
Abstract
To examine the diversity of somatic alterations and clonal evolution according to aggressiveness of disease, nineteen tumor-blood pairs of ‘formerly bronchiolo-alveolar carcinoma (BAC)’ which had been reclassified into preinvasive lesion (adenocarcinoma in situ; AIS), focal invasive lesion (minimally invasive adenocarcinoma; MIA) and invasive lesion (lepidic predominant adenocarcinoma; LPA and non-lepidic predominant adenocarcinoma; non-LPA) according to IASLC/ATS/ERS 2011 classification were explored by whole exome sequencing. Several distinct somatic alterations were observed compare to the lung adenocarcinoma study from the Cancer Genome Atlas (TCGA). There were higher numbers of tumors with significant APOBEC mutation fold enrichment (73% vs. 58% TCGA). The frequency of KRAS mutations was lower in our study (5% vs. 32% TCGA), while a higher number of mutations of RNA-splicing genes, RBM10 and U2AF1, were found (37% vs. 11% TCGA). We found neither mutational pattern nor somatic copy number alterations that were specific to AIS/MIA. We demonstrated that clonal cell fraction was the only distinctive feature that discriminated LPA/non-LPA from AIS/MIA. The broad range of clonal frequency signified a more branched clonal evolution at the time of diagnosis. Assessment of tumor clonal cell fraction might provide critical information for individualized therapy as a prognostic factor, however this needs further study.
Similar content being viewed by others

Introduction
The 2011 pathological classification of lung cancer adenocarcinoma (LUAD) reclassified the previous ‘bronchiolo-alveolar carcinoma’ category (formerly BAC) into adenocarcinoma in situ (AIS), minimally invasive adenocarcinoma (MIA) and invasive adenocarcinoma with lepidic-predominant (LPA) or invasive adenocarcinoma with other predominant subtypes (formerly mucinous BAC; non-LPA). Recent classification1 officially recognizes AIS as a second preinvasive LUAD, after atypical adenomatous hyperplasia, while the complex heterogeneous mixtures of invasive tumors are defined according to the major histologic component. This classification is not solely histologically based, but also incorporates multidisciplinary approaches, including clinical, molecular, radiologic and histologic features. It could assist in predicting outcomes such as AIS and MIA that were correlated with 100% 5-year survival1.
Branched clonal evolution (an evolutionary process that is driven by stepwise, somatic cell mutations with sequential, subclonal selection, derived from Darwinian evolutionary system2) leading to variable intratumor heterogeneity has been found in preinvasive lesions such as in Barrett’s esophagus3, lung adenocarcinoma in situ and atypical adenomatous hyperplasia4. Despite advances in our understanding of lung cancer adenocarcinoma, relatively little is known about the diversity of somatic alterations and the relationship, if there is, between clonal evolution and aggressiveness. Using next-generation sequencing, we conducted a study to examine the genomic characteristics in the exome of in situ to invasive lesions in order to investigate, whether mutation landscape, somatic copy number alterations and clonal evolution could provide clues in the progression from in situ to invasive lesions.
Results
Demographic and pathological review
Demographic data and pathological review are summarized in Table 1. Nineteen cases with tumor previously classified as bronchiolo-alveolar carcinoma (BAC) at all operable stages (IA-IIIA, AJCC 6th) with various outcomes were included. The median age of patients was 65.3 years (range: 54.9–74.8) and median follow-up was 38.1 months (range: 0.2–79.6). All patients underwent surgical resection (84% lobectomy) without either pre-operative chemotherapy or radiotherapy. Last follow-up date (upon December 31, 2015) revealed that 63% of patients were still alive. Upon pathological review (see Methods), five patients were reclassified into adenocarcinoma in situ (AIS) or minimally invasive adenocarcinoma (MIA). The 14 remaining patients were reclassified as invasive adenocarcinoma (8 lepidic predominate (LPA) and 6 non-lepidic predominate (non-LPA)). The LPA and non-LPA cases showed some minor heterogeneity apart from their predominant histologic pattern. Invasive acinar, solid, micropapillary and papillary minor patterns were observed in LPA group. Among the non-LPA cases, only two cases showed a solid minor pattern in addition to predominant acinar morphology (Table 1).
Somatic mutations and copy number alterations
Somatic mutations
We analyzed the exome sequencing data with target sequences of approximately 37 Mb. On average, 6.8 GB sequence read data (about 105 million reads) were generated per sample. The average depth of coverage within targets was 76X. On average, local enrichment of net covered region (protein coding) per exome was 71.3%. In these regions, 93.8% of targeted bases were retained for further analysis and covered by at least one read and 84.8% of targeted bases were covered by at least 10 reads. Using Mutect2 algorithm5 with additional filters for variants with frequency more than 0.01 in 1000 Genomes Project6 and ESP6500 database7 (NHLBI GO exome sequencing project), we detected a total of 13,316 somatic mutations (SNVs and short insertion/deletion) at a median of 15.5 Mb−1 (range: 8.6–39.5). Samples that were reclassified as AIS/MIA had a lower frequency of all somatic mutations than LPA and non-LPA samples with a median of 10.6 Mb−1 (range: 8.8–15.5), 19.5 Mb−1 (range: 8.6–39.5, Mann-Whitney’ test, p-value = 0.3) and 15.5 Mb−1 (range: 14–31.1, Mann-Whitney’ test, p-value = 0.05), respectively. The non-synonymous mutation rate had a median of 12.6 Mb−1 (range: 6.8–32.9) and showed a similar trend compared with all somatic mutations. The frequency of deleterious non-synonymous mutations per Mb, which are considered as potential driver mutations, was consistent among all subgroups (Supplementary Fig. S1). The proportions of variant type, variant classification of all somatic mutations and SNVs allelic fractions are shown in Supplementary Fig. S2.
Next, the frequency of mutation spectrum and base context surrounding mutations in all 19 samples were explored (Fig. 1a). We observed a mutation pattern with a high rate of C > T transitions (G > A, in complementary sequences), especially in the context of a TCW motif (where W corresponds to either A or T), consistent with APOBEC cytidine deaminase activity, which had been reported to play a potential role in carcinogenesis8,9 and correlated with APOBEC mutagenesis activity in many cancer types, including lung cancer adenocarcinoma10. This mutation pattern was observed similarly in AIS/MIA and LPA/non-LPA (Fig. 1b,c). NMF mutation signature analysis uncovered two mutation signatures (Supplementary Fig. S3), similar to signature 5 (common signature found in all cancer types) and signature 2 (attributed to activity of the AID/APOBEC family of cytidine deaminases) with maximal cosine similarities of 0.90 and 0.53, respectively. Despite the low cosine similarity of signature 2, individual sample analysis of APOBEC mutation fold10 (see Methods) showed a significant enrichment (q-value ≤ 0.05) in 73% of our specimens including all AIS/MIA and the majority of invasive lesions (Fig. 1d). Mutation spectrum according to smoking status revealed a similar pattern as identified in previous reports11,12. Current smoker status was associated with cytosine to adenine (C > A, or G > T in complementary sequences) transversions (Supplementary Fig. S4), however no strand bias with C > A transversions between transcribed and non-transcribed strand was observed (Supplementary Fig. S5). The frequency of total mutations tended to be higher in current smokers (n = 8) than non-smokers (n = 11), with a median of 19.1 Mb−1 (range: 8.6–36.6) and 14 Mb−1 (range: 8.8–39.5) (Mann-Whitney’ test, p-value = 0.2), respectively (Supplementary Fig. S4).

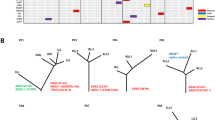
Lego plot of average mutation frequency across 19 lung adenocarcinoma specimens with prior diagnosed bronchiolo-alveolar carcinoma (a) revealed significant cluster mutation pattern of C > T, especially TCW motif. Average mutation frequency according to AIS/MIA (b) and LPA/non-LPA (c) showed a similar pattern therefore AIS/MIA had more predominant than LPA/non-LPA. APOBEC-mediated mutagenesis fold enrichment according to individual lesion was determined. P-values for significance of the APOBEC mutation pattern were corrected using the Benjamini-Hochberg method. Samples with q-value more than 0.05 were considered non-significant (d). All AIS/MIA and the majority of LPA/non-LPA cases had a significant APOBEC-mediated mutagenesis pattern. The dashed line divides the samples with q-values more than 0.5 (below the line) and samples with q-value < 0.05 (above the line).
Somatic copy number alterations (SNCAs)
We identified a total of 3,500 SCNAs at a median of 148 SCNAs per sample, which was similar across the three groups (range: 27–557) (Supplementary Fig. S6). The heat map of normalized raw segmented copy number profile is shown in Supplementary Fig. S7. Consistent with a prior large-scale copy number TCGA LUAD study13, the most common arm-level genomic alteration was gain of chromosome 5p, which occurred in 61% of cases in our study (Supplementary Table S1). We then identified focal SCNAs using GISTIC2.014 with a high amplitude threshold (0.848 and −0.737) that could identify relevant regions of focal SCNAs. GISTIC2.0 identified 16 gains and 6 losses that were contributed from three groups (Table 2) (Supplementary Fig. S6). Despite using different platforms, two focal gains overlapped with the TCGA LUAD study: chromosome 5p15.33 and chromosome 7p11.2. Gain of chromosome 5p15.33, which encodes TERT and CLPTM1L, was demonstrated as the most frequent genetic event in early stage non-small cell lung cancer15. Sequence variants of these two genes were significant susceptibility loci not only in lung cancer16,17, but also for other various types of cancer17,18,19,20. Chromosome 7p11.2 contains the epidermal growth factor receptor (EGFR) gene. Target amplification of these genes had been shown their substantial oncogenic role in a unified pan-cancer analysis across all lineages21.
We identified two novel focal gains: chromosome 12q14.1 that contains proto-oncogene AGAP2 and chromosome 16p12.1 that contained potential proto-oncogene RBBP6. AGAP2 (also known as CENTG1 or PIKE) is a proliferation activator that promotes E2F and CDK2-dependent cell cycle progression. Overexpression of AGAP2 renders cancer cells more resistant to apoptotic stimuli22. RBBP6, retinoblastoma binding protein 6, is an E3 ubiquitin ligase that promotes the degradation of p53, thereby increasing cell proliferation. Higher expression of RBBP6 in ‘formerly BAC’ than other subtypes of lung cancer had been explored, especially in the cells undergoing mitosis. This indicates specific proliferative role in a particular subtype23. Recurrent focal deletion in our study identified chromosome 13q12.11, which contains TPTE2 (also known as TPIP), which belongs to the PTEN family and is a well-known tumor suppressor gene with a potential inhibitory role in cell proliferation and inducing cell apoptosis24,25. The remaining 12 focal gains and 5 focal losses did not include any known proto-oncogene or tumor suppressor genes. The GISTIC2.0 amplification and deletion region (Supplementary Fig. 7) with known or potential proto-oncogene or tumor suppressor gene as defined by COSMIC26, CGP Census27 or elsewhere were summarized according to each significant SCNA (Table 2). All genes according to each focal event are listed in Supplementary Table S2 and Table S3.
Integrated analysis of somatic mutations and copy number variations
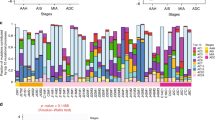
The mutated genes and related key pathways from the previous large-scale sequencing TCGA LUAD study12, including SNVs and SCNAs (Fig. 2), were analyzed in our study (see Methods). There was no apparent difference in the pattern of mutated genes or SNCAs among the three groups. RTK/RAS/RAF pathway activation (84%) was the most frequent alteration followed by various chromatin/RNA splicing factors (68%), p53 alteration (53%), PI(3)K-mTOR pathway activation (53%), alteration of oxidative stress pathway (53%) and alteration of cell cycle pathway (26%). Even though the rate of RTK/RAS/RAS pathway activation was similar to that of the TCGA LUAD study (76%), we found a different proportion of KRAS mutations and alteration. Considering only KRAS mutations which comprised 32% of TCGA lung adenocarcinomas, we found that only one case out of 19 ‘formerly BAC’ in our study was KRAS mutated (5%). A high fraction of EGFR amplification (42% in our study) was observed. A previous study showed that EGFR mutations correlated with response to receptor tyrosine kinase inhibitors and had a potential prognostic role28. The EGFR amplification was associated with EGFR mRNA expression29; however, its potential predictive ability for tyrosine kinase inhibitor response is still controversial30. The results also showed frequent amplification of RIT1 (37%), which encodes a RAS-family small GTPase with significant domain and sequence homology to KRAS, HRAS and NRAS that was recently defined as an oncogenic driver in lung adenocarcinoma to induce cellular transformation in vitro and in vivo31. RIT1 overexpression due to amplification was reported in 25% of hepatocellular carcinoma cases32. Mutations of RNA splicing factors were also frequent including U2AF1 (16%) and RBM10 (21%). The proportion of smokers was lower in our series than in the TCGA LUAD series (42% vs. 76%). After correcting for smoking status, the frequency of mutations in the key pathways (Fig. 2) was significantly different between the two series only for RNA splicing factors (37% vs. 11% TCGA LUAD, Chi-squared test, p-value = 0.0007). There was a U2AF1 S34F missense mutation (n = 2) in the zinc finger domain and R203H (n = 1) in the RS domain. The S34F mutation has been correlated with exon skipping in 3′ UTR splice site alterations such as in proto-oncogene CTNNB133. RBM10 mutation represses Notch signaling and cell proliferation through regulation of NUMB alterative splicing34. We also observed frequent deletion of tumor suppressor genes KEAP1 (47%), STK11 (32%) and SMARCA4 (53%), which are located in the same locus chr19p13.2–13.3. The SCNA correlation of these genes was also observed in the TCGA LUAD study (n = 230) (Spearman’s correlation 0.81 for KEAP1 and STK11; 0.99 for KEAP1 and SMARCA4)35.

Somatic alteration plots.
(a) of genes in multiple key pathways, including genes for lung adenocarcinoma identified in the previous TCGA large-scale sequencing study [8]. Comparative pathway alterations with the large-scale sequencing study are shown (b).
Clonality
Clonal expansion is a crucial part of cancer progression. We evaluated clonal architecture by exploring cancer cell fraction (CCF), the proportion of tumor cells harboring each somatic mutation, using PyClone. This is a Bayesian Dirichlet process clustering method that integrates mutant allele fractions, normal contamination and locus-specific copy number (see Methods). Distribution of CCF per sample was evenly distributed in all synonymous, non-synonymous and deleterious non-synonymous mutations (Supplementary Fig. S8). The median CCF of AIS/MIA, LPA and non-LPA were 0.96, 0.58 and 0.61 respectively. Consistent with the density plot of CCF in all somatic mutations (Fig. 3b), a heat map representing a group of deleterious non-synonymous mutations within different clonal frequencies revealed a distinct pattern between AIS/MIA and LPA/non-LPA cases (Fig. 3a). A higher number of clones per tumor in LPA/non-LPA (median 2, range 1–22) than AIS/MIA (median 1, range 1–5) was also observed. We also validated clonal architecture by CCF in 20 TCGA ‘formerly BAC’ with invasive feature36; 13 LPA and 7 non-LPA (Supplementary Table S4). The density plot of CCF in all somatic mutations revealed a broad range of CCF in both TCGA LPA and TCGA non-LPA (supplementary Fig S9). LPA/non-LPA (invasive lesions) had a wide distribution of CCF suggesting more complex clonal branching at the time of diagnosis than AIS/MIA.

Cancer cell fraction (CCF) of top deleterious non-synonymous mutations (55 genes).
(a) Revealed a pattern of clonal frequency among the three groups. More diverse clonal frequency was found in both adenocarcinoma with predominant lepidic pattern (LPA) and adenocarcinoma with predominant histologic subtype other than lepidic pattern (non-LPA) while homogeneous clonal frequency was found in AIS/MIA. (b) Density plot of posterior probability distribution of CCF of all mutations according to group revealed the same finding as deleterious non-synonymous mutations.
We explored the impact of histological heterogeneity on clonality by comparing LPA and non-LPA cases with none or one invasive pattern in addition to the lepidic pattern, (n = 8) to these with more than one additional invasive pattern (n = 6). The frequencies of SNVs per Mb and copy number variations were not significantly different between the two groups (Mann-Whitney’ test, p-value = 0.6 and 0.1, respectively). The density plot of CCF for LPA and non-LPA by the number of invasive morphologic patterns also showed similar profiles (Supplementary Fig. S9). Finally, significantly mutated genes and related pathways from Fig. 3 were analyzed for significant deviation from the distribution of overall clonal frequency. Only mutations in EGFR and RBM10 had a borderline significantly skewed distribution (q-value = 0.05) towards higher clonal frequencies (median CCF for EGFR and RBM10 were 0.98 and 0.96, respectively). Ubiquitous mutations, these having high clonal frequency, likely reflect the processes involved in tumorigenesis and early development.
Discussion
Genomic characteristics of ‘formerly BAC’ could represent overall variation of LUAD. No specific pattern of mutation landscape or somatic copy number alteration was detected that could discriminate AIS/MIA from LPA/non-LPA. We noticed several characteristics among ‘formerly BAC’ cases from the previous large-scale sequencing LUAD study12,29 or LUAD as part of a pan-cancer study10. First, we observed slightly higher numbers of tumors with significant APOBEC mutation fold enrichment in ‘formerly BAC’ (73% vs. 58% TCGA LUAD study). The proportion of deleterious non-synonymous mutations containing APOBEC mutation pattern was not different between AIS/MIA and LPA (Mann-Whitney’ test, p-value = 0.2) nor between AIS/MIA and non-LPA (p-value = 0.9). Second, the majority (77%) of significant focal events did not include any known or potential proto-oncogene or tumor suppressor gene. Using Reactome pathway-based analysis, we could identify the significant biological theme of the genes on focal event gains. The overrepresented pathways for focal gains (false discovery rate <0.001) (Supplementary Table S5) were mostly related to the adaptive immune system. HLA class II histocompatibility antigen genes HLA-DQA1, HLA-DQA2, HLA-DQB1 and HLA-DRB1, which are located on chromosome 6p21.32, were the important identifiers. Third, although the RTK/RAS/RAF pathway was the significant pathway in ‘formerly BAC’ cases, KRAS mutations had a lower frequency (5% vs. 32% TCGA LUAD) which might be due to the lower proportion of smoker patients in our series, while predominant mutations of RNA-splicing genes, RBM10 and U2AF1, were found (37% vs. 11% TCGA LUAD). A high proportion of SMARCA4, KEAP1 and STK11 deletions were observed in our study compared with the TCGA LUAD study. This could be due to the ReCapSeg pipeline that could not well discriminate deep deletion which represented homozygous deletions from heterozygous deletions, as acknowledged by TCGA LUAD study35. The frequency of pathway alterations that contained gene deletions, such as oxidative stress pathway, PI(3)K-mTOR and various chromatic and RNA splicing factors, were affected by this limitation. There was no apparent difference in mutation landscape or somatic alterations between AIS/MIA and LPA/non-LPA.
CCF was the only distinctive feature to distinguish AIS/MIA from LPA/non-LPA. Branched clonal evolution could be evaluated ideally in either multiple single-cell37 or multi-section mutation analysis38,39. However, evaluation of CCF in a single DNA sample per tumor is more feasible in clinical practice and warrants further evaluation. High clonal diversity was correlated with more incidence of progression from Barrett’s esophagus to esophageal adenocarcinoma3. A larger distribution of clonal frequency in LPA/non-LPA than AIS/MIA cases signified more clonal evolution of LPA/non-LPA than AIS/MIA at the time of diagnosis. This finding showed a consistent pattern for overall mutations and for, deleterious non-synonymous mutations alone and did not depend on the number of invasive histological patterns observed in the tissue samples. Unfortunately the computerized tomography (CT) imaging which has been implemented to augment lung adenocarcinoma classification, in addition to the histological patterns, was not available for our study. Progression of cancer through accumulation of somatic alterations had broad characteristic features, from extensive clonal genetic similarity to share few genetic alterations with primary tumor40. Subclonal genetic diversity might be an indicator of success or failure of treatment, especially if there are specific genomic profiles of sensitivity or resistance to therapeutic targets. Several studies have provided some initial supporting evidence; for example, high subclonal mutations in primary lung adenocarcinoma patients were correlated with high relapse rate39 and genomic characteristics of metastatic/relapsed lesions that have a similar patterns as subclones of primary tumors40,41. Recently, pan-cancer analysis revealed the increased risk of mortality associated with increasing number of clones up to four clones but declined risk at more than four clones, questioning trade-off risk and benefit of tumor heterogeneity42. Assessment of clonal frequency at the time of diagnosis might provide important information for individualized therapy as a prognostic factor however more studies are required.
In conclusion, more branched clonal evolution was the only significant characteristics of aggressiveness from in situ to invasive lesion, while the somatic mutation landscape or somatic alterations did not reveal any significant pattern. Our finding emphasized the importance of clonal assessment at the time of diagnosis.
Methods
Sample selection
Nineteen fresh-frozen lung cancer and matched blood specimens were obtained from cases, previously diagnosed as bronchio-alveolar carcinoma based on local routine protocol, that were recruited from the Moscow City Clinical Oncology Dispensary serving Moscow and the surrounding regions. The study was conducted according to the recommendations of the Declaration of Helsinki. Informed consent was obtained from all cases and the study was approved by the Ethics Committees of the International Agency for Research on Cancer and local institute review board (IRB). Fresh-frozen samples were obtained at the time of surgery with the diagnosis of BAC lung cancer and without neoadjuvant chemo- or radiotherapy. An independent pathological review was performed by B.A.A (prior to any data analysis) based on tissue sections prepared from all fresh-frozen specimens included in the study, as well as slides from a representative formalin-fixed paraffin-embedded (FFPE) block for six cases. Based on IASLC/ATS/ERS 2011 classification1, we categorized the ‘formerly BAC’ cases into three categories. Examples of H&E stained tissue section are provided in Supplementary Fig. S10.
Group 1
adenocarcinoma in situ (AIS) or minimally invasive adenocarcinoma (MIA); (a) AIS was defined as a small solitary adenocarcinoma (≤3 cm) with pure lepidic pattern; restricted along pre-existing alveolar structure, lacking stromal, vascular or pleural invasion and with no papillary or micropapillary patterns. AIS was defined as the second preinvasive lesion of lung adenocarcinoma after adenomatous hyperplasia; (b) Minimally invasive adenocarcinoma (MIA) was defined as a small solitary adenocarcinoma (≤3 cm) with predominant lepidic pattern with ≤5 mm invasion. MIA lacks lymphatic, blood vessel or pleural invasion and tumor necrosis.
Group 2
lepidic predominant adenocarcinoma (LPA); LPA was defined as non-mucinous invasive adenocarcinomas with a predominant lepidic growth but with at least one of the following: focus of invasion (non-lepidic) measuring >5 mm, gross tumor size >3 cm, invasion of lymphatic, blood vessels or pleura, or tumor necrosis.
Group 3
invasive mucinous adenocarcinoma and other; this group includes predominant histologic subtype other than lepidic pattern (non-LPA).
AIS and MIA may be separated from the bulk of T1 tumors and regarded as Tis and Tmi in term of subsequent TNM edition. Therefore in this study, we used the 6th AJCC staging system at the diagnostic period. It should be noted that this classification relied on the pathological review of a single tissue section and we could not exclude the presence of invasion elsewhere in the tumor. Clinical and pathological information was obtained from medical records, including clinical and pathological stages, tumor size, histological type and treatment. Follow-up data was collected from local coordinators from medical records, cancer registry, death certification or active follow-up with the provider physician.
Previously diagnosed ‘formerly BAC’ criterion had been used to select cases form TCGA LUAD clinical data36. We retrieved both diagnosis images (used by the hospital to diagnose participants) and tissue images (used for TCGA analyses) in SVS. format and had been reviewed by co-author B.A.A using Aperio ImageScope43. The twenty ‘formerly BAC’ with invasive features cases tumor-normal pairs’ bam files were retrieved to validate clonality analysis via http://cghub.ucsc.edu [10/06/2016 accessed].
Somatic SNVs, short insertion and deletion variant detection
The preparation of library and exome sequencing and data processing are described in the supplementary information. High confidence somatic substitutions were identified and restricted to just the capture targets (expected exome sequence coverage) from SOLiD® which included 37,256,743 bases (1.29% whole genome sequences, total 195,282 regions). We applied MuTect2 algorithm5 ( version) which has shown ability to detect low allele fractions and is as such suitable for calling somatic SNVs and indels in impure and heterogeneous tumor materials. Additional filters for variants with frequency more than 0.01 in 1000 Genomes Project6 and ESP6500 database (NHLBI GO exome sequencing project) were applied7. All variants were annotated using ANNOVAR v.2015-6-17 (ref. 37)44 and Oncotator v.1.545. Significant genes, previously identified by a large-scale lung cancer sequencing study (q-value < 0.05)12, published MutSig gene analyses across 21 tumor types46 and cosmic gene census v.7047 variants were retrieved from our data. We defined a gene to be mutated if at least one non-synonymous mutation was found in the coding sequence of that gene and were subsequently determined the amino acid substitution and functional prediction scores (SIFT, polyphen2, LRT and mutation Taster) implemented with dbNSFP v.2.648. Variant mutations were scored as ‘deleterious’ when at least two out of the four predictors classified the mutation as deleterious.
version) which has shown ability to detect low allele fractions and is as such suitable for calling somatic SNVs and indels in impure and heterogeneous tumor materials. Additional filters for variants with frequency more than 0.01 in 1000 Genomes Project6 and ESP6500 database (NHLBI GO exome sequencing project) were applied7. All variants were annotated using ANNOVAR v.2015-6-17 (ref. 37)44 and Oncotator v.1.545. Significant genes, previously identified by a large-scale lung cancer sequencing study (q-value < 0.05)12, published MutSig gene analyses across 21 tumor types46 and cosmic gene census v.7047 variants were retrieved from our data. We defined a gene to be mutated if at least one non-synonymous mutation was found in the coding sequence of that gene and were subsequently determined the amino acid substitution and functional prediction scores (SIFT, polyphen2, LRT and mutation Taster) implemented with dbNSFP v.2.648. Variant mutations were scored as ‘deleterious’ when at least two out of the four predictors classified the mutation as deleterious.
Somatic copy-number alterations
Genome-wide copy-ratio profiles were inferred using ReCapSeg49. Depth of coverage at capture targets in tumor samples was normalized by read coverage spanning a target segment with the total number of aligned reads and proportionally calibrated to estimate the copy ratio using depths observed in a panel of normal (non-cancer) diploid genomes. The resulting copy-ratio profiles were normalized to decrease noise and then segmented using the circular binary segmentation (CBS) algorithm50. All segments were called using ReCapSeg Caller with default parameters and classified as amplified, deleted or copy neutral. Significant recurrent somatic copy-number alteration (SCNA) was conducted using GISTIC2.0, a method that identifies likely driver SCNAs by evaluating the frequency and amplitude of observed events14. We used threshold approaches: a board length cutoff of 0.5 chromosome arms, a confidence interval of 95%, a copy-ratio cap of 1.5 and noise thresholds at 0.848 and −0.737 for detection of high amplification and deep deletion, respectively. Biological pathways of genes located on significant focal event gains or losses that do not include known/potential proto-oncogenes or tumor suppressor genes were identified using pathway enrichment analysis from ReactomePA/R package51. Reactome, a manually curated resource that describes chemical reactions, biological processes and pathway database that links proteins to molecular functions, was used to identify significantly altered pathways52.
Mutation signature analysis
We used nonnegative matrix factorization (NMF) as the mathematical approach to decompose a complex multi-dimensional data set to define underlying signatures by NMF/R package53. In brief, all mutation data were converted into a matrix W that was made up of 96 features of single-based substitutions comprising mutation counts for each mutation type (C > A, C > G, C > T, T > A, T > C and T > G; somatic mutations presented in a pyrimidine context) using each possible 5′ and 3′ (C, A, G, T) context defined by the original data. The contribution of each sample to the signatures was defined as Matrix H. The main approach to NMF estimated matrices W and H as a local minimum of the following optimization problem:

The algorithm of Kullback-Leibler divergence penalty was applied to the matrix, where D was a loss function that measures the quality of the approximation. R was an optional regularization function to enforce desirable properties on matrices W and H. The estimated numbers of signatures were based on Brunet’s algorithm, deciphering the minimal set of mutation signatures that optimally explained the proportion of each mutation type. After extraction, the unknown signatures (signature A, B) were compared to the catalog of 30 consensus signatures published in the COSMIC database v.7254 using the cosine similarity method as previously described55. The pattern of mutations of signature B showed a maximal cosine similarity of 0.90 with the pattern of mutations of signature 5, which is the common signature found in all cancer types. The pattern of mutations of signature A showed a maximal cosine similarity of 0.53 with the pattern of mutation of signature 2. This pattern has been attributed to activity of the AID/APOBEC family of cytidine deaminases.
Detection of an APOBEC mutation pattern
We checked for the frequency of an APOBEC mutation pattern using a previously described method that demonstrated effective correlation between exome and genome sequencing data10. The enrichment ETCW characterizing the strength of mutagenesis at TCW motif in mutation clusters was calculated as

where mutationTCW is the number of mutated cytosines (and guanines) falling in a TCW (or WGA) motif, mutationC(or G) is the total number of mutated cytosines (or guanines), contextTCW is the total number of TCW (or WGA) motif within a 41-nucleotides region centered on the mutated cytosines (and guanines) and contextC(or G) is the total number of cytosines (or guanines) within the 41-nucleotide region centered on the mutated cytosines (or guanines). Only specific base substitutions were included (TCW to TTW or TGW, WGA to WAA or WCA, C to T or G and G to A or C). Over-representation of APOBEC signature mutation in each sample was analyzed using a one-sided Fisher’s exact test comparing the ratio of the number of cytosine-to-thymine or cytosine-to-guanine substitutions and guanine-to-adenine or guanine-to-cytosine substitutes that occurred in and out of the APOBEC target motif (TCW or WGA) to an analogous ratio for all cytosines and guanines that reside inside and outside of TCW or WGA motif. P-values were corrected using Benjamin-Hochberg multiple testing correction. A threshold of q-value < 0.05 was used to define the significant level of the APOBEC signature.
Clonality analysis
Tumor purity and allelic copy number, as a pre-requisite for CCF (clonal cell fraction) estimation, were analyzed using Sequenza (v2.1.1), which had shown good correlation with the SNP array-based ASCAT algorithm56. We generated input mpileup format from SAMtools v.0.1.1857 for tumor and matched normal tissues with minimum coverage requirement and restricted to the capture targets. The algorithm uses both binned coverage ratio data and allelic ratio as input and computes the sufficient sequencing depth (phred base quality > 20). The purity and allelic copy number were analyzed based on a probabilistic model, using maximum a posteriori estimation applied to segmented data.
Estimation of CCF was performed using PyClone v.0.12.7, using the Bayesian model and Dirichlet process clustering method for grouping sets and estimating cellular prevalence58. For each tumor, PyClone was run on all somatic mutations using the parental copy number method and pyclone binomial density. The Markov chain Monte Carlo (MCMC) step of PyClone was run for 10,000 iterations with burn-in and thinning parameters set to 1,000 and 10, respectively, resulting in 9000 independent samples from the posterior distribution of CCF per mutation. Otherwise, default options for PyClone were used.
Significantly mutated genes and related pathways were tested for skewing of clonal frequency by Wilcoxon test. Clonal frequency estimation was adjusted by maximum clonal frequency value to remove the effect of normal cell contamination. The adjusted clonal frequencies were tested as to whether the distributions of the mutations were different from than the background distribution of all clonal frequency estimates using Wilcoxon test (R package). Resultant p-values were then adjusted using Benjamini & Hochberg method.
Additional Information
How to cite this article: Vinayanuwattikun, C. et al. Elucidating Genomic Characteristics of Lung Cancer Progression from In Situ to Invasive Adenocarcinoma. Sci. Rep. 6, 31628; doi: 10.1038/srep31628 (2016).
References
Witt, C. European respiratory society/american thoracic society/international association for the study of lung cancer international multidisciplinary classification of lung adenocarcinoma: state of the art. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer 6, 1451, doi: 10.1097/JTO.0b013e318224643b (2011).
Greaves, M. & Maley, C. C. Clonal evolution in cancer. Nature 481, 306–313, doi: 10.1038/nature10762 (2012).
Maley, C. C. et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nature genetics 38, 468–473, doi: 10.1038/ng1768 (2006).
Izumchenko, E. et al. Targeted sequencing reveals clonal genetic changes in the progression of early lung neoplasms and paired circulating DNA. Nature communications 6, 8258, doi: 10.1038/ncomms9258 (2015).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology 31, 213–219, doi: 10.1038/nbt.2514 (2013).
Genomes Project, C. et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65, doi: 10.1038/nature11632 (2012).
Exome Variant Server NHLBI GO Exome Sequencing Project (ESP) Seattle, WA (URL: http://evs.gs.washington.edu/EVS/) [22/12/14 accessed].
Suspene, R. et al. Somatic hypermutation of human mitochondrial and nuclear DNA by APOBEC3 cytidine deaminases, a pathway for DNA catabolism. Proceedings of the National Academy of Sciences of the United States of America 108, 4858–4863, doi: 10.1073/pnas.1009687108 (2011).
Shinohara, M. et al. APOBEC3B can impair genomic stability by inducing base substitutions in genomic DNA in human cells. Scientific reports 2, 806, doi: 10.1038/srep00806 (2012).
Roberts, S. A. et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nature genetics 45, 970–976, doi: 10.1038/ng.2702 (2013).
Imielinski, M. et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150, 1107–1120, doi: 10.1016/j.cell.2012.08.029 (2012).
Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature511, 543-550, doi: 10.1038/nature13385 (2014).
Weir, B. A. et al. Characterizing the cancer genome in lung adenocarcinoma. Nature 450, 893–898, doi: 10.1038/nature06358 (2007).
Mermel, C. H. et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome biology 12, R41, doi: 10.1186/gb-2011-12-4-r41 (2011).
Kang, J. U., Koo, S. H., Kwon, K. C., Park, J. W. & Kim, J. M. Gain at chromosomal region 5p15.33, containing TERT, is the most frequent genetic event in early stages of non-small cell lung cancer. Cancer genetics and cytogenetics 182, 1–11, doi: 10.1016/j.cancergencyto.2007.12.004 (2008).
McKay, J. D. et al. Lung cancer susceptibility locus at 5p15.33. Nature genetics 40, 1404–1406, doi: 10.1038/ng.254 (2008).
Wang, Z. et al. Imputation and subset-based association analysis across different cancer types identifies multiple independent risk loci in the TERT-CLPTM1L region on chromosome 5p15.33. Human molecular genetics 23, 6616–6633, doi: 10.1093/hmg/ddu363 (2014).
Haiman, C. A. et al. A common variant at the TERT-CLPTM1L locus is associated with estrogen receptor-negative breast cancer. Nature genetics 43, 1210–1214, doi: 10.1038/ng.985 (2011).
Kote-Jarai, Z. et al. Seven prostate cancer susceptibility loci identified by a multi-stage genome-wide association study. Nature genetics 43, 785–791, doi: 10.1038/ng.882 (2011).
Petersen, G. M. et al. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nature genetics 42, 224–228, doi: 10.1038/ng.522 (2010).
Zack, T. I. et al. Pan-cancer patterns of somatic copy number alteration. Nature genetics 45, 1134–1140, doi: 10.1038/ng.2760 (2013).
Qi, Q. & Ye, K. The roles of PIKE in tumorigenesis. Acta pharmacologica Sinica 34, 991–997, doi: 10.1038/aps.2013.71 (2013).
Motadi, L. R., Bhoola, K. D. & Dlamini, Z. Expression and function of retinoblastoma binding protein 6 (RBBP6) in human lung cancer. Immunobiology 216, 1065–1073, doi: 10.1016/j.imbio.2011.05.004 (2011).
Mishra, R. R., Chaudhary, J. K. & Rath, P. C. Cell cycle arrest and apoptosis by expression of a novel TPIP (TPIP-C2) cDNA encoding a C2-domain in HEK-293 cells. Molecular biology reports 39, 7389–7402, doi: 10.1007/s11033-012-1571-6 (2012).
Mishra, R. R., Chaudhary, J. K., Bajaj, G. D. & Rath, P. C. A novel human TPIP splice-variant (TPIP-C2) mRNA, expressed in human and mouse tissues, strongly inhibits cell growth in HeLa cells. PloS one 6, e28433, doi: 10.1371/journal.pone.0028433 (2011).
Bamford, S. et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. British journal of cancer 91, 355–358, doi: 10.1038/sj.bjc.6601894 (2004).
Futreal, P. A. et al. A census of human cancer genes. Nature reviews. Cancer 4, 177–183, doi: 10.1038/nrc1299 (2004).
Sholl, L. M. et al. Lung adenocarcinoma with EGFR amplification has distinct clinicopathologic and molecular features in never-smokers. Cancer research 69, 8341–8348, doi: 10.1158/0008-5472.CAN-09-2477 (2009).
Ding, L. et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–1075, doi: 10.1038/nature07423 (2008).
Zhang, X. & Chang, A. Molecular predictors of EGFR-TKI sensitivity in advanced non-small cell lung cancer. International journal of medical sciences 5, 209–217 (2008).
Berger, A. H. et al. Oncogenic RIT1 mutations in lung adenocarcinoma. Oncogene 33, 4418–4423, doi: 10.1038/onc.2013.581 (2014).
Li, J. T. et al. [Mutation and amplification of RIT1 gene in hepatocellular carcinoma]. Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical genetics 21, 43–46 (2004).
Brooks, A. N. et al. A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PloS one 9, e87361, doi: 10.1371/journal.pone.0087361 (2014).
Hernandez, J. et al. Tumor suppressor properties of the splicing regulatory factor RBM10. RNA biology, 0, doi: 10.1080/15476286.2016.1144004 (2016).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling 6, pl1, doi: 10.1126/scisignal.2004088 (2013).
National Research Institute (NCI) and National Human Genome Research Institute (NHGRI), The Cancer Genome Atlas (TCGA) URL; http://tcga-data.nci.nih.gov/tcga/ [03/03/2015 accessed].
Navin, N. et al. Tumour evolution inferred by single-cell sequencing. Nature 472, 90–94, doi: 10.1038/nature09807 (2011).
de Bruin, E. C. et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 346, 251–256, doi: 10.1126/science.1253462 (2014).
Zhang, J. et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 346, 256–259, doi: 10.1126/science.1256930 (2014).
Kim, H. et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome research 25, 316–327, doi: 10.1101/gr.180612.114 (2015).
Eleveld, T. F. et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nature genetics 47, 864–871, doi: 10.1038/ng.3333 (2015).
Andor, N. et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nature medicine 22, 105–113, doi: 10.1038/nm.3984 (2016).
Leica Biosystems, Aperio ImageScope URL; www.leicabiosystems.com/digital-pathology/digital-pathology-management/imagescope/ [03/03/2015 accessed].
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research 38, e164, doi: 10.1093/nar/gkq603 (2010).
Ramos, A. H. et al. Oncotator: cancer variant annotation tool. Human mutation 36, E2423–2429, doi: 10.1002/humu.22771 (2015).
Lawrence, M. S. et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501, doi: 10.1038/nature12912 (2014).
Forbes, S. A. et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic acids research 39, D945–950, doi: 10.1093/nar/gkq929 (2011).
Liu, X., Jian, X. & Boerwinkle, E. dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Human mutation 34, E2393–2402, doi: 10.1002/humu.22376 (2013).
Broad Institute, MA, ReCapSeg (URL: http://gatkforums.broadinstitute.org/gatk/discussion/5640/recapseg-overview#latest) [23/01/2016 accessed].
Venkatraman, E. S. & Olshen, A. B. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics 23, 657–663, doi: 10.1093/bioinformatics/btl646 (2007).
Yu, G. & He, Q. Y. ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Molecular bioSystems 12, 477–479, doi: 10.1039/c5mb00663e (2016).
Fabregat, A. et al. The Reactome pathway Knowledgebase. Nucleic acids research 44, D481–487, doi: 10.1093/nar/gkv1351 (2016).
Gaujoux, R. & Seoighe, C. A flexible R package for nonnegative matrix factorization. BMC bioinformatics 11, 367, doi: 10.1186/1471-2105-11-367 (2010).
Wellcome Trust Sanger Institute, COSMIC Catalogue of somatic mutations in cancer http://cancer.sanger.ac.uk/cosmic/signatures [31/03/2015 accessed].
Alexandrov, L. B., Nik-Zainal, S., Wedge, D. C., Campbell, P. J. & Stratton, M. R. Deciphering signatures of mutational processes operative in human cancer. Cell reports 3, 246–259, doi: 10.1016/j.celrep.2012.12.008 (2013).
Favero, F. et al. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO 26, 64–70, doi: 10.1093/annonc/mdu479 (2015).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993, doi: 10.1093/bioinformatics/btr509 (2011).
Roth, A. et al. PyClone: statistical inference of clonal population structure in cancer. Nature methods 11, 396–398, doi: 10.1038/nmeth.2883 (2014).
Acknowledgements
We are indebted to the patients included in this study who donated tumor and blood specimens. We acknowledge the help of Peng Li, Matthieu Foll and Chatchawit Aporntewan for computational technical support and Estelle Chanudet-van den Brink for methodological support. The work carried out by CV was undertaken during the tenure of a postdoctoral fellowship from the International Agency for Research on Cancer, partially supported by the European Commission FP7 Marie Curie Actions-people-Co-funding of regional, national and international programs (COFUND).
Author information
Authors and Affiliations
Contributions
J.M., P.B. and G.S. directed the study. G.S., F.L.C. and C.V. conceived the study, A.M. and D.Z. recruited study participant and provided clinical data, M.V., C.Vo., G.B. and C.V. performed bioinformatics analysis, C.V. was responsible for integrity of the data, interpretation and drafting of the manuscript. B.A.A. conducted pathological review of specimens. D.P., N.F. and G.D. optimized exome sequencing protocols, prepared samples and performed experiments. All authors read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/ http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Vinayanuwattikun, C., Le Calvez-Kelm, F., Abedi-Ardekani, B. et al. Elucidating Genomic Characteristics of Lung Cancer Progression from In Situ to Invasive Adenocarcinoma. Sci Rep 6, 31628 (2016). https://doi.org/10.1038/srep31628
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep31628
This article is cited by
-
Candidate mechanisms of acquired resistance to first-line osimertinib in EGFR-mutated advanced non-small cell lung cancer
Nature Communications (2023)
-
Gene expression profiling using targeted RNA-sequencing to elucidate the progression from histologically normal lung tissues to non-invasive lesions in invasive lung adenocarcinoma
Virchows Archiv (2022)
-
Ambrisentan, an endothelin receptor type A-selective antagonist, inhibits cancer cell migration, invasion, and metastasis
Scientific Reports (2020)
-
Genomic and immune profiling of pre-invasive lung adenocarcinoma
Nature Communications (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
