
Abstract
Among the diverse cytokines involved in osteoclast differentiation, interleukin (IL)-3 inhibits RANKL-induced osteoclastogenesis. However, the mechanism underlying IL-3-mediated inhibition of osteoclast differentiation is not fully understood. Here we demonstrate that the activation of signal transducers and activators of transcription 5 (STAT5) by IL-3 inhibits RANKL-induced osteoclastogenesis through the induction of the expression of Id genes. We found that STAT5 overexpression inhibited RANKL-induced osteoclastogenesis. However, RANKL did not regulate the expression or activation of STAT5 during osteoclast differentiation. STAT5 deficiency prevented IL-3-mediated inhibition of osteoclastogenesis, suggesting a key role of STAT5 in IL-3-mediated inhibition of osteoclast differentiation. In addition, IL-3-induced STAT5 activation upregulated the expression of Id1 and Id2, which are negative regulators of osteoclastogenesis. Overexpression of ID1 or ID2 in STAT5-deficient cells reversed osteoclast development recovered from IL-3-mediated inhibition. Importantly, microcomputed tomography and histomorphometric analysis revealed that STAT5 conditional knockout mice showed reduced bone mass, with an increased number of osteoclasts. Furthermore, IL-3 inhibited RANKL-induced osteoclast differentiation less effectively in the STAT5 conditional knockout mice than in the wild-type mice after RANKL injection. Taken together, our findings indicate that STAT5 contributes to the remarkable IL-3-mediated inhibition of RANKL-induced osteoclastogenesis by activating Id genes and their associated pathways.
Similar content being viewed by others


Introduction
Proper development and activity of osteoblasts and osteoclasts, which is responsible for bone formation and resorption, respectively, is crucial for healthy bone homeostasis. An imbalance between the numbers of the two cell types causes pathological problems such as osteopetrosis or osteoporosis, and many studies have aimed to understand the molecular mechanisms underlying osteoblast/osteoclast development and function.
Osteoblasts originate from mesenchymal stem cells, whereas osteoclasts differentiate from monocyte/macrophage lineage cells originating from hematopoietic stem cells. In in vitro culture systems, macrophage-colony stimulating factor (M-CSF) and receptor-activated nuclear factor kappa-B ligand (RANKL) can sufficiently aid in the development of osteoclasts1. In vivo studies revealed that RANKL overexpression causes osteoporosis. Conversely, RANKL knockout mice showed a striking osteopetrotic phenotype in vivo. Upon RANKL stimulation, RANK (RANKL receptor) recruits the adaptor protein TRAF6 and signal cascades are activated via the activation of signaling molecules such as ERK, JNK, P38 MAPK, and NF-κB2,3. Furthermore, RANKL increases the expression of the early osteoclastic gene, c-Fos, which in turn upregulates a master regulator of osteoclastogenesis, NFATc14. NFATc1 can cooperate with other transcription factors such as PU.1 and Mitf, resulting in the induction of the expression of osteoclastic genes including Oscar, Acp5, Ctsk, and Atp6v0d25,6,7.
Osteoclasts have been reported to be closely related to the immune system7. Osteoclast differentiation and function can be affected not only directly by immune cells but also indirectly via many cytokines produced by immune cells or other cell types. Pro-resorptive cytokines such as interleukin-1 (IL-1), TNF-α, and IL-17 strongly induce osteoclast formation and function. T cells produce various cytokines including interferon (IFN)-γ, IL-3, IL-4, and IL-10, which exert potent inhibitory effects on osteoclast differentiation8. IL-3, a cytokine secreted predominantly by activated T lymphocytes, serves as a link between the immune system and the hematopoietic system. Although studies using either organ culture or whole bone marrow cultures have revealed IL-3 to be a stimulator of osteoclastogenesis in vitro9,10, recent studies have shown that IL-3 irreversibly inhibits RANKL-induced osteoclast differentiation by downregulation of c-Fos expression, prevention of NF-κB signaling, and inhibition of RANK expression11,12. In addition, inhibitors of differentiation and DNA binding (Ids) are involved in the inhibition of RANKL-induced osteoclast differentiation and are known to be induced by IL-3. Thus, IL-3 negatively regulates osteoclast differentiation by regulating Ids and c-Fos expression13,14.
Signal transducers and activators of transcription (STATs) are a family of latent cytoplasmic proteins that are activated to participate in gene control when cells encounter various cytokines15. Seven known mammalian STAT proteins such as STAT1, 2, 3, 4, 5A, 5B, and 6 exist. STATs are activated by tyrosine phosphorylation following the binding of specific ligands to cognate receptors, leading to dimerization and subsequent translocation of the STATs to the nucleus15. Activated STATs bind to specific DNA motifs in regulatory regions and thereby control the transcription of genes that regulate cell proliferation, differentiation, apoptosis, and immune responses.
Two members of the STAT family, STAT5A and STAT5B (collectively called STAT5), have gained prominence owing to the fact that they are activated by a wide variety of cytokines such as IL-3 and granulocyte-macrophage colony-stimulating factor (GM-CSF), which are known to play important roles in the growth and differentiation of hematopoietic precursors16,17,18. Emerging evidence suggests that the STAT signaling pathway plays an important role in bone development and metabolism19. Recently, it was demonstrated that STAT5 negatively regulates bone resorption of osteoclasts by inducing Dusp gene expression20. However, although STAT5 has been shown to play a role in bone metabolism, the contribution of STAT5 to osteoclast differentiation has not yet been revealed.
In this study, the role of STAT5 in osteoclast differentiation and the underlying mechanism was investigated. The effect of IL-3 on osteoclast development were investigated in Mx1-Cre transgenic mice in which floxed Stat5 was specifically deleted. We discovered that the inhibitory effect of IL-3 on RANKL-induced osteoclastogenesis was dependent on STAT5 activation. In addition, STAT5 activation induced Id gene expression, resulting in the inhibition of osteoclast differentiation. Thus, we concluded that STAT5 inhibits osteoclast differentiation by controlling negative regulators upon IL-3 stimulation.
Results
STAT5 activation attenuates RANKL-induced osteoclast differentiation
To understand the role of STAT5 in osteoclast differentiation, we initially examined Stat5 levels during RANKL-induced osteoclastogenesis using RT-PCR. When bone marrow-derived macrophage-like cells (BMMs) were cultured with M-CSF and RANKL, the expression of NFATc1, a master transcription factor for osteoclast differentiation, strongly increased. The mRNA expression of Stat5a and Stat5b was observed throughout osteoclastogenesis (Fig. 1A). Next, the effect of STAT5 on osteoclast differentiation was examined by overexpressing a constitutively active form of STAT5A (STAT5A1*6) in osteoclast precursors. STAT5A1*6 overexpression strongly attenuated RANKL-induced osteoclastogenesis (Fig. 1B) and resulted in a significant reduction in the number of TRAP-positive multinucleated cells (Fig. 1C). Consistently, the expression of RANKL-inducible osteoclastic genes including c-fos, Nfatc1, Acp5, and Oscar was significantly reduced by ectopic STAT5A1*6 expression (Fig. 1D). Correspondingly, c-Fos and NFATc1 protein levels were also decreased (Fig. 1E). Moreover, activated STAT5 attenuated pERK and p38 levels in the early stages of RANKL-induced osteoclast differentiation (Fig. 1F). These results suggest that active STAT5A inhibits osteoclastogenesis, which is accompanied by the reduced expression of genes associated with osteoclast differentiation.

(A) BMMs were cultured with M-CSF and RANKL for the indicated times. Total RNA was collected at each time point. RT-PCR was performed to detect expression of the indicated genes. All gels run under the same experimental conditions and the representative images were cropped and shown. (B,C) BMMs were transduced with pMX-IRES-EGFP (control) or constitutively active STAT5A (STAT5A1*6) retrovirus and cultured with M-CSF in the presence or absence of RANKL for three days. (B) Cultured cells were stained for TRAP. (C) The number of TRAP-positive MNCs per well was counted. Data are represented as the mean ± SD. ***P < 0.001 vs. control; n = 4. (D–F) BMMs were transduced with pMX-IRES-EGFP (control) or STAT5A1*6 retrovirus and cultured with M-CSF and RANKL for the indicated times. (D) mRNA levels of c-fos, Nfatc1, Acp5 and Oscar were assessed by quantitative real-time PCR. Data represent mean ± SD of triplicate samples. **P < 0.01; ***P < 0.001 vs. control. (E,F) Whole cell lysates were harvested from cultured cells and were immunoblotted with the indicated antibodies. All gels run under the same experimental conditions and the representative images were cropped and shown. Bar: 100 μm. All results are representative of at least three independent experiments. Statistical analyses were implemented in TTEST.
RANKL-induced osteoclast differentiation is not affected by STAT5 deficiency
Osteoclast differentiation in the absence of STAT5 was examined in cells obtained from MX1-Cre recombinase-mediated Stat5 conditional knockout (cKO) mice, targeting both STAT5 isoforms in osteoclasts. We first confirmed the almost complete absence of Stat5a and Stat5b mRNA and STAT5 protein in the BMMs (Fig. 2A). Unexpectedly, upon differentiation from BMMs lacking STAT5, osteoclast formation was unimpaired and comparable with that observed with Stat5fl/fl cells (Fig. 2B,C). Furthermore, the mRNA levels of c-fos, Nfatc1, Acp5, and Oscar were not altered by the absence of STAT5 (Fig. 2D). In addition, c-Fos and NFATc1 protein levels were comparable between the Stat5fl/fl and Stat5 cKO samples (Fig. 2E). These data suggested the possibility that RANKL signaling does not regulate STAT5 expression. We therefore examined STAT5 phosphorylation upon RANKL stimulation. Upon the stimulation of BMMs with RANKL for a short period of time following starvation, no STAT5 phosphorylation was observed at any time over the duration of RANKL stimulation, whereas IκB degradation was clearly evident (Fig. 2F), indicating that BMMs had been sufficiently stimulated by RANKL. Furthermore, STAT5 phosphorylation was never observed during RANKL-mediated osteoclastogenesis, whereas NFATc1 expression was strongly induced during osteoclast differentiation (Fig. 2G). In summary, these data show that STAT5 deficiency did not alter RANKL-induced osteoclast differentiation, suggesting that STAT5 activation does not occur via the RANKL signaling pathway.

(A–E) Bone marrow cells were harvested from long bones of STAT5 conditional knockout (Stat5 cKO) mice or Stat5fl/fl littermates. (A) Total RNA was isolated from BMMs and RT-PCR was performed to detect expression of the indicated genes (upper panel). Whole cell lysates were harvested from BMMs and were immunoblotted with antibodies against STAT5A or actin (lower panel). All gels run under the same experimental conditions and the representative images were cropped and shown. (B,C) BMMs were cultured with M-CSF in the presence or absence of RANKL for three days. (B) Cultured cells were stained for TRAP. (C) The number of TRAP-positive MNCs per well was counted. Data are represented as the mean ± SD. n.s., not significant; n = 4. (D,E) BMMs were cultured with M-CSF and RANKL for the indicated times. (D) mRNA levels of c-fos, Nfatc1, Acp5 and Oscar were assessed by quantitative real-time PCR. Data represent mean ± SD of triplicate samples. n.s., not significant. (F) BMMs were stimulated with RANKL for the indicated times. (G) BMMs were cultured with M-CSF and RANKL for the indicated times. (E–G) Whole cell lysates were harvested from cultured cells and were immunoblotted with the indicated antibodies. All gels run under the same experimental conditions and the representative images were cropped and shown. Bar: 100 μm. All results are representative of at least three independent experiments. Statistical analyses were implemented in TTEST.
IL-3 attenuates RANKL-induced osteoclast differentiation
Although STAT5 activation is not regulated by RANKL, it did result in significant suppression of osteoclast differentiation. To address this conundrum, we searched for other cytokines capable of activating STAT5 and thereby inhibiting RANKL-induced osteoclast differentiation. IL-3 has been shown to activate STAT5 in a variety of cells21, including by BMMs, as shown in this study (Supplementary Fig. 1A). We observed osteoclast differentiation in the presence of increasing concentrations of IL-3 from 10 pg/mL to 1 ng/mL; IL-3 also suppressed RANKL-induced osteoclast differentiation in a dose-dependent manner (Supplementary Fig. 1B,C). Thus, a negative effect of IL-3 on osteoclast development would be consistent with reports of STAT5-mediated inhibition of osteoclastogenesis.
STAT5 deficiency prevents IL-3-mediated inhibition of osteoclastogenesis
Next, we determined whether the inhibitory effect of IL-3 on osteoclast differentiation is attributable to STAT5 activation. To this end, we tested whether the inhibitory effect of IL-3 on osteoclast differentiation was abrogated by STAT5 deficiency. When osteoclast differentiation from Stat5fl/fl cells and Stat5 cKO cells in the presence of IL-3 was compared, it was found that STAT5 deficiency partially restored osteoclast formation that was inhibited by IL-3 in the Stat5fl/fl cells (Fig. 3A,B). This was confirmed by mRNA expression of the osteoclastic genes c-fos, Nfatc1, Acp5, and Oscar. In the Stat5fl/fl cells, the expression of RANKL-inducible c-fos, Nfatc1, Acp5, and Oscar was strongly suppressed by IL-3 in pre-osteoclasts (pOC). In contrast, in Stat5 cKO cells, the expression of c-fos was recovered to almost the level observed in the absence of IL-3, while the expression of Nfatc1, Acp5, and Oscar was lower than that in the absence of IL-3. However, the expression was still higher than that in the IL-3–treated pOC from the Stat5fl/fl cells (Fig. 3C). Taken together, these results strongly suggest that STAT5 is a key modulator of the IL-3-mediated inhibition of osteoclast development.

Bone marrow cells were harvested from long bones of Stat5 cKO mice or Stat5fl/fl littermates. (A,B) BMMs were cultured with M-CSF and RANKL for three days in various concentrations of IL-3. (A) Cultured cells were stained for TRAP. (B) The number of TRAP-positive MNCs per well was counted. Data are represented as the mean ± SD. ***P < 0.001 vs. Stat5fl/fl control; #P < 0.05 vs. Stat5 cKO control; n.s., not significant; n = 4. (C) BMMs were treated with IL-3 (1 ng/mL). Cells were further cultured in the presence (pOC) or absence (BMM) of RANKL for two days, and subjected to semi-quantitative real-time PCR for the indicated genes. Data represent mean ± SD of triplicate samples. ***P < 0.001; n.s., not significant. Bar: 100 μm. All results are representative of at least three independent experiments. All data were analyzed using ANOVA.
Activation of STAT5A or STAT5B alone is sufficient to inhibit osteoclast differentiation
Since redundancy between STAT5A and STAT5B has been proposed in other cell types, we tested this possibility in osteoclastogenesis. Overexpression of either wild-type STAT5A or STAT5B caused a similar reduction in osteoclastogenesis, although the extent of inhibition was weaker than that observed upon overexpression of the constitutive active STAT5A1*6 (Supplementary Fig. 2A,B). In addition, downregulation of RANKL-mediated NFATc1 induction in osteoclastogenesis was also comparable when STAT5A and STAT5B were ectopically expressed, but this was significantly less than that observed in the control (Supplementary Fig. 2C). Furthermore, IL-3 inhibited osteoclast differentiation by approximately 23% and 30% following overexpression of STAT5A and STAT5B, respectively (Supplementary Fig. 2D,E), indicating overlapping functions of STAT5A and STAT5B in the inhibition of osteoclast differentiation. Since STAT5A and STAT5B exhibited an overlapping inhibitory effect on osteoclast differentiation with similar potentials, we determined whether the absence of either isoform would still inhibit RANKL-induced osteoclast differentiation. To address this, a constitutively active form of STAT5A (STAT5A1*6) was overexpressed in the BMMs obtained from Stat5fl/fl and Stat5 cKO mice. Expression of the activated STAT5A in Stat5 cKO BMMs led to the attenuation of TRAP positive multi-nucleated cells to a degree comparable to that observed in the Stat5fl/fl samples (Supplementary Fig. 3A,B). This provides compelling evidence indicating that activation of either isoform alone is sufficient to inhibit osteoclast differentiation.
ID1 and ID2 are responsible for the STAT5-mediated inhibition of osteoclast differentiation
To understand the mechanism underlying the inhibition of osteoclast differentiation by STAT5, we searched for STAT5 target genes that were regulated during both RANKL and IL-3 stimulation. RNA sequencing was performed on the BMMs obtained from the Stat5fl/fl and Stat5 cKO mice stimulated with M-CSF and RANKL, in the presence or absence of IL-3 (GSE76988). The RNA sequencing data indicated that the mRNA expression of Id1 and Id2 was attenuated upon RANKL stimulation and that the levels were recovered by IL-3 stimulation, but not in the absence of STAT5 (Supplementary Table 1). Therefore, we proposed that Id1 and Id2 are the target genes of STAT5. It has also been previously reported that Id1 and Id2 function as negative regulators of osteoclast differentiation13. To examine whether Id1 and Id2 are involved in the STAT5-mediated inhibition of osteoclast differentiation, STAT5A1*6 was overexpressed during osteoclast differentiation of the BMMs. Although this led to a significant increase in the mRNA expression of Id1 and Id2 (Fig. 4A), STAT5A1*6 did not affect the decrease in Id gene expression observed following RANKL stimulation. In both BMMs and pOC, the expression of Id1 and Id2, but not that of Id3, was significantly increased in the presence of IL-3 (Fig. 4B). In STAT5-deficient cells, however, IL-3 failed to increase the expression of Id1 and Id2 (Fig. 4B), suggesting that IL-3 induces the expression of Id1 and Id2 through STAT5.

(A) BMMs were transduced with pMX-IRES-EGFP (control) or STAT5A1*6 retrovirus and cultured with M-CSF and RANKL for the indicated times. Levels of Id1 and Id2 mRNA were assessed by quantitative real-time PCR. Data represent mean ± SD of triplicate samples. **P < 0.01; ***P < 0.001 vs. control. (B–D) Bone marrow cells were harvested from long bones of Stat5 cKO mice or Stat5fl/fl littermates. (B) BMMs were treated with IL-3 (1 ng/mL) and cells were further cultured in the presence (pOC) or absence (BMM) of RANKL for two days. Levels of Id1 and Id2 mRNA were assessed by quantitative real-time PCR. Data represent mean ± SD of triplicate samples. **P < 0.01; ***P < 0.001 vs. control; n.s., not significant. (C,D) Bone marrow cells were harvested from long bones of Stat5 cKO mice or Stat5fl/fl littermates. BMMs were transduced with pMX-IRES-EGFP (control), Id1 or Id2 retrovirus and cultured with M-CSF and RANKL for three days in the presence or absence of IL-3, as indicated. (C) Cultured cells were stained for TRAP. (D) The number of TRAP-positive MNCs per well was counted. Data are represented as the mean ± SD. ***P < 0.001 vs. control; n = 4. Bar: 100 μm. All results are representative of at least three independent experiments. Data were analyzed using TTEST (A) or ANOVA (B,C).
Next, we examined whether the ectopic expression of ID1 and ID2 could restore the IL-3-mediated inhibition of osteoclast differentiation that was abrogated by STAT5 deficiency. Consistent with our previous report13, ID1 or ID2 overexpression suppressed osteoclast development (data not shown), and the osteoclasts in STAT5-deficient cells were almost fully developed, even in the presence of IL-3 (Fig. 4C). Strikingly, quantification of the number of osteoclasts revealed that the osteoclast formation rescued from IL-3-mediated inhibition in STAT5-deficient cells was abolished by the ectopic expression of ID1 or ID2, where osteoclast differentiation was returned to almost the levels observed in the controls (Fig. 4C,D).
It has been reported that IL-3 induces dendritic cell differentiation22, and it therefore seemed possible that the increase in Id1 and Id2 gene expression by IL-3-mediated inhibition of osteoclast differentiation that occurs via STAT5 activation, as shown in the present study, was due to a change in the fate of the cells. In order to examine the effect of STAT5 activation on dendritic cell differentiation, dendritic cell surface markers, including CD80, CD86, MHC class II, and CD11c were analyzed during the osteoclast differentiation of BMMs overexpressing either an empty vector (pMX-IRES-EGFP) or the constitutively active from of STAT5A (STAT5A1*6) using fluorescence-activated cell sorting (FACS). Increase in the CD80+, CD86+, MHC class II+, and CD11c+ cell populations was observed in the presence of activated STAT5, indicating a conversion of cell fate from osteoclast precursors to dendritic cells (Supplementary Fig. 4). These results suggest that STAT5 activation by IL-3 inhibits RANKL-induced osteoclast differentiation via the induction of Id1 and Id2 expression, while converting the cell fate of osteoclast precursors to dendritic cells.
STAT5 conditional knockout mice exhibit osteoporotic bone phenotype
To evaluate the physiological functions of STAT5 in mice, the bone quality of the Stat5fl/fl and Stat5 cKO mice was compared. Microcomputed tomography (μCT) analysis with three-dimensional reconstruction of the trabecular bones of the distal femurs of 16-week-old male mice revealed a relatively lower bone mass in the Stat5 cKO than in the Stat5fl/fl mice, but there was no difference between 8-week-old Stat5 cKO and Stat5fl/fl mice (Fig. 5A). The reduced bone mass in 16-week-old Stat5 cKO mice was accompanied by 32.8%, 10.6%, and 21.5% reductions in the bone volume, trabecular thickness, and trabecular numbers, respectively, and an increase of 15.8% in trabecular separation (Fig. 5B). Meanwhile, there was no significant difference in μCT parameters between Stat5 cKO and Stat5fl/fl mice at the age of 8 weeks (Fig. 5B). These results indicate that the Stat5 cKO mice were more osteoporotic than the Stat5fl/fl mice, and this effect was age-dependent. Furthermore, the number of osteoclasts and osteoblasts in the trabecular bones of the proximal tibia of the same mice at the age of 16 weeks analyzed with μCT were subjected to quantification using TRAP and H&E staining, respectively. The Stat5 cKO mice exhibited a 5% increase in the number of osteoclasts and exhibited an increasing tendency without significance in the osteoclast surface. However, the number of osteoblasts in the trabecular bones was comparable between the Stat5fl/fl and Stat5 cKO mice, while the osteoblast surface exhibited decreasing tendency without significance (Fig. 5C,D), demonstrating that STAT5 deficiency in osteoclasts does not affect osteoblast differentiation. These results suggest that the reduced bone mass in the Stat5 cKO mice is likely due to a reduced inhibitory effect of IL-3 on osteoclast differentiation via the STAT5-Id axis, which is different from that observed in vitro.

(A,B) Long bones obtained from 8-week-old and 16-week-old male Stat5 cKO mice or Stat5fl/fl littermates were subjected to μCT analysis. (A) Representative three-dimensional images of femurs in Stat5 cKO mice or Stat5fl/fl littermates. (B) Bone volume per tissue volume, trabecular bone thickness, trabecular separation and trabecular number were assessed from the μCT measurements. Data are represented as the mean ± SD. *P < 0.05; ***P < 0.001 vs. control; n.s., not significant; 8-week-old Stat5fl/fl, n = 5; 8-week-old Stat5 cKO, n = 5; 16-week-old Stat5fl/fl, n = 9; 16-week-old Stat5 cKO, n = 7. (C,D) Long bones obtained from 16-week-old male Stat5 cKO mice or Stat5fl/fl littermates were subjected to histomorphometric analyses. (C) Hematoxylin and eosin (H&E) and TRAP staining of histological sections of proximal tibiae. (D) Osteoclast surface per bone surface, osteoclast number per bone surface, osteoblast surface per bone surface, and osteoblast number per bone surface were assessed. Data are represented as the mean ± SD. *P < 0.05 vs. control; n.s., not significant; Stat5fl/fl, n = 7; Stat5 cKO, n = 5. Bars: (A) 500 μm; (C) 100 μm. Statistical analyses were implemented in TTEST.
Administration of IL-3 has no inhibitory effect on RANKL-induced bone destruction in STAT5 conditional knockout mice
Having demonstrated the inhibitory effect of IL-3 on osteoclast differentiation, which occurs via STAT5 in vitro, we investigated the same effect in vivo using Stat5fl/fl and Stat5 cKO littermates following an intraperitoneal injection of RANKL with or without IL-3, as illustrated in Fig. 6A. μCT analysis of 8-week-old mice revealed that the bone volumes were reduced by 41.6% and 35.9% in the Stat5 cKO and Stat5fl/fl littermates, respectively, after RANKL injection. The bone loss observed in the RANKL-injected Stat5fl/fl mice was recovered by 19.89% after intraperitoneal IL-3 administration. On the contrary, there was almost no bone volume recovery in the Stat5 cKO mice, which continued to exhibit an osteoporotic phenotype, even after intraperitoneal IL-3 administration (Fig. 6B,C).

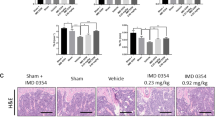
(A–G) 8-week-old Stat5 cKO mice or Stat5fl/fl littermates were intraperitoneally administrated PBS, RANKL, with or without IL-3. Long bones obtained were subjected to μCT and histomorphometric analysis (A) Illustration of IL-3 administration strategy. (B) Representative three-dimensional images of femurs in Stat5 cKO mice or Stat5fl/fl littermates. (C) Bone volume per tissue volume was assessed from the μCT measurements. Data are represented as the mean ± SD. **P < 0.01; ***P < 0.001 vs. control; n.s., not significant; (PBS Stat5fl/fl n = 8, Stat5 cKO n = 8, RANKL Stat5fl/fl n = 10, Stat5 cKO n = 10, RANKL and IL-3 Stat5fl/fl n = 14, Stat5 cKO n = 11). (D) TRAP staining of histological section of proximal tibiae. (E) Osteoclast number per bone surface was assessed. Data are represented as the mean ± SD. ***P < 0.001 vs. control; **P < 0.01 vs. control; *P < 0.05 vs. control. n.s. not significant; n = 8. (F) Hematoxylin and eosin (H&E) stain of proximal tibiae. (G) Osteoblast number per bone surface was assessed. Data are represented as the mean ± SD. n.s., not significant; n = 8. Bars: (B) 500 μm; (C,D) 100 μm. All data were analyzed using ANOVA.
Next, histological analysis was performed to confirm bone loss recovery by intraperitoneal IL-3 administration at the cellular level in the tibiae of the Stat5fl/fl and Stat5 cKO littermate mice. After RANKL injection, there was an increase in the number of TRAP-positive cells, which indicated that the number of osteoclasts was increased by 116.09% and 54.78% in the Stat5 cKO and Stat5fl/fl littermates, respectively. Following IL-3 injection, the number of TRAP-positive cells was decreased by 22.41% in the Stat5fl/fl mice, while there was no significant change in the osteoclast number in the Stat5 cKO mice (Fig. 6D,E). In addition, there was no significant difference in the osteoblast numbers between the RANKL and IL-3-injected groups (Fig. 6F,G). These results indicate that bone loss by RANKL injection and bone mass recovery by IL-3 injection in the control mice were due to an increase and a decrease in the osteoclast numbers, respectively, rather than due to changes in the osteoblast numbers. However, the number of osteoclasts remained relatively high even when IL-3 was intraperitoneally administered in the Stat5 cKO mice. In summary, IL-3 had an inhibitory effect on osteoclast differentiation in vivo in the presence of STAT5, whereas it failed to inhibit osteoclast differentiation in the absence of STAT5, indicating that STAT5 is a critical factor in the IL-3-mediated inhibition of osteoclast differentiation in vivo as well as in vitro.
Discussion
This study showed that IL-3 negatively regulates RANKL-induced osteoclast differentiation through the transcription factors STAT5 and ID1/ID2 and shed light on the underlying mechanism. Although previous studies had reported that IL-3 inhibits human osteoclastogenesis and bone resorption through the downregulation of c-Fms22 and prevents RANKL-induced nuclear translocation of NF-κB11, the transcriptional circuit was not understood. Our study closes this gap and demonstrates that IL-3-induced STAT5 activation in osteoclast precursors reduces c-Fos expression and osteoclast differentiation. However, the induction of osteoclast differentiation by RANKL was independent of IL-3. Our study provides evidence indicating a direct contribution of STAT5 in the negative effect of IL-3 on osteoclast differentiation.
Previously, we had demonstrated that ID transcription factors are responsible for the negative regulation of RANKL-induced osteoclast differentiation13. In addition, IL-3-and STAT5-dependent activation of Id1 transcription has been reported23. We chose to focus on IDs, as we believed that they might be directly responsible for the inhibition of osteoclast differentiation. Notably, the expression of Id1 and Id2, but not that of Id3, was induced by IL-3 through STAT5 in the osteoclasts. In support of a critical function of ID1 or ID2, their overexpression significantly inhibited RANKL-induced osteoclast differentiation despite STAT5 deficiency. This indicates that IL-3 negatively regulates osteoclast differentiation through the activation of STAT5 and the subsequent induction of Id1 and Id2 expression. These findings are consistent with those of a previous study showing that IL-3 plays an inhibitory role in osteoclast differentiation by regulating the expression of c-Fos and Ids14.
Two independent studies suggested that IL-3 inhibits osteoclast differentiation by diverting the osteoclast precursor cells to either a macrophage or a dendritic cell lineage11,22. In the current study, we expanded on these findings and demonstrated that STAT5A1*6 overexpression increased the expression of dendritic cell markers, such as CD80, CD86, MHC class II, and CD11c. These findings solidify the notion that IL-3-induced STAT5 activation is responsible for the conversion of osteoclast precursor cells into dendritic cells.
Previously, Hirose et al. reported that conditional osteoclast-specific STAT5 mutant mice exhibited an osteoporotic phenotype20. Consistent with this, an osteoporotic bone phenotype was observed in the Stat5 cKO mice in this study, indicating that STAT5 is essential for bone homeostasis under physiological conditions. While Hirose et al. demonstrated a significant increase in osteoclast surface but not osteoclast number the present study provided evidence of an increased osteoclast number but no significant change in osteoclast surface. These discrepancies are possibly because of the differences in the method used to delete STAT5 from the cells between the two studies. In this study, MX1-Cre was used to remove Stat5 from hematopoietic stem cells and therefore STAT5 was absent from macrophage precursor cells from a very early stage, further confirming that STAT5 deficiency could mitigate cellular development, proliferation, and differentiation in vivo. In contrast, Hirose et al. utilized Cathepsin K-Cre to eliminate STAT5 in pre-osteoclast cells, which might have been more effective at a later stage, such as during resorption activity, than during cell proliferation and development. We suggest that the cell differential elimination of STAT5 elimination is a primary reason for the different lesions determined in these two studies.
It has been suggested that STAT5 deficiency increases the bone-resorbing activity of osteoclasts, which is related to the activation of ERK through the regulation of the expression of Dusp1 and Dusp220. It is well known that ERK positively regulates osteoclast differentiation as well as bone resorptive activity24. Similarly, RANKL-induced ERK was slightly reduced upon overexpression of constitutively active STAT5 in the BMMs. Although further studies are needed to elucidate whether STAT5 activation regulates osteoclast differentiation and function by inhibiting RANKL-induced ERK activation, we propose that the reduced osteoclast differentiation caused by STAT5 activation is at least in part caused by decreased ERK activation.
The bone loss observed in the Stat5 cKO mice seemed to be age-related. When the bone volumes of 8-week-old Stat5 cKO and wild-type littermates were analyzed, there was no significant difference in bone volumes between the Stat5 cKO and wild-type littermates. However, when the bone phenotype was analyzed between the Stat5 cKO and wild-type littermates at the age of 16 weeks, the bone mass in the Stat5 cKO mice was found to be reduced. Therefore, these observations imply that the bone loss observed in Stat5 deficiency may be due to aging. In addition, the reduced bone mass observed in the absence of STAT5 at the age of 16 weeks appears to be associated with increased osteoclast differentiation rather than with the increased bone resorbing activity of osteoclasts. Although osteoclast differentiation remained unchanged when BMMs lacking STAT5 were cultured with RANKL, our in vivo study revealed a significant increase in the number of osteoclasts and a moderate increase in osteoclast surface. Collectively, these results suggest that STAT5 acts as a negative factor in osteoclast differentiation under the control of an existing endogenous signaling pathway in vivo, which may stimulate STAT5 activation and block osteoclast differentiation, rather than directly inhibiting the RANKL signaling pathway. IL-3 is a potent cytokine for the activation of STAT5 and the inhibition of osteoclast differentiation. Our results provided evidence suggesting that RANKL-induced bone loss was rescued by IL-3 administration only in the presence of STAT5. We hypothesize that RANKL increases IL-3 expression in osteoclasts and thus stimulates STAT5 activation, which ensures normal osteoclast differentiation and thus maintains bone homeostasis. In addition to IL-3, GM-CSF may contribute to bone homeostasis. Based on the in vivo mouse model, we propose that the IL-3/STAT5 pathway has a therapeutic value for age-related bone diseases.
Methods
Reagents
Recombinant human RANKL was purified from bacteria. Recombinant human M-CSF was a gift from Dr. Daved Fremont (Washington University, St. Louis, MO, USA). Recombinant mouse IL-3 and GM-CSF were purchased from R&D systems (Abingdon, UK).
Mice
All animal experiments were approved by the Chonnam National University Medical School Research Institutional Animal Care and Use Committee and were carried out in accordance with the approved guidelines. STAT5 floxed (Stat5flox/flox) mice, targeting both Stat5a and 5b, were generated as previously described25. To generate Stat5 cKO mice (Stat5flox/flox;Mx1-Cre+/‒), STAT5fl/fl mice were crossed with Mx1-Cre+/‒ mice expressing Cre recombinase under the control of a type I interferon-inducible Mx1 promoter. Stat5flox/flox mice were crossed with Mx1-Cre+/‒ mice to produce Stat5flox/+;Mx1-Cre+/‒ offspring, which were subsequently backcrossed with Stat5flox/flox mice to generate Stat5 cKO mice. Genotyping was performed by PCR on tail genomic DNA with specific primers as follows; Stat5 WT forward, 5′-GAA AGC ATG AAA GGG TTG GAG-3′; Stat5 WT reverse, 5′-AGC AGC AAC CAG AGG ACT TAC-3′; Stat5 fl2 forward, 5′-TAC CCG CTT CCA TTG CTC AG-3′; Stat5 fl2 reverse, 5′-AGC AGC AAC CAG AGG ACT AC-3′; Mx1-Cre forward, 5′-GTG TTG CCG CGC CAT CTG C-3′; Mx1-Cre reverse, 5′-CAC CAT TGC CCC TGT TTC ACT ATC-3′. To induce Stat5 excision by Cre expression, STAT5flox/flox;Mx1-Cre+/‒ mice were intraperitoneally injected with 0.25 mL of 1 mg/mL poly I:C (Sigma-Aldrich, St. Louis, MO, USA) three times (at two-day intervals) from postnatal day 10. Stat5flox/flox littermates, that were identically administrated with poly I:C, were used as controls.
Plasmid DNA constructs
The expression vector for constitutively active Stat5a (Stat5a1*6) has been described previously26. The full-length coding sequences of mouse Stat5a (NM_011488) and Stat5b (MN_011489) were purchased from 21C Frontier Human Gene Bank (Daejeon, Korea). The genes were subcloned into a retroviral vector (pMX-IRES-EGFP) that included a C-terminal Flag tag. Retrovirus vectors encoding ID1 and ID2 were described previously13.
Cell cultures
Plat E cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (HyClone Laboratories, Logan, UT, USA) supplemented with 10% FBS, puromycin (1 μg/mL) and blasticidin (10 μg/mL) every three days. Cells were plated onto either 100 mm dishes (1.3 × 106 cell/dish) or 6-well plates (3 × 105 cells/well) one day prior to transfection.
Retroviral gene transduction
Plat E cells were transfected for retrovirus packaging with FuGENE 6 (Promega, Madison, WI, USA) according to the manufacturer instructions. Retroviral supernatant was collected from the culture media 48 hours after transfection. Murine bone marrow-derived macrophages (BMMs) were subsequently infected with the supernatant for eight hours in the presence of 10 μg/mL of polybrene (Sigma-Aldrich).
Osteoclast formation
Osteoclasts were generated from murine bone marrow cells as previously described27. Bone marrow cells obtained via flushing the long bones of 6-week-old mice were cultured in α-MEM (HyClone Laboratories) containing 10% FBS, 100 U/mL penicillin and 100 mg/mL streptomycin (Life Technologies, Carlsbad, CA, USA) in the presence of M-CSF (30 ng/mL) for 3 days. After non-adherent cells were removed, adherent BMMs were plated onto 96-well plates (1 × 104 cells/well) and further cultured with M-CSF (30 ng/mL) and RANKL (100 ng/mL). Cultured cells were fixed and stained for TRAP. TRAP-positive multinuclear cells (>3 nuclei/cell) [TRAP+ MNCs] were counted as osteoclasts. Cells were observed using a Leica DMIRB microscope equipped with an N Plan 10 × 0.25 numerical aperture objective lens (Leica Microsystems, Wetzler, Germany). Images were captured using ProgRes® Capture Pro software (Jenoptik, Jena, Germany).
RT-PCR
Cells were lysed in Qiazol (QIAGEN GmbH, Hilden, Germany) and total RNA was isolated from cultured cells according to the manufacturer instructions. RNA (2 μg) was reverse transcribed into cDNA using Superscript II Reverse Transcriptase (Life Technologies). PCR with specific primers was used to determine mRNA expression level. Primer sequences were as follows: Gapdh forward, 5′-TGA CCA CAG TCC ATG CCA TCA CTG-3′; Gapdh reverse, 5′-CAG GAG ACA ACC TGG TCC TCA GTG-3′; Stat5a forward, 5′-AGT ATT ACA CTC CTG TAC TTG CGA AAG-3′; Stat5a reverse, 5′-GGA GCT TCT AGC GGA GGT GAA GAG ACC-3′; Stat5b forward, 5′-GGT CCC CTG TGA GCC CGC AAC-3′; Stat5b reverse, 5′-TGA CTG TGC GTG AGG GAT CCA CTG ACT-3′; Nfatc1 forward, 5′-CTC GAA AGA CAG CAC TGG AGC AT-3′; Nfatc1 reverse, 5′-CGG CTG CCT TCC GTC TCA TAG-3′.
Real-time PCR
Quantitative real-time PCR analysis was performed in triplicate with SYBR Green (Qiagen) and with specific primers using Rotor-Gene Q (Qiagen). mRNA expression levels were normalized to Gapdh. The relative quantitation value for each target gene compared to the calibrator for that target was expressed as 2−(Ct-Cc) (Ct and Cc are the mean threshold cycle differences after normalizing to Gapdh). The relative expression levels for each sample were presented by semi-log plot. Primer sequences were as follows: Gapdh forward, 5′-TGA CCA CAG TCC ATG CCA TCA CTG-3′; Gapdh reverse, 5′-CAG GAG ACA ACC TGG TCC TCA GTG-3′; c-fos forward, 5′-ATG GGC TCT CCT GTC AAC ACA CAG-3′; c-fos reverse, 5′-TGG CAA TCT CAG TCT GCA ACG CAG-3′; Nfatc1 forward, 5′-CTC GAA AGA CAG CAC TGG AGC AT-3′; Nfatc1 reverse, 5′-CGG CTG CCT TCC GTC TCA TAG-3′; Acp5 forward, 5′-CTG GAG TGC ACG ATG CCA GCG ACA-3′; Acp5 reverse, 5′-TCC GTG CTC GGC GAT GGA CCA GA-3′; Oscar forward, 5′-TGC TGG TAA CGG ATC AGC TCC CCA GA-3′; Oscar reverse, 5′-CCA AGG AGC CAG AAC CTT CGA AAC T-3′; Id1 forward, 5′-AAG TGA GCA AGG TGG AGA TC-3′; Id1 reverse, 5′-GGG CTG GAG TCC ATC TGG TC-3′; Id2 forward, 5′-AAC ATG AAC GAC TGC TAC TC-3′; Id2 reverse, 5′-AGA GTA CTT TGC TAT CAT TC-3′.
Western blotting
Cells were washed with PBS and lysed in extraction buffer [50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40, 1 mM PMSF and protease inhibitor cocktails]. Cell lysates were separated by SDS-PAGE and transferred to a PVDF membrane (Millipore corporation, Billerica, MA, USA). Membranes were blocked with 5% skim milk in TBS-T [10 mM Tri-HCl (pH 7.6), 150 mM NaCl, 0.1% Tween 20] and immunoblotted with antibodies against c-Fos (Santa Cruz Biotechnology, Dallas, TX, USA), NFATc1 (Santa Cruz Biotechnology), Phospho-STAT5 (Cell Signaling Technology), STAT5A (Cell Signaling Technology, Beverly, MA), IκB (Cell Signaling Technology), Flag-HRP (Sigma-Aldrich), Actin-HRP (Sigma-Aldrich), mouse-IgG-HRP (Abcam, Cambridge, UK) and rabbit-IgG-HRP (Abcam). Signals were detected with ECL solution (Millipore corporation) and analyzed using a LAS3000 luminescent image analyzer (GE Healthcare, Piscataway, NJ, USA).
RNA sequencing
BMMs were isolated from Stat5fl/fl and Stat5 cKO littermates and differentiated to osteoclasts under the following conditions: Stat5fl/fl BMMs with M-CSF (30 ng/mL) only; Stat5fl/fl BMMs with M-CSF (30 ng/mL) and RANKL (100 ng/mL); Stat5fl/fl BMMs with M-CSF (30 ng/mL), RANKL (100 ng/mL) and IL-3 (1 ng/mL); and Stat5 cKO BMMs with M-CSF (30 ng/mL), RANKL (100 ng/mL) and IL-3 (1 ng/mL). Total RNA was extracted and mouse RNA Transcriptome Sequencing Analysis was performed using a HiSeq2000 system (Macrogen Inc., Korea). RNA sequencing data in this article have been deposited in Gene Expression Omnibus (GEO accession no. GSE76988).
Micro-computed tomography (μCT)
Micro-computed tomography (μCT) images were taken using a high-resolution Skyscan 1172 system (Skyscan, Kontich, Belgium) with an X-ray source at 50 kV and 201 μA with a 0.5 mm aluminum filter. Images were captured every 0.7 degree angle with 11 μm·pixel−1. Raw images were reconstructed into serial cross-sectional images with identical thresholds (0 to 6,000 in Hounsfield units) for all samples using Recon software (Skyscan). A region of interest (ROI) was manually designated with CTAn software (Skyscan), within 300 steps of the trabecular bone of the distal femur, starting from 80 steps away from the epiphyseal plate. Three-dimensional morphometry was characterized by measuring the bone volume over the tissue volume (BV/TV), trabecular thickness (Tb.Th), trabecular separation (Tb.Sp), and trabecular number (Tb.N). Three-dimensional images of trabecular bones were remodeled with a window density from 90 to 165 using Ant software (Skyscan).
Histomorphometric analysis
Tibiae collected from 8-week-old or 16-week-old mice were fixed overnight in 4% paraformaldehyde and decalcified in 5.5% EDTA in 3.8% formaldehyde buffer for two weeks at 4 °C. After samples were gradually dehydrated and embedded in paraffin, paraffin blocks were cut into 4 μm-thick longitudinal sections. Following deparaffinization with xylene, two consecutive sections were stained with tartrate-resistant acid phosphatase (TRAP) and hematoxylin & eosin (H&E) for determining osteoclasts and osteoblasts, respectively.
Fluorescence-activated cell sorting
Bone marrow-derived macrophages were transduced with a control vector (pMX-FLAG-IRES-EGFP) or the constitutively active form of STAT5A (pMX-FLAG-IRES-EGFP-STAT5A1*6), differentiated to osteoclasts in the presence of M-CSF (30 ng/mL) and RANKL (100 ng/mL), and collected. The collected cells were stained with PE-anti-mouse CD80 (BD PharmingenTM, San Diego, CA, USA), PE-anti-mouse CD 86 (BD PharmingenTM), PE-anti-mouse MHC class II (eBioscience, San Diego, CA, USA), and APC-anti-mouse CD11c (eBioscience) for 20 min at 4 °C. Stained cells were analyzed using a Navios flow cytometer with Kaluza software (Beckman Coulter, Brea, CA, USA).
Statistics
All values are expressed as the mean ± SD. Statistically significant differences were determined using a two-tailed Student’s t-test for two independent samples. Differences with a P value less than 0.05 were considered statistically significant. For statistical analyses of multiple group comparisons, analysis of variance (ANOVA) with post-hoc Tukey HSD test was performed to calculate P values. P < 0.05 was considered as statistically significant.
Additional Information
How to cite this article: Lee, J. et al. STAT5 is a key transcription factor for IL-3-mediated inhibition of RANKL-induced osteoclastogenesis. Sci. Rep. 6, 30977; doi: 10.1038/srep30977 (2016).
References
Suda, T. et al. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocrine reviews 20, 345–357, doi: 10.1210/edrv.20.3.0367 (1999).
Lee, Z. H. & Kim, H. H. Signal transduction by receptor activator of nuclear factor kappa B in osteoclasts. Biochemical and biophysical research communications 305, 211–214 (2003).
Kim, J. H. & Kim, N. Signaling Pathways in Osteoclast Differentiation. Chonnam medical journal 52, 12–17, doi: 10.4068/cmj.2016.52.1.12 (2016).
Kim, J. H. & Kim, N. Regulation of NFATc1 in Osteoclast Differentiation. J Bone Metab 21, 233–241, doi: 10.11005/jbm.2014.21.4.233 (2014).
Danks, L. & Takayanagi, H. Immunology and bone. Journal of biochemistry 154, 29–39, doi: 10.1093/jb/mvt049 (2013).
Kim, K. et al. Nuclear factor of activated T cells c1 induces osteoclast-associated receptor gene expression during tumor necrosis factor-related activation-induced cytokine-mediated osteoclastogenesis. The Journal of biological chemistry 280, 35209–35216, doi: 10.1074/jbc.M505815200 (2005).
Walsh, M. C. et al. Osteoimmunology: interplay between the immune system and bone metabolism. Annual review of immunology 24, 33–63, doi: 10.1146/annurev.immunol.24.021605.090646 (2006).
Takayanagi, H. Osteoimmunology and the effects of the immune system on bone. Nature reviews. Rheumatology 5, 667–676, doi: 10.1038/nrrheum.2009.217 (2009).
Barton, B. E. & Mayer, R. IL-3 induces differentiation of bone marrow precursor cells to osteoclast-like cells. Journal of immunology 143, 3211–3216 (1989).
Hattersley, G. & Chambers, T. J. Effects of interleukin 3 and of granulocyte-macrophage and macrophage colony stimulating factors on osteoclast differentiation from mouse hemopoietic tissue. Journal of cellular physiology 142, 201–209, doi: 10.1002/jcp.1041420125 (1990).
Khapli, S. M., Mangashetti, L. S., Yogesha, S. D. & Wani, M. R. IL-3 acts directly on osteoclast precursors and irreversibly inhibits receptor activator of NF-kappa B ligand-induced osteoclast differentiation by diverting the cells to macrophage lineage. Journal of immunology 171, 142–151 (2003).
Khapli, S. M. et al. Irreversible inhibition of RANK expression as a possible mechanism for IL-3 inhibition of RANKL-induced osteoclastogenesis. Biochemical and biophysical research communications 399, 688–693, doi: 10.1016/j.bbrc.2010.07.143 (2010).
Lee, J. et al. Id helix-loop-helix proteins negatively regulate TRANCE-mediated osteoclast differentiation. Blood 107, 2686–2693, doi: 10.1182/blood-2005-07-2798 (2006).
Oh, J. et al. Inhibitory regulation of osteoclast differentiation by interleukin-3 via regulation of c-Fos and Id protein expression. Journal of cellular physiology 227, 1851–1860, doi: 10.1002/jcp.22913 (2012).
Darnell, J. E. Jr. STATs and gene regulation. Science 277, 1630–1635 (1997).
Feldman, G. M. et al. STAT5A-deficient mice demonstrate a defect in granulocyte-macrophage colony-stimulating factor-induced proliferation and gene expression. Blood 90, 1768–1776 (1997).
Shelburne, C. P. et al. Stat5: an essential regulator of mast cell biology. Molecular immunology 38, 1187–1191 (2002).
Hennighausen, L. & Robinson, G. W. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes & development 22, 711–721, doi: 10.1101/gad.1643908 (2008).
Li, J. JAK-STAT and bone metabolism. Jak-Stat 2, e23930, doi: 10.4161/jkst.23930 (2013).
Hirose, J. et al. Bone resorption is regulated by cell-autonomous negative feedback loop of Stat5-Dusp axis in the osteoclast. The Journal of experimental medicine 211, 153–163, doi: 10.1084/jem.20130538 (2014).
Verweij, M. M. et al. STAT5 in human basophils: IL-3 is required for its FcepsilonRI-mediated phosphorylation. Cytometry B Clin Cytom 82, 101–106, doi: 10.1002/cyto.b.20629 (2012).
Gupta, N. et al. IL-3 inhibits human osteoclastogenesis and bone resorption through downregulation of c-Fms and diverts the cells to dendritic cell lineage. J Immunol 185, 2261–2272, doi: 10.4049/jimmunol.1000015 (2010).
Xu, M., Nie, L., Kim, S. H. & Sun, X. H. STAT5-induced Id-1 transcription involves recruitment of HDAC1 and deacetylation of C/EBPbeta. Embo j 22, 893–904, doi: 10.1093/emboj/cdg094 (2003).
He, Y. et al. Erk1 positively regulates osteoclast differentiation and bone resorptive activity. PLos One 6, e24780, doi: 10.1371/journal.pone.0024780 (2011).
Cui, Y. et al. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Molecular and cellular biology 24, 8037–8047, doi: 10.1128/MCB.24.18.8037-8047.2004 (2004).
Onishi, M. et al. Identification and characterization of a constitutively active STAT5 mutant that promotes cell proliferation. Molecular and cellular biology 18, 3871–3879 (1998).
Kim, K. et al. MafB negatively regulates RANKL-mediated osteoclast differentiation. Blood 109, 3253–3259, doi: 10.1182/blood-2006-09-048249 (2007).
Acknowledgements
This research was supported by grants from the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (MRC, 2011-0030132 and NRF-2014R1A2A2A01003966). LH was supported by the IRP of the NIDDK/NIH.
Author information
Authors and Affiliations
Contributions
J.L. and S.S. performed most experiments. J.H.K. and K.K. performed the animal experiments. I.K. prepared and performed microCT. B.-C.J. and K.-I.N. performed histologic analyses. K.K. provided critical protocols and tips for many experimental procedures. L.H. provided STAT5 KO mice and provided critical protocols. N.K. designed the experiments and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Lee, J., Seong, S., Kim, J. et al. STAT5 is a key transcription factor for IL-3-mediated inhibition of RANKL-induced osteoclastogenesis. Sci Rep 6, 30977 (2016). https://doi.org/10.1038/srep30977
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep30977
This article is cited by
-
Alternative regulatory mechanism for the maintenance of bone homeostasis via STAT5-mediated regulation of the differentiation of BMSCs into adipocytes
Experimental & Molecular Medicine (2021)
-
IL-3 inhibits rat osteoclast differentiation induced by TNF-α and other pro-osteoclastogenic cytokines
Journal of Biosciences (2021)
-
A Conditional Knockout Mouse Model Reveals a Critical Role of PKD1 in Osteoblast Differentiation and Bone Development
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
