
Abstract
Protein-protein docking methods have been widely used to gain an atomic-level understanding of protein interactions. However, docking methods that employ low-resolution energy functions are popular because of computational efficiency. Low-resolution docking tends to generate protein complex structures that are not fully optimized. GalaxyRefineComplex takes such low-resolution docking structures and refines them to improve model accuracy in terms of both interface contact and inter-protein orientation. This refinement method allows flexibility at the protein interface and in the overall docking structure to capture conformational changes that occur upon binding. Symmetric refinement is also provided for symmetric homo-complexes. This method was validated by refining models produced by available docking programs, including ZDOCK and M-ZDOCK, and was successfully applied to CAPRI targets in a blind fashion. An example of using the refinement method with an existing docking method for ligand binding mode prediction of a drug target is also presented. A web server that implements the method is freely available at http://galaxy.seoklab.org/refinecomplex.
Similar content being viewed by others


Introduction
Protein-protein interactions play critical roles in various biological processes, including enzyme catalysis1, cellular signal transduction2, and macromolecular assembly3. Three-dimensional protein-protein complex structures can provide atomic-level insights that can improve our understanding of protein-protein interactions and facilitate the engineering of proteins or small molecules with desired binding properties4. However, the number of co-crystalized protein-protein complex structures is still limited due to difficulties posed by experimental approaches. For example, experimentally determined three-dimensional protein-protein complex structures in the human proteome cover less than 10% of known interactions5,6. Therefore, accurate prediction of protein complex structures via in silico methods can be an effective alternative approach.
In silico prediction of protein-protein interactions was initially approached using rigid-body docking methods in which only the relative orientation between two proteins, represented by six translational and rotational degrees of freedom, is treated explicitly. The rigid-body docking problem can be solved efficiently using fast-Fourier transformation (FFT)7 or geometric hashing8 techniques. The internal flexibility of each protein structure is considered only implicitly, and low-resolution energy functions that allow some atomic overlaps are used, assuming that atomic overlaps or locally unfavorable interactions can be relaxed by local side-chain or backbone movement. Therefore, complex model structures generated by rigid-body docking may have some atomic clashes and may not have precise interface contacts9. Rigid-body docking methods are still effective for the generation of globally correct complex structures when conformational changes induced by binding are limited to local regions. Further refinement of the models generated by rigid-body docking using more computationally extensive flexible docking methods can therefore produce useful predictions for practical applications9,10,11,12,13,14,15,16,17.
Several methods for refinement of rigid-body docking model structures have been developed with some success by applying energy minimization or molecular dynamics simulations. RDOCK9 performs local energy minimizations on the protein-protein complex model structures generated by ZDOCK7, a rigid-body docking program. RDOCK was also extended by combining with ZRANK18. RosettaDock12 can refine protein-protein complex structures by the optimization technique of Monte Carlo with minimization. RosettaDock can optimize inter-protein orientation, side-chain conformation, and backbone conformation, such as loops15. Zhang et al.16 achieved effective conformational sampling with RosettaDock by applying an advanced sampling technique called well-tempered ensemble two dimensional Hamiltonian Replica Exchange Monte Carlo (WTE-H-REMC). HADDOCK19 performs energy optimization of interface side chains and backbone in torsion angle space first and then runs simulated annealing molecular dynamics (MD) simulations in Cartesian space with explicit waters. Król et al. applied nanosecond MD simulations11 to the refinement problem. The method iATTRACT10 performs energy minimization of the protein-protein complex structures generated by ATTRACT20 considering interface side-chain degrees of freedom for selected residues and rigid-body degrees of freedom. FiberDock13 and SymmRef14 employ normal modes to describe global backbone structure changes induced by binding for hetero-complexes and homo-complexes, respectively. Overall, the methods developed so far have been more successful in refining relatively high-accuracy models, and refinement of less accurate complex models or those involving inaccurate monomer structures, such as predicted structures, has yet to be achieved.
In this study, we introduce a new refinement method that improves less accurate protein-protein complex model structures compared to previous methods, for both hetero- and homo-complexes. This method, called GalaxyRefineComplex, was developed by extending the GalaxyRefine method for protein monomer structures21,22. GalaxyRefine successfully improved homology model structures in the blind protein structure prediction experiment Critical Assessment of techniques for protein Structure Prediction (CASP)23,24. GalaxyRefineComplex adapts the effective sampling method of GalaxyRefine by performing repetitive repacking of interface side chains followed by short MD relaxations. This sampling procedure mimics a protein-protein binding process in which side-chain interactions between two approaching proteins drive changes in the inter-protein orientation and intra-protein backbone conformation. The method was validated by refining models generated by ZDOCK7, M-ZDOCK25, and various methods used in the previous Critical Assessment of Prediction of Interactions (CAPRI)26,27 and CASP experiments. GalaxyRefineComplex was also successfully tested in a blind fashion in the CAPRI round 3026 (http://www.ebi.ac.uk/msd-srv/capri/round30/results/), which was held jointly with CASP11 (http://www.ebi.ac.uk/msd-srv/capri/round30/CAPRI_R30_v20141224.SW.pdf).
Methods
Overall procedure
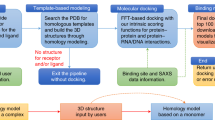
A flowchart of the GalaxyRefineComplex method is provided in Fig. 1. This method is based on the GalaxyRefine method21,22 for structure refinement of single protein chains and was extended to protein-protein complexes. The refinement calculation starts with a local energy minimization and a 1.2-ps MD relaxation with a 4-fs time step, as in GalaxyRefine. According to our observation, energy minimization tends to generate very compact structures, and a short relaxation of 1.2 ps can create some physical space for atomic fluctuations, facilitating further conformational sampling. A major difference of GalaxyRefineComplex from GalaxyRefine is in the treatment of protein interfacial residues. Interfacial residues are defined here as those within 8 Å Cα-Cα distance from any residue of the interaction partner. Relaxation of the input complex structure is driven by side-chain repacking of interfacial residues as follows. Interfacial residues are first repacked by three Monte Carlo (MC) steps of replacing the side-chain conformation of a cluster of up to five interfacial residues with a non-clashing rotamer conformation for three different clusters. Local side-chain conformation could be reasonably optimized by this short MC. A short MD relaxation of 0.6 ps is then performed with a 4-fs time step to allow overall conformational changes, including changes in the backbone and inter-protein orientation. During the Monte Carlo steps, the van der Waals radius is reduced to 70% to allow a small amount of clashes, which was proved effective in GalaxyRefine. The clashes can be relieved in subsequent relaxation steps. Side-chain repacking and relaxation is repeated 22 times (13.2-ps) because the relaxation was observed to converge after 10–15 ps. During the relaxation, the temperature is set to 300 K and is gradually decreased to 50 K for the last six steps (3.6 ps) as in simulated annealing to drive convergence to lower energy minimum without being trapped to nearby higher energy local minimum. Finally, local energy minimization is performed. For symmetric refinement of homo-complexes, symmetry transformation matrices for input chain structures are used to recover the symmetry after each MD step. The energy used for the MD relaxation and the criterion for model selection is explained in the following sections.

Flowchart of the GalaxyRefineComplex method.
GALAXY energy for complex refinement
The energy function used for MD relaxation (Eq. 1) is a linear combination of physics-based energy terms, knowledge-based energy terms, and restraint energy terms, with the relative weights of GalaxyRefine energy21,22, except for the restraint terms, as explained below.

The physics-based terms include molecular mechanics bonded energy (Ebanded) and Lennard-Jones (Evdw) and Coulomb (ECoulomb) non-bonded interaction energy terms of CHARMM2228 with FACTS solvation free energy (EFACTS,pol for the polar term and EFACTS,SA for non-polar surface area term)29. The knowledge-based terms include hydrogen bond energy (EHBond)30, dipolar-DFIRE potential energy (EdDFIRE)31, and side-chain (ERotamer) and backbone (ERama) torsion angle energy32. The restraint energy terms include the following two components:


in which the reference distances  and the reference positions
and the reference positions  are taken from the input structure. The distance restraint of Eq. 2 is applied to all interface Cα-Cα and N-O atom pairs with distances dij < 10 Å with the same weight of
are taken from the input structure. The distance restraint of Eq. 2 is applied to all interface Cα-Cα and N-O atom pairs with distances dij < 10 Å with the same weight of  as used in GalaxyRefine for non-interface residues and with a much smaller weight of
as used in GalaxyRefine for non-interface residues and with a much smaller weight of  for interfacial residues to allow more structural changes on interface regions. Weak position restraint of Eq. 3 with a weight of
for interfacial residues to allow more structural changes on interface regions. Weak position restraint of Eq. 3 with a weight of  (compared to
(compared to  in GalaxyRefine) is applied to all Cα atoms to allow global changes in inter-protein orientation.
in GalaxyRefine) is applied to all Cα atoms to allow global changes in inter-protein orientation.
Model generation and selection
Each of the two relaxation protocols, i.e., protocol 1, which applies only distance restraints (Eq. 2), and protocol 2, which applies both distance and position restraints (Eqs 2 and 3), is used to generate 16 structures by performing the relaxations described above 16 times. The five lowest-energy models out of the 16 models for each of protocols 1 and 2 are returned as 10 refined models. The five lowest-energy models from protocol 1 (and protocol 2) are ranked 1–5 (and 6–10) in the order of energy. This scheme for determining ranking among the 10 models, and in particular for selecting model 1, was determined by examination of the refinement results on the training set (constructed as explained in the next subsection) in terms of ligand RMSD (L-RMSD), interface RMSD (I-RMSD), fraction of predicted native contacts (Fnat), and the MolProbity score (MolP), as summarized in Supplementary Table S1.
Training and test sets
The refinement method was extensively tested on model structures of varying accuracies. First, models generated by ZDOCK7 for the ZDOCK benchmark 4.0 set complexes33 with unbound monomer structures were used to test the hetero-oligomer refinement method. Those generated by M-ZDOCK25 for the PISA benchmark set complexes34 with bound monomer structures were used to test the symmetric homo-oligomer refinement method. Only those complexes with less than 1,000 residues were considered for computational efficiency. For each complex, up to 1, 3, and 3 structures among models with high, medium, and acceptable accuracies, respectively (classified according to the CAPRI criterion)27, and up to three structures with the lowest L-RMSD among incorrect models with less than 15 Å L-RMSD were selected randomly. The number of model structures selected for each complex could be less than 10 because the number of models satisfying the above accuracy criteria could be less than the maximum number that could be selected.
Each of the ZDOCK and M-ZDOCK models was randomly divided into two subsets, and one subset was used as a training set to select model 1, as described in the previous subsection, and the other subset was used as a test set. The training set was composed of 643 ZDOCK models for 89 hetero-complex targets and 452 M-ZDOCK models for 46 homo-complex targets. The ZDOCK benchmark test set consisted of 677 models for 90 hetero-complex targets, and the PISA benchmark test set consisted of 445 models for 46 homo-complex targets.
Two additional test sets were constructed by collecting the model structures submitted during CAPRI blind prediction experiments. For each of the hetero-complex targets of CAPRI rounds 22, 24, 26, and 30 and the homo-complex targets of CAPRI round 30, up to 10 models showing varying accuracies were selected as described above. As a result, 34 models for five hetero-complex targets and 60 models for 13 homo-complex targets were selected. The models submitted by our own group (“Seok”) were excluded in these test sets because they were already refined by GalaxyRefineComplex. The blind prediction results of GalaxyRefineComplex on the CAPRI round 30 targets are presented separately.
Comparison with existing methods
For performance comparison, available refinement docking methods, including RosettaDock, FiberDock, and SymmRef, were tested on the same sets. RosettaDock and FiberDock were used for refinement of hetero-complex models, and RosettaDock with the symmetry option and SymmRef were used for refinement of homo-complex models. For RosettaDock refinement12,17, the “docking_local_refine” protocol was applied to generate 1,000 structures with the extra χ1 (-ex1) and aromatic χ2 (-ex2aro) side-chain rotamer options for side-chain optimization. Among the 1,000 generated models, 10 models with the best energy values were selected. For homo-complex refinement by RosettaDock, a symmetry definition file generated by the “make_symmdef_file.pl” script for the initial complex structure was used to maintain symmetry. FiberDock and SymmRef were run with default parameters13,14. These methods generated a single refined model for each initial complex structure.
Results and Discussion
Performance comparison in terms of the CAPRI model accuracy criterion
The performance of GalaxyRefineComplex was compared with those of RosettaDock and FiberDock on the two hetero-complex sets (ZDOCK benchmark set and CAPRI set) and with those of RosettaDock run with a symmetry option and SymmRef, a symmetric version of FiberDock, on the two homo-complex sets (PISA benchmark set and CAPRI set). The accuracies of the initial models and refined models were classified using the CAPRI model quality criterion. The CAPRI criterion reflects the biological relevance of the model structures, and model qualities are classified as high (***), medium (**), acceptable (*), and incorrect considering L-RMSD and I-RMSD from the experimental structure and the Fnat. The detailed criterion is as follows27: ‘high’ if L-RMSD or I-RMSD is lower than 1.0 Å with Fnat higher than 0.5, ‘medium’ if L-RMSD is lower than 5.0 Å or I-RMSD is lower than 2.0 Å with Fnat higher than 0.3, ‘acceptable’ if L-RMSD is lower than 10.0 Å or I-RMSD is lower than 4.0 Å with Fnat higher than 0.1, and incorrect for all other cases.
As shown in Table 1, GalaxyRefineComplex improved 114 of 263 incorrect models to acceptable or higher quality for the 677 ZDOCK models of the ZDOCK benchmark set by hetero-complex refinement, while RosettaDock improved 68 models for the same set when the best of 10 refined models was considered. When model 1’s were considered, only GalaxyRefineComplex succeeded in increasing the number of models with acceptable or higher quality, while RosettaDock and FiberDock failed. The numbers of high- and medium-quality models were also increased by GalaxyRefineComplex. RosettaDock and FiberDock were slightly better than GalaxyRefineComplex for refining models to high accuracy; both of the former methods improved five models to high accuracy, while GalaxyRefineComplex improved three.
When applied to the 34 hetero-complex models submitted during CAPRI experiments, GalaxyRefineComplex and RosettaDock improved eight and seven models, respectively, to acceptable or higher quality out of 15 incorrect models when the best of 10 refined models was considered (see Table 1). GalaxyRefineComplex also improved a larger number of incorrect models to acceptable or higher quality than RosettaDock and FiberDock when model 1’s were considered. Overall, GalaxyRefineComplex performed better than RosettaDock and FiberDock for improving incorrect or acceptable models to acceptable or medium accuracy and slightly worse for improving models to high accuracy when using the two hetero-complex test sets.
In the homo-complex refinement test on the 445 M-ZDOCK models of the PISA benchmark set, GalaxyRefineComplex showed a refinement performance that was similar to that of the hetero-complex refinement when both the best of 10 models and model 1’s were considered, as shown in Table 1. GalaxyRefineComplex improved a larger number of incorrect models to acceptable or higher quality than RosettaDock and SymmRef model 1’s were considered. However, RosettaDock and SymmRef performed much better than in the hetero-complex refinement test, improving more than 200 of 400 models to high accuracy, while GalaxyRefineComplex improved only 24 models. This seemingly different behavior on homo-complex refinement may be explained by the fact that the complex models of the PISA benchmark set were generated using the “bound” monomer structures because of the unavailability of unbound monomer structures. Therefore, this refinement set does not represent real case problems and can instead be considered an artificial set. Such problems may be relatively easy if shape complementarity is exploited intensively.
Refining of the 60 homo-complex models submitted during the CAPRI experiment was used to represent real case problems in which the initial models were generated by homology modeling. This test set, based on homology, is easier than the CAPRI hetero-complex set, with 58 of 60 initial models already having acceptable or higher quality. None of the tested methods could increase the number of acceptable or higher quality models in this case. However, GalaxyRefineComplex succeeded in refining five models to medium accuracy, while the other two methods failed.
Refinement results in terms of ligand RMSD, interface RMSD, fraction of native contacts, and MolProbity score
Refinement results on the four test sets were analyzed in more detail using the three model quality measures of L-RMSD, I-RMSD, and Fnat used in CAPRI and MolP35, which measures physical incorrectness, such as the existence of steric clashes, Ramachandran outliers, and side-chain rotamer outliers. The results are summarized in Table 2. More detailed results for GalaxyRefineComplex are provided in Supplementary Tables S2–S4. The distributions of the values for the accuracy measures are also presented in Fig. 2 and Supplementary Figure S1.

Quality comparison of the best out of 10 refined models generated by GalaxyRefineComplex (red) and RosettaDock (green) and the single refined models generated by FiberDock (blue in (a,b)) and SymmRef (blue in (c,d)) when the initial models were (a) ZDOCK models and (b) CAPRI models for hetero-complexes and (c) M-ZDOCK models and (d) CAPRI models for homo-complexes. The refinement results for different target complexes are depicted in boxplots, which present first and third quartiles as boxes and the median as the band inside the boxes. The minimum and maximum data within 1.5 interquartile range of the lower and upper quartile are represented as the bottom and top ends of whiskers, respectively, and data points outside of this range are shown with black dots as outliers. Note that only single models, not top ten models, were evaluated for FiberDock and SymmRef because these programs generate only single models.
When applied to the 677 ZDOCK models of the ZDOCK benchmark set, only GalaxyRefineComplex could improve model quality in all four measures on average. In contrast, RosettaDock only improved MolP, and FiberDock did not improve any models when model 1’s or the mean of final 10 models were considered. When the best of 10 refined models were considered, RosettaDock could improve models on average, but the extents of improvement for all measures were smaller than those of GalaxyRefineComplex. The statistical significance of the differences in L-RMSD and I-RMSD improvements by the two methods was not substantial, with p-values of 0.024 and 0.16, respectively, whereas that in Fnat and MolP was greater, with p-values of 6.5 × 10−34 and 1.1 × 10−253, respectively. GalaxyRefineComplex could improve initial models in 82%, 85%, 91%, and 100% of the cases in terms of L-RMSD, I-RMSD, Fnat, and MolP, respectively, when the best of 10 models were considered.
In the refinement test on the 34 hetero-complex CAPRI models, GalaxyRefineComplex showed consistent improvement in all four quality measures on average, while RosettaDock and FiberDock did not, as shown in Table 2. The success of GalaxyRefineComplex for improving CAPRI models is more notable, considering that the CAPRI models may have already been refined by CAPRI predictors. Based on the data presented in the table, we concluded that complex models generated by the current docking methods could be easily improved at least in interfacial contacts and physical correctness. L-RMSD and I-RMSD may also be improved, but to a lesser extent.
For the 445 M-ZDOCK models of the PISA benchmark set that were generated with “bound” monomer structures, GalaxyRefineComplex could improve models in all measures, while RosettaDock could not improve in I-RMSD and SymmRef could not improve in MolP when the mean of the 10 models were considered (see Table 2). However, SymmRef showed the best performance in terms of the other three measures. The performance of RosettaDock was similar to that of GalaxyRefineComplex when model 1’s were considered, but significantly better when the best of 10 models were considered. This implied that RosettaDock scoring may need to be improved. The relatively poor performance of the GalaxyRefineComplex compared with that of the other two methods on model complex structures of “bound” monomer structures could be ascribed to the fact that only inter-protein orientations between receptor and ligand proteins needed to be adjusted in this case. GalaxyRefineComplex samples only interfacial residue conformations explicitly and allowed inter-protein orientations follow the conformational change of interfacial residues by short MD relaxation.
For the 60 homo-complex CAPRI models, all three methods tended to perform worse than for the other three test sets, as shown in Table 2. This set was the most difficult to refine because the monomer structures, based on homology, deviated more from the native structures than those of the other sets. Initial models of the ZDOCK benchmark set were constructed from unbound monomer structures resolved experimentally, those of hetero-complex CAPRI set from either unbound experimental structures or homology models, and those of the PISA set from bound structures. The overall quality of the initial complex models was better than in the other sets, as discussed in the previous subsection. Therefore, it may be difficult to improve the model quality without accounting for structural flexibilities of monomers at the interface. Despite this difficulty, GalaxyRefineComplex performed the best in all four measures among the compared methods, implying that the explicit sampling of interfacial residues and subsequent structural relaxation was effective.
Successful refinement examples by GalaxyRefineComplex are illustrated in Fig. 3. A hetero-complex model generated by ZDOCK for the target TA12 of the ZDOCK benchmark 4.033 was refined from acceptable to medium accuracy with improvement in L-RMSD from 7.24 to 1.87 Å, as shown in Fig. 3A. The initial model had 41% of native contacts with some voids at the interface and was refined to cover 90% of the native contacts. The hetero-complex model submitted in CAPRI round 26 for target 54 as P38_M07 was refined from acceptable to medium accuracy, as shown in Fig. 3B. Although improvements in RMSDs were small (<1 Å), native contacts increased by more than 20% through refinement. A homo-complex model generated by M-ZDOCK for one of the PISA benchmark set targets (PDB ID: 1MOQ) was refined dramatically from incorrect to medium accuracy, as shown in Fig. 3C. In another example, the model for target 87 of CAPRI round 30 submitted by group TS417 was refined from acceptable to medium accuracy, with improvement in L-RMSD from 5.17 to 4.19 Å (Fig. 3D).

Successful refinement examples for hetero-complexes, i.e., (a) a model for TA12 of the ZDOCK benchmark 4.0 and (b) the model for CAPRI round 26 T54 submitted as P38_M07, and for homo-complexes, i.e., (c) a model for 1MOQ of the PISA benchmark set and (d) a model for CAPRI round 30 T87 submitted as TS417_2. In each panel, experimentally resolved structures are shown on the left, and model structures before and after refinement are shown in the middle and on the right, respectively. Receptor protein structures are depicted in green, ligand protein structures in experimental structures and in models before and after refinement are shown in yellow, pink, and violet, respectively. Red arrows indicate directions of changes in relative orientation made by refinement (from the pink to violet structures).
Blind prediction results of GalaxyRefineComplex in CAPRI round 30
GalaxyRefineComplex was used in CAPRI round 30 in a blind fashion under the group name “Seok”. Initial models generated using GalaxyGemini36, GalaxyLoop37, and other methods were subjected to refinement. Improvement by refinement is summarized in Table 3, and results for individual targets are provided in Supplementary Table S5. As in the test results reported in the previous subsection, L-RMSD and I-RMSD were improved by small magnitudes on average, whereas Fnat and MolP were improved more substantially.
A practical example: accurate prediction of ligand binding mode of HIV-1 integrase by refinement
GalaxyRefineComplex was applied to the binding mode prediction of an inhibitor to HIV-1 integrase to illustrate the impact of complex refinement on practical applications such as drug discovery. HIV-1 integrase mediates integration of viral DNA into human DNA38 by forming a complex with a crucial co-factor, the human protein lens epithelium-derived growth factor (LEDGF)/p75. The co-factor binds at the dimer interface of HIV-1 integrase catalytic core domains, and inhibitors that bind at the same dimer interface can interfere with the co-factor binding. We first modeled the dimer structure by using M-ZDOCK with the monomer structure of HIV-1 integrase (PDB ID: 4DMN)39. GalaxyRefineComplex was then applied to generate a refined complex model. An inhibitor of HIV-1 integrase (PDB ID: 4NYF)40 was then docked by using the GalaxyDock41,42 protein-ligand docking program to the dimer model before and after refinement. Although the RMSD improvement of the model by refinement was rather mild (from 3.17 Å/1.40 Å/0.727 to 1.55 Å/1.09 Å/0.841 in L-RMSD/I-RMSD/fnat), the refinement lead to more dramatic improvement in the binding mode prediction (from 1.70 Å/36% to 0.60 Å/92% in ligand RMSD/percentage of protein-ligand contacts <5.0 Å). Moreover, the key interactions38,40 including the hydrogen bonds between protein backbone and ligand carboxyl group that were not predicted with the initial model could be predicted accurately with the refined model, as shown in Fig. 4.

The full structure of the HIV-1 integrase dimer (PDB ID: 4NYF) and the ligand binding site at the dimer interface are shown in (a). Ligand docking results obtained with the dimer models before and after refinement are presented in (b,c), respectively. In (b,c), the crystal structure is colored in yellow. The predicted complex structures are colored in pink and blue, respectively. It can be seen that the predicted hydrogen bonds (cyan lines, generated using UCSF Chimera43) match perfectly with those of crystal structure in (c) but not in (b).
Origin of the effective refinement by GalaxyRefineComplex
GalaxyRefineComplex effectively refined hetero- and homo-complex model structures on the test sets, showing better results than the other refinement docking methods. Moreover, GalaxyRefineComplex consistently improved models constructed from monomer structures derived from either unbound experimental structures or homology models, unlike the compared methods. This requires additional computational cost, and the computer time as a function of protein size is provided in Supplementary Figure S2. GalaxyRefineComplex can improve docking models despite errors in the input monomer or interface structures due to the following two features. First, it uses a hybrid energy that consists of both physics-based and knowledge-based energy components. The physics-based energy terms contribute to improving the physical correctness of models, such as that measured by MolP. Knowledge-based energy, terms such as dipolar-DFIRE, makes the energy landscape smoother, allowing effective conformational sampling under an erroneous structural environment37. Knowledge-based energy also tends to better discriminate “native-like” conformations from decoys than physics-based energy31. Therefore, we believe that including those knowledge-based energy terms contributes to effective model selection. Second, intensive interface side-chain sampling before overall structure relaxation contributes to effective flexible refinement of docking models. According to our analysis, a refinement protocol without such intensive interface residue sampling performs much worse than the current protocol (see Supplementary Table S6 for detailed results). GalaxyRefineComplex mimics an actual process of conformational change induced by binding in which repacking of interfacial side chains drives further change to the conformation of the backbone.
Conclusions
In this work, we presented a method for refining protein-protein complex structures generated by other docking programs. This method, called GalaxyRefineComplex, was compared with FiberDock, SymmRef, and RosettaDock on several sets of docking models with a range of initial model qualities. GalaxyRefineComplex showed consistent improvement, particularly for models of acceptable quality and for incorrect models. The method was able to improve model quality not only for unbound/bound structures but also for homology model structures, while the other methods were not. High-accuracy models could be improved mainly in contacts, whereas lower accuracy models could be refined both in contacts and relative inter-protein orientation. Repetitive side-chain repacking at the interface allows prediction of side-chain conformational change upon binding, contributing to improving contacts between interacting proteins. The knowledge-based energy terms of the GALAXY energy makes the method less sensitive to the accuracy of initial model qualities. The current method may be applied to various applications in which low- to medium-accuracy models are available but high-quality models are not. The program is freely available on the GalaxyWEB server at http://galaxy.seoklab.org/refinecomplex.
Additional Information
How to cite this article: Heo, L. et al. GalaxyRefineComplex: Refinement of protein-protein complex model structures driven by interface repacking. Sci. Rep. 6, 32153; doi: 10.1038/srep32153 (2016).
References
Negri, A. et al. Protein-protein interactions at an enzyme-substrate interface: characterization of transient reaction intermediates throughout a full catalytic cycle of Escherichia coli thioredoxin reductase. Proteins 78, 36–51 (2010).
Pawson, T. & Nash, P. Protein-protein interactions define specificity in signal transduction. Genes Dev. 14, 1027–1047 (2000).
Russell, R. B. et al. A structural perspective on protein-protein interactions. Curr Opin Struct Biol 14, 313–324 (2004).
Kastritis, P. L. & Bonvin, A. M. On the binding affinity of macromolecular interactions: daring to ask why proteins interact. J R Soc Interface 10, 20120835 (2013).
Kundrotas, P. J., Zhu, Z., Janin, J. & Vakser, I. A. Templates are available to model nearly all complexes of structurally characterized proteins. Proc Natl Acad Sci USA 109, 9438–9441 (2012).
Mosca, R., Ceol, A. & Aloy, P. Interactome3D: adding structural details to protein networks. Nat Methods 10, 47–53 (2013).
Chen, R., Li, L. & Weng, Z. ZDOCK: an initial-stage protein-docking algorithm. Proteins 52, 80–87 (2003).
Schneidman-Duhovny, D., Inbar, Y., Nussinov, R. & Wolfson, H. J. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic acids research 33, W363–367 (2005).
Li, L., Chen, R. & Weng, Z. RDOCK: refinement of rigid-body protein docking predictions. Proteins 53, 693–707 (2003).
Schindler, C. E., de Vries, S. J. & Zacharias, M. iATTRACT: simultaneous global and local interface optimization for protein-protein docking refinement. Proteins 83, 248–258 (2015).
Krol, M., Tournier, A. L. & Bates, P. A. Flexible relaxation of rigid-body docking solutions. Proteins 68, 159–169 (2007).
Gray, J. J. et al. Protein-protein docking predictions for the CAPRI experiment. Proteins 52, 118–122 (2003).
Mashiach, E., Nussinov, R. & Wolfson, H. J. FiberDock: Flexible induced-fit backbone refinement in molecular docking. Proteins 78, 1503–1519 (2010).
Mashiach-Farkash, E., Nussinov, R. & Wolfson, H. J. SymmRef: a flexible refinement method for symmetric multimers. Proteins 79, 2607–2623 (2011).
Wang, C., Bradley, P. & Baker, D. Protein-protein docking with backbone flexibility. Journal of molecular biology 373, 503–519 (2007).
Zhang, Z., Schindler, C. E., Lange, O. F. & Zacharias, M. Application of Enhanced Sampling Monte Carlo Methods for High-Resolution Protein-Protein Docking in Rosetta. PLoS One 10, e0125941 (2015).
Chaudhury, S. et al. Benchmarking and analysis of protein docking performance in Rosetta v3.2. PLoS One 6, e22477 (2011).
Pierce, B. & Weng, Z. ZRANK: reranking protein docking predictions with an optimized energy function. Proteins 67, 1078–1086 (2007).
Dominguez, C., Boelens, R. & Bonvin, A. M. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. Journal of the American Chemical Society 125, 1731–1737 (2003).
May, A. & Zacharias, M. Accounting for global protein deformability during protein-protein and protein-ligand docking. Biochim Biophys Acta 1754, 225–231 (2005).
Heo, L., Park, H. & Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic acids research 41, W384–388 (2013).
Lee, G. R., Heo, L. & Seok, C. Effective protein model structure refinement by loop modeling and overall relaxation. Proteins (2015).
Nugent, T., Cozzetto, D. & Jones, D. T. Evaluation of predictions in the CASP10 model refinement category. Proteins 82 Suppl 2, 98–111 (2014).
Modi, V. & Dunbrack, R. L. Assessment of refinement of template-based models in CASP11. Proteins (2016).
Pierce, B., Tong, W. & Weng, Z. M-ZDOCK: a grid-based approach for Cn symmetric multimer docking. Bioinformatics 21, 1472–1478 (2005).
Lensink, M. F. et al. Prediction of homo- and hetero-protein complexes by protein docking and template-based modeling: a CASP-CAPRI experiment. Proteins (2016).
Lensink, M. F. & Wodak, S. J. Docking, scoring, and affinity prediction in CAPRI. Proteins 81, 2082–2095 (2013).
MacKerell, A. D. et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. The journal of physical chemistry. B 102, 3586–3616 (1998).
Haberthur, U. & Caflisch, A. FACTS: Fast analytical continuum treatment of solvation. Journal of computational chemistry 29, 701–715 (2008).
Kortemme, T., Morozov, A. V. & Baker, D. An orientation-dependent hydrogen bonding potential improves prediction of specificity and structure for proteins and protein-protein complexes. Journal of molecular biology 326, 1239–1259 (2003).
Yang, Y. & Zhou, Y. Specific interactions for ab initio folding of protein terminal regions with secondary structures. Proteins 72, 793–803 (2008).
Canutescu, A. A., Shelenkov, A. A. & Dunbrack, R. L., Jr. A graph-theory algorithm for rapid protein side-chain prediction. Protein science: a publication of the Protein Society 12, 2001–2014 (2003).
Hwang, H., Vreven, T., Janin, J. & Weng, Z. Protein-protein docking benchmark version 4.0. Proteins 78, 3111–3114 (2010).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. Journal of molecular biology 372, 774–797 (2007).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta crystallographica. Section D, Biological crystallography 66, 12–21 (2010).
Lee, H., Park, H., Ko, J. & Seok, C. GalaxyGemini: a web server for protein homo-oligomer structure prediction based on similarity. Bioinformatics 29, 1078–1080 (2013).
Park, H., Lee, G. R., Heo, L. & Seok, C. Protein loop modeling using a new hybrid energy function and its application to modeling in inaccurate structural environments. PLoS One 9, e113811 (2014).
Engelman, A., Kessl, J. J. & Kvaratskhelia, M. Allosteric inhibition of HIV-1 integrase activity. Curr Opin Chem Biol 17, 339–345 (2013).
Kessl, J. J. et al. Multimode, cooperative mechanism of action of allosteric HIV-1 integrase inhibitors. J Biol Chem 287, 16801–16811 (2012).
Fader, L. D. et al. Discovery of BI 224436, a Noncatalytic Site Integrase Inhibitor (NCINI) of HIV-1. ACS Med Chem Lett 5, 422–427 (2014).
Shin, W. H. & Seok, C. GalaxyDock: protein-ligand docking with flexible protein side-chains. J Chem Inf Model 52, 3225–3232 (2012).
Shin, W. H., Kim, J. K., Kim, D. S. & Seok, C. GalaxyDock2: protein-ligand docking using beta-complex and global optimization. Journal of computational chemistry 34, 2647–2656 (2013).
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. Journal of computational chemistry 25, 1605–1612 (2004).
Acknowledgements
This study was funded by grants from the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, & Future Planning (2016R1A2A1A05005485 and 2012M3C1A6035362), and Korea Institute of Science and Technology Information supercomputing center (KSC-2015-C2-045).
Author information
Authors and Affiliations
Contributions
L.H. and C.S. designed the work and wrote the manuscript, L.H., H.L. and C.S. analyzed and interpreted the data. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Heo, L., Lee, H. & Seok, C. GalaxyRefineComplex: Refinement of protein-protein complex model structures driven by interface repacking. Sci Rep 6, 32153 (2016). https://doi.org/10.1038/srep32153
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep32153
This article is cited by
-
Structural insights into cardiolipin replacement by phosphatidylglycerol in a cardiolipin-lacking yeast respiratory supercomplex
Nature Communications (2023)
-
Structure of UBE2K–Ub/E3/polyUb reveals mechanisms of K48-linked Ub chain extension
Nature Chemical Biology (2022)
-
Sclerostin inhibits Wnt signaling through tandem interaction with two LRP6 ectodomains
Nature Communications (2020)
-
Mixed lineage kinase 3 promotes breast tumorigenesis via phosphorylation and activation of p21-activated kinase 1
Oncogene (2019)
-
SHBG141–161 Domain-Peptide Stimulates GPRC6A-Mediated Response in Leydig and β-Langerhans cell lines
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
