R4 (2013).
Mohammed, H. et al. Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Rep 3, 342–349 (2013).
Ghosh, M. G., Thompson, D. A. & Weigel, R. J. PDZK1 and GREB1 are estrogen-regulated genes expressed in hormone-responsive breast cancer. Cancer Res 60, 6367–6375 (2000).
Yamamoto, M., Cid, E., Bru, S. & Yamamoto, F. Rare and frequent promoter methylation, respectively, of TSHZ2 and 3 genes that are both downregulated in expression in breast and prostate cancers. PLos One 6, e17149 (2011).
Sato, N., Fukushima, N., Chang, R., Matsubayashi, H. & Goggins, M. Differential and Epigenetic Gene Expression Profiling Identifies Frequent Disruption of the RELN Pathway in Pancreatic Cancers. Gastroenterology 130, 548–565 (2006).
Laviolette, L. A., Hodgkinson, K. M., Minhas, N., Perez-Iratxeta, C. & Vanderhyden, B. C. 17beta-estradiol upregulates GREB1 and accelerates ovarian tumor progression in vivo. Int J Cancer 135, 1072–1084 (2014).
Johansson, J., Tabor, V., Wikell, A., Jalkanen, S. & Fuxe, J. TGF-beta1-Induced Epithelial-Mesenchymal Transition Promotes Monocyte/Macrophage Properties in Breast Cancer Cells. Front Oncol 5, 3 (2015).
Lin, C. W., Lin, P. Y. & Yang, P. C. Noncoding RNAs in Tumor Epithelial-to-Mesenchymal Transition. Stem Cells Int 2016, 2732705 (2016).
Nath, A., Li, I., Roberts, L. R. & Chan, C. Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci Rep 5, 14752 (2015).
Obacz, J. et al. The role of AGR2 and AGR3 in cancer: similar but not identical. Eur J Cell Biol 94, 139–147 (2015).
Yuan, Y., Chen, H., Ma, G., Cao, X. & Liu, Z. Reelin is involved in transforming growth factor-beta1-induced cell migration in esophageal carcinoma cells. PLos One 7, e31802 (2012).
Brodin, B. A. CINSARC: a new look into an old concept gives hope for new treatments for synovial sarcomas. Transl Pediatr 2, 70–72 (2013).
Xue, Z. et al. Mesenchymal stem cells promote epithelial to mesenchymal transition and metastasis in gastric cancer though paracrine cues and close physical contact. J Cell Biochem 116, 618–627 (2015).
Bonomi, S. et al. Oncogenic alternative splicing switches: role in cancer progression and prospects for therapy. Int J Cell Biol 2013, 962038 (2013).
Rose, M. et al. Epigenetic inactivation of ITIH5 promotes bladder cancer progression and predicts early relapse of pT1 high-grade urothelial tumours. Carcinogenesis 35, 727–736 (2014).
Riku, M. et al. Down-regulation of the zinc-finger homeobox protein TSHZ2 releases GLI1 from the nuclear repressor complex to restore its transcriptional activity during mammary tumorigenesis. Oncotarget 7, 5690–5701 (2016).
Acknowledgements
The authors would like to thank the Pathologist Dr. Ricardo Vilain for the review of all tumour and normal tissue specimens used in this study, as well as Ms Sarah Nielsen and Dr. Mythily Mariasegaram for their assistance with archival specimens. We would also like to acknowledge, that the MBDCap data was kindly provided by Dr. Clare Stirzaker and Prof. Sue Clark from the Garvan Institute, Sydney. Dr. Andrea Mathe is supported by a University Postgraduate Award. This study was funded by the National Breast Cancer Foundation and the Hunter Medical Research Institute, through donations from Hunter Breast Cancer Foundation, PULSE, Brad Webb and Paul Craven along with the Wests Group Australia.
Author information
Authors and Affiliations
Contributions
A.M. study concept and design, carried out experiments, analysis and interpretation of data, drafting of manuscript. M.W.B. carried out experiments of the validation cohort with A.M. W.J.L. performed analysis of MBDcap seq data. C.S. provided data for MBDcap seq data. S.G.B. patient collection, material support. J.F.F. study design, obtained funding. S.J.C. study design of MBDcap study. K.A.A.-K. study concept, carried out experiments of validation cohort with A.M., design and supervision, interpretation of data, obtained funding, critical revision of the manuscript. R.J.S. study design, obtained funding, critical revision of the manuscript. All authors have read and approved the final version of this manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Mathe, A., Wong-Brown, M., Locke, W. et al. DNA methylation profile of triple negative breast cancer-specific genes comparing lymph node positive patients to lymph node negative patients. Sci Rep 6, 33435 (2016). https://doi.org/10.1038/srep33435
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep33435
This article is cited by
-
Minimalist approaches to cancer tissue-of-origin classification by DNA methylation
Modern Pathology (2020)
-
Studying Lymphatic Metastasis in Breast Cancer: Current Models, Strategies, and Clinical Perspectives
Journal of Mammary Gland Biology and Neoplasia (2020)
-
A two-gene epigenetic signature for the prediction of response to neoadjuvant chemotherapy in triple-negative breast cancer patients
Clinical Epigenetics (2019)
-
Characterisation of DNA methylation changes in EBF3 and TBC1D16 associated with tumour progression and metastasis in multiple cancer types
Clinical Epigenetics (2019)
-
Identification of differentially expressed genes regulated by molecular signature in breast cancer-associated fibroblasts by bioinformatics analysis
Archives of Gynecology and Obstetrics (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
