
Abstract
Host genetic variability may contribute to susceptibility of bacterial meningitis, but which genes contribute to the susceptibility to this complex disease remains undefined. We performed a genetic association study in 469 community-acquired pneumococcal meningitis cases and 2072 population-based controls from the Utrecht Health Project in order to find genetic variants associated with pneumococcal meningitis susceptibility. A HumanExome BeadChip was used to genotype 102,097 SNPs in the collected DNA samples. Associations were tested with the Fisher exact test. None of the genetic variants tested reached Bonferroni corrected significance (p-value <5 × 10−7). Our strongest signals associated with susceptibility to pneumococcal meningitis were rs139064549 on chromosome 1 in the COL11A1 gene (p = 1.51 × 10−6; G allele OR 3.21 [95% CI 2.05–5.02]) and rs9309464 in the EXOC6B gene on chromosome 2 (p = 6.01 × 10−5; G allele OR 0.66 [95% CI 0.54–0.81]). The sequence kernel association test (SKAT) tests for associations between multiple variants in a gene region and pneumococcal meningitis susceptibility yielded one significant associated gene namely COL11A1 (p = 1.03 × 10−7). Replication studies are needed to validate these results. If replicated, the functionality of these genetic variations should be further studied to identify by which means they influence the pathophysiology of pneumococcal meningitis.
Similar content being viewed by others

Introduction
Acute bacterial meningitis is a life-threatening disease associated with substantial morbidity and mortality and ranks among the top 10 infectious causes of death1,2. Streptococcus pneumoniae is the leading causative pathogen and is identified in 70% of adult patients3,4. Despite early antibiotic and anti-inflammatory therapy, case fatality rates of pneumococcal meningitis range from 18–30% and neurological sequelae, including hearing loss, focal neurological deficits and cognitive impairment, occur in 30–55% of surviving patients4,5,6,7,8,9,10.
Several factors have been identified that contribute to the susceptibility to pneumococcal meningitis11. For example, patients with an immunocompromised state, organ transplantation, splenectomy or anatomical defects with cerebrospinal fluid leakage are at increased risk of pneumococcal meningitis12,13,14,15. Genetic variation leading to difference in immune response has also been identified to influence susceptibility to pneumococcal meningitis. Extreme phenotype studies looking at recurrent pneumococcal disease have shown that genetic variation in complement factor 2 (C2), interleukin-1 receptor-associated kinase 4 (IRAK4) and nuclear factor κB essential modulator protein (NEMO) were the cause of this increased susceptibility16. Furthermore, case-control studies mostly looking at variants in genes involved in the innate immune system have pointed out genetic variants associated with acquiring invasive pneumococcal disease16,17,18,19,20. In a Dutch case-control study genetic variations in the beta2-adrenoreceptor (ADRB2) and mannose binding lectin (MBL) genes were associated with susceptibility to pneumococcal meningitis17,18. These genetic association studies evaluated one or only few single-nucleotide polymorphisms within one gene in relatively small cohorts. A hypothesis-free approach in larger groups of patients and controls is needed to unravel new genetic risk factors for the susceptibility to pneumococcal meningitis. The aim of our study was to identify new genetic variants involved in susceptibility to pneumococcal meningitis, genotyping over 240,000 genetic variants of which the majority is exonic or otherwise functional.
Results
Between 2006 and 2011, 656 patients with bacterial meningitis were included in the MeninGene study21. Of these patients, 469 (72%) had pneumococcal meningitis. After quality control filters 408 pneumococcal meningitis patients and 2072 controls were included in the genetic associations analysis for meningitis disease susceptibility. Demographic and clinical data of the successfully genotyped patients can be found in Table 1. After exclusion of all monomorf and sex chromosome variants a total of 100,464 single nucleotide polymorphisms (SNPs) passed quality control thresholds of >95% call rate and Hardy Weinberg equilibrium and were incorporated in the association analysis.
The genomic control parameter λ was 0.31 when all variations were included. This was driven by the rare variants and exclusion of variants with a minor allele frequency below 0.01%, increased λ to 0.93 (Supplementary Fig. 1). For our single marker analysis we therefore included those with a minor allele frequency higher than 0.01%.
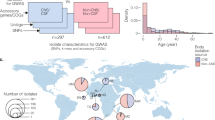
None of the tested genetic variants reached the Bonferroni corrected significance threshold (p-value <5 × 10−7). Six genetic variants associated with pneumococcal meningitis reached p-values lower than 1 × 10−4 (Fig. 1). Our strongest signal was the missense rs139064549 in the collagen type XI alpha 1 (COL11A1) gene (p = 1.51 × 10−6; G allele OR 3.21 [95% CI 2.05–5.02]) and the second strongest was the intron variant rs9309464 in the exocyst complex component 6B (EXOC6B) gene (p = 6.01 × 10−5; G allele OR 0.66 [95% CI 0.54–0.81]; Table 2). Of these six variants with a p-value lower than 1 × 10−4, three variants were located in the fibrous sheath CABYR binding protein (FSCB) gene, namely rs3809429 (p = 6.80 × 10−5; A-allele OR 1.65 [95% CI 1.30–2.09]), rs3825630 (p = 6.80 × 10−5; G allele OR 1.65 [95% CI 1.30–2.09]) and rs1959379 (p = 8.81 × 10−5; A allele OR 1.64 [95% CI 1.30–2.09]). The sixth variant rs617169 (p = 7.33 × 10−5; G allele OR 0.72 [95% CI 0.61–0.85]) was not located within a gene, but the nearest gene was protein kinase N2 (PKN2) located on chromosome 2.

Manhattan plot of p-values.
The y-axis indicates the –log10 (p-values) of the SNPs in the association analysis and the x-axis indicates the chromosomal position. The horizontal blue line indicates the threshold of p = 1 × 10−4. All markers of the three genes with the highest association signal, namely COL11A1 (rs139064549, p-value = 1.51 × 10−6, location chromosome 1), EXOC6B (rs9309464, p-value = 6.01 × 10−5, location chromosome 2) and FSCB (with rs3809429, rs3825630 rs1959379, p-value = 6.80 × 10−5, 6.80 × 10−5, 8.81 × 10−5 location chromosome 14), have been colored green.
In the single marker analysis we did not include variants with a MAF below 0.01%. To assess the role of these rare variants in the association testing of pneumococcal susceptibility we used the region based sequence kernel association test (SKAT) allowing us to include all common and rare variations within a gene region. By using the SKAT analysis, we found one significant associated gene namely COL11A1 (p = 1.03 × 10−7). In this analysis we tested a total of 12968 gene sets and after correcting for these multiple tests COL11A1 reaches a significance of p = 0.001. No difference in the p-value of COL11A1 was observed when correcting for the proportion of case-control imbalance. The second strongest signal in the SKAT analysis was the polymerase (DNA directed) lambda (POLL) gene (p = 8.10 × 10−5), which was not significant after multiple testing correction. When corrected this p-value for proportion of case-control imbalance the association was slightly more significant (p = 7.05 × 10−5). We also looked if the portion of common and rare variants drove the associations with pneumococcal meningitis susceptibility when the rare variants threshold was defined as a function of the total sample size22. This analysis showed only for POLL (p = 1.90 × 10−5) that association was based on the proportion of common and rare variants, although this did not reach genome-wide significance after correction for multiple testing.
Discussion
We performed the first hypothesis free exome-wide association study on disease susceptibility in a large homogeneous group of pneumococcal meningitis patients. We did not identify a significant association in our single marker analysis after correction for multiple testing. The strongest associations of single nucleotide variations with pneumococcal meningitis susceptibility are found within genes that have not been related to pneumococcal meningitis or pneumococcal disease previously. Our strongest signal (rs139064549) is located in the COL11A1 gene, which encodes for one component of type XI collagen that adds structure and strength to connective tissues. COL11A1 has a fundamental role in the control of fibrillogenesis and mutations in this gene are associated with Stickler syndrome23,24. Stickler syndrome is a genetic disorders affecting connective tissue and is characterized by distinctive facial abnormalities, ocular problems, hearing loss and joint problems24. Related gene ontologies of the COL11A1 gene are extracellular matrix structural constituents and extracellular binding. The association of the COL11A1 gene could indicate that differences in the extracellular matrix or anatomical defects can alter the susceptibility of pneumococcal meningitis. The second strongest association (rs9309464) is located in an intronic region of the EXOC6B gene. The EXOC6B gene is part of a multiprotein complex required for targeted exocytosis. This complex is highly expressed in the central nervous system and is important for cell polarity, growth and neuronal cell migration25. In previous studies, genetic variations affecting the EXOC6B gene have been associated with cerebral disorders, like intellectual disability, but associations with infectious diseases have not been reported26. The three genetic associations found in the FSCB gene are all intron variants. FSCB is a testis specific protein and in mice it is suggested be involved in the later stages of fibrous sheath assembly of spermiogenesis27. The FSCB gene is not described to be associated with neurological or infectious diseases in earlier studies. Since rs617169, the fifth strongest associated variant is located outside any gene and therefore we could not explain the possible involvement during pneumococcal meningitis susceptibility. The closest gene to rs617169 is the PKN2 gene encoding for a multifunctional protein kinase N228.
The SKAT analysis, performed to identify differences between patients and controls in the prevalence of a set of variants within genes, showed the association of one gene; COL11A1. As expected, most rare variants were found in the largest group, namely the control group. This finding suggests that the fivefold difference in amount of patients and controls is responsible for the significant differences we have found. We tried to determine this effect by applying the hybrid SKAT resampling method to test the proportion of case-control imbalance. This did not result in a noteworthy change of the p-value. Consequently, this indicates that a larger number of patients is needed to determine the true role of rare variants in pneumococcal meningitis susceptibility. As described above, we found a marginally significant association of the COL11A1 gene with pneumococcal meningitis susceptibility. The second highest association is found in the POLL gene and was not significance after correction for multiple testing. POLL encodes for a DNA polymerase, which participates in V(D)J recombination to generate B-cell and T-cell receptor diversity. In animal models POLL seems to be important for light-chain rearrangements29. The potential role of POLL in B-cell and T-cell differentiation could explain that genetic variation in this gene influences pneumococcal meningitis susceptibility.
Previous genetic association studies in pneumococcal meningitis susceptibility have used a hypothesis driven approach in relatively small cohorts16,17,18,19,20,30. For example, association of a set of polymorphisms in the MBL2 gene resulted in a p-value of 0.017 (OR 8.21 [95% CI 1.05–64.09]) and the highest association of a SNP in the ADRB2 gene had a p-value of 0.007 (OR 1.52 [95% CI 1.12–2.07]) were reported. In our data the set of SNPs in MBL2 gene reached a p-value of 0.09 (OR 1.65 [95% CI 0.91–2.98]) and the same genetic variation in the ADRB2 gene a p-value of 0.164 (OR 1.12 [95% CI 0.1–1.3]). The associations found in those studies were not very strong and both studies concluded validation in larger populations is needed17,18. The challenge in genetic association studies looking at infectious diseases is the reproducibility of the data. The strength of our unique pneumococcal meningitis patient cohort is the homogenous group of patients but we are limited in replication of our findings in a similar group of patients. Especially since we found one gene with exome wide significance, replication of this association within another patient population is essential to validate this result.
Another limitation of our study is that by using the exome SNP array, it remains unclear if the associated genetic variants are also the causal variants with a biological function. Potentially the identified variants are in linkage disequilibrium -pair wise correlation between nearby genetic variants- with the causal variants.
We observed minimal deviation from the null for variants with MAF > 0.2 with a p-value < 0.01 (see supplement Fig. 1A). Permutation-based approach did increase lambda to 1.02, but did not affect the early deviation from the null. Likely causes of the early deviation are the technical (genotyping) and population differences between the cases and controls, or missing covariates such as antibiotic use or occurrence of co-infections (supplement Fig. 1C).
Our current study did not detect previously reported associations. This could be explained by our homogenous category of pneumococcal meningitis patients. Susceptibility to invasive pneumococcal infections is strongly dependent on host-pathogen interaction, therefore specifying the causing pathogen and type of infection can be crucial in determining specific risk genes for a complex trait like pneumococcal meningitis.
Conclusion
Our study reveals new genetic variants associated with the susceptibility to pneumococcal meningitis. These genetic variations were just below exome-wide significance. We found one gene significantly associated when looking for associations between multiple common and rare variants with pneumococcal meningitis susceptibility, namely the COL11A1 gene. These findings need to be replicated in an independent pneumococcal meningitis patient cohort. Following replication studies, the role of these genes in the pathogenesis of pneumococcal meningitis needs to be determined. Increasing our biological insight into the pathogenesis of bacterial meningitis brings us one step closer to understanding the disease and discovering new therapeutic and preventive strategies.
Methods
Study cohort and data collection
Patients included in this study are part of a prospective nationwide observational cohort study in the Netherlands; the MeninGene study4. For this study, all patients of 16 years and older with a community acquired bacterial meningitis were included from 2006 to 2011. Bacterial meningitis was defined by positive culture of a bacterial pathogen in the cerebrospinal fluid or a positive polymerase chain reaction test for a bacterial pathogen in the cerebrospinal fluid in combination with specific cerebrospinal fluid findings indicative of bacterial meningitis31. The Netherlands Reference Laboratories for Bacterial Meningitis (NRLBM) provided researchers of the MeninGene study with a daily update of where patients with bacterial meningitis have been admitted in the preceding days. Furthermore, physicians could contact the researchers to include patients with a 24/7 telephone service. Patients with hospital acquired bacterial meningitis, severe head trauma or neurosurgery in the previous month, or with a neurosurgical device in the central nervous system were excluded. Online case-record forms were used to collect data on patients’ history, symptoms and laboratory findings during admission, treatment, clinical course during hospitalized period and outcome at discharge.
Blood for DNA extraction was drawn from the patients and collected in sodium/EDTA tubes. Isolation of the DNA was performed with the Gentra Puregene isolation kit (Qiagen) according to manufacturer’s protocol. Thereafter the yield and quality of the extractions were determined to ensure appropriate conditions for genotyping.
The control population comprised 2072 individuals from the Utrecht Health project32. The Utrecht Health Project (UHP) is an ongoing dynamic population study initiated in a newly developed large residential area in Leidsche Rijn, part of the city of Utrecht. All new inhabitants were invited by their general practitioner to participate in the UHP. Of all participants an individual health profile (IHP) was made by dedicated research nurses. In a sample of the UHP participants genomic data has been collected.
Genotyping and quality control
All bacterial meningitis patients and controls were genotyped using the Illumina Exome array v1.1 in collaboration with the Human Genome Facility and the department of Epidemiology, Erasmus MC, the Netherlands as part of the Netherlands ExomeChip Project. After genotyping we performed calling on our MeninGene samples and to improve calling accuracy samples were joined with UHP and other Utrecht BBMRI-NL cohorts comprising N = 9844 samples. The samples from the other Utrecht BBMRI-NL cohorts were excluded after quality control (QC). All autosomal SNPs were called using GenomeStudio software, from Illumina, using the GenTrain 2.0 cluster algorithm using the protocol as described in Guo et al.33.
Called data was exported in PLINK format for final sample and SNP QC34. High quality, independent SNPs with less than 1% missing values, no significant differences in missing values between cases and controls, minor allele frequency (MAF) > 5% and Hardy-Weinberg equilibrium (HWE) > 1 × 10−3 were used to determine relatedness, heterozygostiy and ancestry. Individuals with a minimal second degree of relatedness, or higher degree relatedness and heterozygosity above or below three standard deviations were left out. To avoid population stratification we used multidimensional scaling (MDS), implemented in PLINK, on all meningitis cases, controls and HapMap samples to select only those patients that clustered with the European CEU HapMap samples (Supplementary Fig. 2)35. Finally we performed QC by removing all SNPs that had >5% missing values and HWE p-value <1 × 10−6.
Statistical analyses
Differences between cases and controls were calculated using the Fisher’s exact test on allelic association using PLINK 1.936. To correct for multiple testing we applied the Bonferroni correction (P < 0.05/number of analyzed SNPs). To investigate if permutation-adjustment removed the inflation permutation adjusted p-values after 1,000,000 permutations have been calculated using PLINK. Because we excluded all the variants with a frequency below 1% we also wanted to test if there was a significant association between pneumococcal meningitis patients and healthy controls with the inclusion of these rare variants. To test this we applied the gene-based test Sequence Kernel Association Test (SKAT) adaptive sum to test the association of the phenotype and SNP sets per gene with all variations having the same weight22,37. Secondly we tested all genes with the combined test of rare and common variants to look at the effect of rare variants22,37. For the gene-based SKAT test we corrected for any population stratification by including the first four MDS dimensions. To determine the proportion of case-control imbalance we used implemented hybrid-resampling method built in SKAT22,37.
Ethics statement
The MeninGene study was approved by the ethics committee of the Academic Medical Center, Amsterdam, The Netherlands. Ethical approval was granted in all participating centers and written informed consent was obtained from all participating individuals or legally authorized representatives. The study was conducted according to the principles of the Declaration of Helsinki (version of 2013, Fortaleza, Brazil) and in accordance with the Medical Research Involving Human Subjects Act (WMO) and other guidelines, regulations and acts. The UHP study was approved by the Medical Ethical Committee of the University Medical Center, Utrecht, The Netherlands and written informed consent was obtained from all participants.
Additional Information
How to cite this article: Kloek, A. T. et al. Exome Array Analysis of Susceptibility to Pneumococcal Meningitis. Sci. Rep. 6, 29351; doi: 10.1038/srep29351 (2016).
References
McIntyre, P. B., O’Brien, K. L., Greenwood, B. & van de Beek, D. Effect of vaccines on bacterial meningitis worldwide. Lancet 380, 1703–1711, doi: 10.1016/S0140-6736(12)61187-8 (2012).
Lozano, R. et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2095–2128, doi: 10.1016/S0140-6736(12)61728-0 (2012).
Brouwer, M. C., Tunkel, A. R. & van de Beek, D. Epidemiology, diagnosis and antimicrobial treatment of acute bacterial meningitis. Clinical microbiology reviews 23, 467–492, doi: 10.1128/CMR.00070-09 (2010).
Bijlsma, M. W. et al. Community-acquired bacterial meningitis in adults in the Netherlands, 2006-14: a prospective cohort study. Lancet Infect Dis, doi: 10.1016/S1473-3099(15)00430-2 (2015).
Weisfelt, M., van de Beek, D., Spanjaard, L., Reitsma, J. B. & de Gans, J. Clinical features, complications and outcome in adults with pneumococcal meningitis: a prospective case series. The Lancet. Neurology 5, 123–129, doi: 10.1016/S1474-4422(05)70288-X (2006).
Weisfelt, M. et al. Dexamethasone and long-term outcome in adults with bacterial meningitis. Annals of neurology 60, 456–468, doi: 10.1002/ana.20944 (2006).
van de Beek, D. et al. Clinical features and prognostic factors in adults with bacterial meningitis. The New England journal of medicine 351, 1849–1859, doi: 10.1056/NEJMoa040845 (2004).
Hoogman, M., van de Beek, D., Weisfelt, M., de Gans, J. & Schmand, B. Cognitive outcome in adults after bacterial meningitis. J.Neurol.Neurosurg.Psychiatry 78, 1092–1096 (2007).
de Gans, J., van de Beek, D. & European Dexamethasone in Adulthood Bacterial Meningitis Study, I. Dexamethasone in adults with bacterial meningitis. The New England journal of medicine 347, 1549–1556, doi: 10.1056/NEJMoa021334 (2002).
van de Beek, D., Brouwer, M. C., Thwaites, G. E. & Tunkel, A. R. Advances in treatment of bacterial meningitis. Lancet 380, 1693–1702, doi: 10.1016/S0140-6736(12)61186-6 (2012).
van de Beek, D., de Gans, J., Tunkel, A. R. & Wijdicks, E. F. Community-acquired bacterial meningitis in adults. The New England journal of medicine 354, 44–53, doi: 10.1056/NEJMra052116 (2006).
Adriani, K. S., Brouwer, M. C. & van de Beek, D. Risk factors for community-acquired bacterial meningitis in adults. The Netherlands journal of medicine 73, 53–60 (2015).
Adriani, K. S., Brouwer, M. C., van der Ende, A. & van de Beek, D. Bacterial meningitis in adults after splenectomy and hyposplenic states. Mayo Clinic proceedings 88, 571–578, doi: 10.1016/j.mayocp.2013.02.009 (2013).
Adriani, K. S., van de Beek, D., Brouwer, M. C., Spanjaard, L. & de Gans, J. Community-acquired recurrent bacterial meningitis in adults. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 45, e46–e51, doi: 10.1086/520682 (2007).
van de Beek, D., Drake, J. M. & Tunkel, A. R. Nosocomial bacterial meningitis. The New England journal of medicine 362, 146–154, doi: 10.1056/NEJMra0804573 (2010).
Brouwer, M. C. et al. Host genetic susceptibility to pneumococcal and meningococcal disease: a systematic review and meta-analysis. Lancet Infect Dis 9, 31–44, doi: 10.1016/S1473-3099(08)70261-5 (2009).
Brouwer, M. C., Baas, F., van der Ende, A. & van de Beek, D. Genetic variation and cerebrospinal fluid levels of mannose binding lectin in pneumococcal meningitis patients. PloS one 8, e65151, doi: 10.1371/journal.pone.0065151 (2013).
Adriani, K. S. et al. Genetic variation in the beta2-adrenocepter gene is associated with susceptibility to bacterial meningitis in adults. PloS one 7, e37618, doi: 10.1371/journal.pone.0037618 (2012).
Lingappa, J. R. et al. Identifying host genetic risk factors in the context of public health surveillance for invasive pneumococcal disease. PloS one 6, e23413, doi: 10.1371/journal.pone.0023413 (2011).
Chapman, S. J. et al. Common NFKBIL2 polymorphisms and susceptibility to pneumococcal disease: a genetic association study. Crit Care 14, R227, doi: 10.1186/cc9377 (2010).
van de Beek, D. Progress and challenges in bacterial meningitis. Lancet 380, 1623–1624, doi: 10.1016/S0140-6736(12)61808-X (2012).
Ionita-Laza, I., Lee, S., Makarov, V., Buxbaum, J. D. & Lin, X. Sequence kernel association tests for the combined effect of rare and common variants. American journal of human genetics 92, 841–853, doi: 10.1016/j.ajhg.2013.04.015 (2013).
Fichard, A., Kleman, J. P. & Ruggiero, F. Another look at collagen V and XI molecules. Matrix biology : journal of the International Society for Matrix Biology 14, 515–531 (1995).
Annunen, S. et al. Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. American journal of human genetics 65, 974–983, doi: 10.1086/302585 (1999).
Heider, M. R. & Munson, M. Exorcising the exocyst complex. Traffic 13, 898–907, doi: 10.1111/j.1600-0854.2012.01353.x (2012).
Evers, C. et al. Mosaic deletion of EXOC6B: further evidence for an important role of the exocyst complex in the pathogenesis of intellectual disability. American journal of medical genetics. Part A 164A, 3088–3094, doi: 10.1002/ajmg.a.36770 (2014).
Li, Y. F. et al. FSCB, a novel protein kinase A-phosphorylated calcium-binding protein, is a CABYR-binding partner involved in late steps of fibrous sheath biogenesis. J Biol Chem 282, 34104–34119, doi: 10.1074/jbc.M702238200 (2007).
Palmer, R. H., Ridden, J. & Parker, P. J. Cloning and expression patterns of two members of a novel protein-kinase-C-related kinase family. Eur J Biochem 227, 344–351 (1995).
Bertocci, B., De Smet, A., Weill, J. C. & Reynaud, C. A. Nonoverlapping functions of DNA polymerases mu, lambda and terminal deoxynucleotidyltransferase during immunoglobulin V(D)J recombination in vivo. Immunity 25, 31–41, doi: 10.1016/j.immuni.2006.04.013 (2006).
Adriani, K. S. et al. Common polymorphisms in the complement system and susceptiblity to bacterial meningitis. The Journal of infection 66, 255–262, doi: 10.1016/j.jinf.2012.10.008 (2013).
Spanos, A., Harrell, F. E., Jr. & Durack, D. T. Differential diagnosis of acute meningitis. An analysis of the predictive value of initial observations. JAMA 262, 2700–2707, doi: DOI 10.1001/jama.262.19.2700 (1989).
Grobbee, D. E. et al. The Utrecht Health Project: optimization of routine healthcare data for research. European journal of epidemiology 20, 285–287 (2005).
Guo, Y. et al. Illumina human exome genotyping array clustering and quality control. Nature protocols 9, 2643–2662, doi: 10.1038/nprot.2014.174 (2014).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics 81, 559–575, doi: 10.1086/519795 (2007).
International HapMap, C. et al. Integrating common and rare genetic variation in diverse human populations. Nature 467, 52–58, doi: 10.1038/nature09298 (2010).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Giga Science 4, 7, doi: 10.1186/s13742-015-0047-8 (2015).
Wu, M. C. et al. Rare-variant association testing for sequencing data with the sequence kernel association test. American journal of human genetics 89, 82–93, doi: 10.1016/j.ajhg.2011.05.029 (2011).
Acknowledgements
This study makes use of data generated by the Netherlands ExomeChip Project. A full list of the investigators is available from www.bbmri.nl. Funding for the project was provided by the Netherlands Organization for Scientific Research under award number 184021007, dated July 9, 2009 and made available as a Rainbow Project of the Biobanking and Biomolecular Research Infrastructure Netherlands (BBMRI-NL). This work was supported by grants from the European Research Council (ERC Starting Grant [proposal/contract 281156]), Netherlands Organization for Health Research and Development (ZonMw; NWO-Vidi grant 2010 [proposal/contract 016.116.358]), both to Diederik van de Beek. The Netherlands Reference Laboratory for Bacterial Meningitis is supported by the National Institute of Public Health and the Environmental Protection, Bilthoven. The Utrecht Health Project received grants from the Ministry of Health, Welfare and Sports (VWS), the University of Utrecht, the Province of Utrecht, the Dutch Organisation of Care Research, the University Medical Centre of Utrecht and the Dutch College of Healthcare Insurance Companies. Folkert W. Asselbergs is supported by a Dekker scholarship-Junior Staff Member 2014T001 – Netherlands Heart Foundation and UCL Hospitals NIHR Biomedical Research Centre.
Author information
Authors and Affiliations
Contributions
A.T.K. and B.F. carried out the experiments, analyzed data and wrote the manuscript. J.v.S. contributed to the quality control and analyzing of the data. A.v.d.E., M.L.B., F.W.A., M.C.B. and D.v.d.B. contributed to the design and cohort collection of this study. M.V.S., M.C.B. and D.v.d.B. advised on the first version of the manuscript. All authors read, revised and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kloek, A., van Setten, J., van der Ende, A. et al. Exome Array Analysis of Susceptibility to Pneumococcal Meningitis. Sci Rep 6, 29351 (2016). https://doi.org/10.1038/srep29351
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep29351
This article is cited by
-
Host genetic variability and pneumococcal disease: a systematic review and meta-analysis
BMC Medical Genomics (2019)
-
Community-acquired bacterial meningitis
Nature Reviews Disease Primers (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
