
Abstract
Orchardgrass (Dactylis glomerata L.) is one of the most economically important perennial, cool-season forage species grown and pastured worldwide. High-density genetic linkage mapping is a valuable and effective method for exploring complex quantitative traits. In this study, we developed 447,177 markers based on SLAF-seq and used them to perform a comparative genomics analysis. Perennial ryegrass sequences were the most similar (5.02%) to orchardgrass sequences. A high-density linkage map of orchardgrass was constructed using 2,467 SLAF markers and 43 SSRs, which were distributed on seven linkage groups spanning 715.77 cM. The average distance between adjacent markers was 0.37 cM. Based on phenotyping in four environments, 11 potentially significant quantitative trait loci (QTLs) for two target traits–heading date (HD) and flowering time (FT)–were identified and positioned on linkage groups LG1, LG3 and LG5. Significant QTLs explained 8.20–27.00% of the total phenotypic variation, with the LOD ranging from 3.85–12.21. Marker167780 and Marker139469 were associated with FT and HD at the same location (Ya’an) over two different years. The utility of SLAF markers for rapid generation of genetic maps and QTL analysis has been demonstrated for heading date and flowering time in a global forage grass.
Similar content being viewed by others

Introduction
Orchardgrass (Dactylis glomerata L.) is one of the top four most economically important perennial forage grasses and is native to northern Africa, Europe and temperate Asia1,2. Tetraploid orchardgrass is the most widespread among the more than 200 cultivars currently available1 and it has been naturalized on almost every continent due to its extensive uses for forage, hay and pasture3,4. Flowering time (FT) is key to the productivity of many flowering plants5. Early-flowering cultivars tend to have higher seed yield, while late-flowering cultivars are desirable in pastures because livestock avoid consuming flowering stems6. Late-heading date cultivars are also used when mixing orchardgrass with legumes7,8. However, because of the large genome and autotetraploid status of orchardgrass, it is difficult for growers and breeders to easily identify the genes controlling FT. Therefore, revealing the placement of FT control genes in linkage groups and developing species-specific markers for marker-assisted selection (MAS) is of significant value in breeding programs.
MAS is in high demand in molecular breeding programs for its independence from the environment and high efficiency for selection of desirable lines. Construction of high-density molecular genetic maps and identification of quantitative trait loci (QTLs) are critical steps for MAS. For example, 23 traits, including developmental, morphological and phenological traits, were detected based on the linkage maps of two major tetraploid switchgrass cultivars (Alamo-A4 and Kanlow-K5)9. High-density genetic maps for two diploid Miscanthus sinensis cultivars were produced and three QTLs for zebra stripe intensity (zbi1, zbi2 and zbi3 on linkage groups 7, 10 and 3) were identified10. A genetic map with 434 restriction-site associated DNA (RAD) markers and simple sequence repeat (SSR) markers in perennial ryegrass allowed the concentrations of palmitic, stearic, linoleic and a-linolenic acids to be identified11. The high-density genetic maps of Zoysia japonica were compared with those of other species and provided insights into genome evolution in the Chloridoideae12.
In the past few years, much progress has been made in the construction of genetic markers and linkage maps in orchardgrass. The first orchardgrass map was constructed using 164 SSRs and 108 sequence-related amplified polymorphism (SRAP) markers for two Chinese diploid cultivars, 01996 and YA02-103, with average distances of 9.6 cM in the male map and 8.9 cM in the female map13. More recent orchardgrass linkage maps were constructed for tetraploid orchardgrass based on either SSR or amplified fragment length polymorphism (AFLP) markers with relatively low marker density14,15. Among these markers, SSRs are the most widely used due to their conservation, synteny and superior transferability16. Recently, 606 polymorphic SSR markers were developed from a Japanese orchardgrass variety, Akimidori II2,17. Orchardgrass expressed sequence tag (EST) libraries constructed from three cultivars–Latar, Paiute and Potomac–were used to develop SSR markers pertaining to salt, drought and cold stress17. Though some species-specific orchardgrass markers were developed17,18, the number of traditional genetic markers is still very limited for high-density map construction.
As next-generation sequencing (NGS) technology has become available, it is now possible to achieve dense marker coverage without a reference genome19. For example, NGS-based technologies such as RAD-seq20 and specific-locus amplified fragment sequencing (SLAF-seq)21 have recently been developed and could provide an ample number of markers for high-density genetic map construction. These methods have been used in many species for map construction, including the use of RAD-seq in perennial ryegrass11 and the use of SLAF-seq in soybean22, sesame23, cucumber24 and walnut25. Combining NGS and reduced representation libraries (RRL), SLAF-seq is a very time- and cost-effective method. The efficiency of SLAF-seq was tested on data from rice and soybean. Results showed that the marker arrangement and order were consistent between the map and genome in rice, with the genetic map consisting of 12 linkage groups corresponding to the 12 rice chromosomes26. A consistent locus on Gm13 was detected by QTL mapping and genome-wide association study (GWAS) mapping approaches in soybean27. SLAF-seq was demonstrated to be an ideal tool with high resolution for large-scale genotyping in QTL mapping or gene discovery studies. In this study, we constructed a high-density genetic map by combining SLAF and SSR markers and we identified QTLs associated with heading date (HD) and FT using these markers. The result is, to our knowledge, the densest genetic map in orchardgrass. In addition, our analysis was based on field evaluations over two years and at two different locations. This map should serve as a platform for future identification and genetic dissection of many other complex and important traits.
Results
Analysis of SLAF-seq data
In total, we obtained 291.21 M reads after SLAF library construction and high-throughput sequencing. The average percentage of Q30 bases (bases with a quality score of 30, indicating a 1% chance of an error and thus 99% confidence) was 89.4% for all reads. The average guanine-cytosine (GC) content was 45%. Arabidopsis thaliana (Columbia ecotype, genome size = 119.48M) was used as a control to estimate the validity of library construction. We generated 93,742 total A. thaliana reads with 89.29% Q30 bases and 46.46% GC content (Supplemental Table 1). A total of 301,898 and 303,836 SLAFs were generated for the orchardgrass male and female parents, respectively. The average number of SLAFs in the progeny was 172,334 (Supplemental Table 2). We detected 447,177 high-quality SLAF markers in total. These SLAF markers could be separated into three groups: 89,038 polymorphic markers (including mapped biallelic markers and unmapped biallelic markers), 8,777 repetitive markers (mutiallelic markers with tag number larger than 4 in parents) and 349,362 non-polymorphic markers (monomorphic markers with only one tag in parents) (Supplemental Table 3). Mutiallelic SLAFs which could not be used for recombination rate calculating were removed from further analysis. Of the 89,038 polymorphic SLAF markers, 52,600 biallelic markers were further classified into eight segregation patterns (Supplemental Fig. 1). The CP (common parent) population was obtained from the F1 hybrid offspring of two fully homozygous parents. Therefore, only the aa × bb segregation pattern, which included 15,891 SLAF markers, could not be used to construct the genetic map. The remaining 36,709 polymorphic SLAFs were therefore used to construct the genetic map.
Linkage map construction
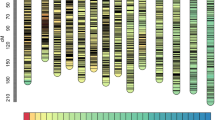
After a five-step filtering process (see Methods), 2,922 polymorphic SLAFs (Supplementary Table 4) and 114 SSR markers that followed the 1:1 Mendelian segregation pattern were used for constructing the orchardgrass genetic map using HighMap software. Two linkage maps, one for each parent, were constructed. The female map spanned 666.80 cM, with 1,594 markers and an average inter-marker spacing of 0.48 cM (Supplementary Table 5). The length of the male parent map was 658.74 cM, with 1,024 markers and an average inter-marker spacing of 0.89 cM (Supplementary Table 6). A combined outbred linkage map was constructed from the two parent maps using 2,467 SLAFs and 43 SSRs (2,510 total markers) arranged into seven linkage groups (LGs) (Fig. 1, Table 1). The length of the combined map was 715.77 cM and the average distance between adjacent markers of the map was 0.37 cM. The number of markers in each LG varied from 161 to 688, with an average of 359 markers per LG. The sizes of the individual LGs ranged from 60.85 to 149.66 cM, with average inter-marker distances of 0.19–0.68 cM. LG1 was the longest group, with 688 loci spanning 149.66 cM, while LG4 was the shortest, with 326 loci spanning 60.85 cM. LG4 was also the densest, whereas LG7 contained the fewest markers. The SSR markers were distributed across LG1, LG2, LG3, LG6 and LG7. Several markers were mapped to the same linkage group when comparing our map and our previous tetraploid map14 (Supplemental Fig. 2). For example, MarkerOG537N2 and MarkerOG677N2, located in LG1 of that previous tetraploid map, were grouped in A-H1 and L-H1 (A and L are the parents and H is the combined linkage group). Similarly, MarkerOG365N1 and MarkerOG365N3, located in LG2, were grouped in A-H2 and MarkerOG402N2, located in LG6, was found in L-H6. This result supports that our current high-density linkage map of orchardgrass can be a reliable reference for mapping important traits and for comparing and combining different genetic maps.

Distribution of SLAF and SSR markers on seven linkage groups.
The linkage group number is shown on the x-axis and genetic distances (cM) are shown on the y-axis.
Comparative genome analysis
The 447,177 high-quality SLAF markers generated from orchardgrass were compared with the genome sequences of four other grass species: perennial ryegrass (Lolium perenne L.), barley (Hordeum vulgare L.), rice (Oryza sativa L.) and the model plant species Brachypodium distachyon. The total numbers of matching marker containing sequences between orchardgrass and each species were 22,448 (5.02%) to perennial ryegrass; 19,207 (4.30%) to barley; 14,478 (3.24%) to B. distachyon; and 5,432 (1.21%) to rice (Table 2). The 2,259 mapped SLAF markers were also assessed in the other four grass species. The mapped markers followed a similar trend to that of all markers, with 124 (5.02%) matching to perennial ryegrass, 73 (2.96%) matching to barley, 63 (2.55%) matching to B. distachyon and 15 (0.61%) matching to rice. These results reflect the fact that orchardgrass is more closely related to perennial ryegrass than to the other three grasses. A Circos plot was used to show the linear relationships between orchardgrass and barley, rice and B. distachyon (Supplemental Fig. 3), illustrating a correspondence between the mapped SLAF markers and their genomic locations. Comparison of the orchardgrass LGs and the barley chromosomes showed that each of the seven LGs was primarily divided into two parts that were distributed across different barley chromosomes. Although there were only 15 mapped markers between orchardgrass and rice, rice chromosomes 1, 2, 4 and 8 with two mapped markers were divided to form LG1, 2, 4, 5, 6 in orchardgrass. For B. distachyon, only one marker in LG3 could be identified on chromosome 3 using BLAST. All markers in LGs were also found across different chromosomes in B. distachyon. Although 461,845 scaffolds have been assembled in perennial ryegrass, only 590 scaffolds were included in its genetic map28. There was no correspondence between the 2,467 mapped orchardgrass SLAF markers and the 590 mapped perennial ryegrass scaffolds.
Phenotypic variation
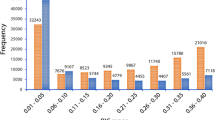
The FT of the orchardgrass population ranged from 72 to 145 days, with an average of 115 days across the four data sets (two years and two locations). The HD ranged from 53 to 120 days, with an average of 88 days. The shortest average FT was 99 days in data set E2 (2015-Ya’an) and the longest was 118 days in E1 (2015-Baoxing) (Table 3). For HD, the shortest average was 53 days in E2 and the longest was 69 in E1. The results showed that FT and HD exhibited similar trends across the different environments, indicating that the environment had almost the same influence on FT and HD. The coefficients of variation of FT ranged from 5.82% (E1) to 11.75% (E2) and the coefficients for HD ranged from 7.15% (E1) to 12.42% (E3, 2014-Hongya), demonstrating that E1 had the smallest influence on the traits and was more stable for genetic study than the other three environments. The spearman correlation coefficients was analyzed by SAS. The expected high correlation coefficients were calculated between FT and HD (Table 4). The two traits were normally distributed among the population over the two years (Fig. 2).

Frequency distributions of orchardgrass heading times and flowering times.
The x-axis indicates the days to heading and flowering. The left y-axis indicates the number of individuals and the right y-axis reflects the frequency distribution.
QTL analysis
An interval mapping model with a LOD (logarithm of odds) score of 2.5 for potential QTLs was used for QTL detection. In total, 11 significant QTLs for HD and FT were found to be distributed on LG1, LG3 and LG5 of the orchardgrass map (Table 5, Fig. 3). For FT, the QTL with the highest LOD score (8.69) was located at 12.01 cM of LG3 near Marker139469 (Table 5). The proportion of phenotypic variation explained by this QTL was 18.30%. For HD, the highest LOD score (12.21) was found at Marker126472, located at 18.31 cM of LG3 and explained 23.60% of the phenotypic variation. Marker167780 and Marker139469 were implicated in both FT and HD at the same experimental location (Ya’an) in both years. In order to explore potential candidate genes, we compared and annotated the significant QTLs for FT and HD. After annotation, Marker250988 was found to belong to the hd1 gene. One candidate gene, VRN1 (Lp_74D14_1 and Lp_7D23_1), was found by analysis of annotated genes.

QTL analysis of heading date and flowering time in linkage group 3 (LG3).
Logarithm of odds (LOD; blue) and percentage of phenotypic variance explained (Expl%; red) curves for heading time and flowering time of orchardgrass markers (x-axis) in LG3. The gray line indicates the threshold LOD score of 2.5.
Discussion
SLAF-seq is a recently developed NGS-based genotyping approach and it has been demonstrated to be highly accurate and cost-effective. SLAF-seq genotyping results have provided important information for genome evolution studies and molecular-assisted breeding. This method has recently been applied successfully to draft genomes29 and can be used to study genome evolution30 and construct high-density genetic maps in several plant species31,32,33,34. Because of its large genome size (4,300 Mb), heterozygosity and ploidy level, sequencing of the complete orchardgrass genome has been challenging35. The low cost and high accurancy of SLAF-seq makes it an approachable method for marker enrichment and molecular plant breeding. Genetic map construction and identification of species-specific markers for MAS are becoming the most feasible methods for development of molecular breeding programs for crop species without a reference genome. Here, we devolped high-throughput markers for orchardgrass. In total, 447,177 SLAF markers were developed and 2,922 filtered polymorphic markers were eventually identified for genetic linkage map construction. BLAST analysis was first performed between the orchardgrass SLAF markers and the genome sequences of four related grass species, confirming that orchardgrass is more closely related to perennial ryegrass than to the other three grass species. As Lolium and Dactylis are both in the Pooideae, perennial ryegrass could be regarded as a model plant for further genome analysis in orchardgrass. Orchardgrass was more closely related to B. distachyon, which has five chromosomes and belongs to the Festucoideae, than to rice, which has twelve chromosomes and belongs to the Oryzoideae, concurring that the Pooideae are more closely related to the Festucoideae than to the Oryzoideae. B. distachyon and barley are different species in same subfamily Festucoideae and we found that orchardgrass was more closely related to barley than to B. distachyon. It has been proposed that the common ancestor of all grass species most likely had seven chromosomes36. Following whole-genome duplication (WGD) or polyploidization, chromosome numbers usually decline and some genetic information may be lost. As a valuable and integral grass species, orchardgrass could be used to study this process in depth in the future.
Genetic maps are also important for genome sequence assembly, QTL mapping and comparative genomic analyses among grasses. The Z. japonica genetic map was also compared with that of other species to provide insights into genome evolution and chromosome number evolution in the Chloridoideae12. In our study, comparisons between mapped SLAF markers in orchardgrass and four other genome sequences showed similar trends to the BLAST results from all high-quality SLAF markers. A WGD/polyploidization event occurred before the divergence of the major cereals37, reflecting the distant relationship between orchardgrass and rice. Only one marker in LG3 was identified on chromosome 3 of B. distachyon with BLAST, whereas Bd21 lacked a member of the CO gene family (CO3) and a vernalization pathway gene (VRN2)37, showing that the genes corresponding to HD and FT QTLs in orchardgrass may not be present in B. distachyon. No correspondence was found between the perennial ryegrass and orchardgrass genetic maps, reflecting the fact that the perennial ryegrass genetic map has a low density of markers. Dactylis and Lolium are core forage and turf grasses. The study and utilization of the Pooideae are becoming more prevalent in grass research. However, this is the first time that the SLAF-seq technique was successfully applied to the development of markers in a forage grass with high specificity and stability.
A high-density genetic linkage map, containing the largest number of molecular markers among previously reported genetic maps23,24,25, was constructed with the SLAF-seq technique. However, because of the limited size of the mapping population and density of markers, many QTLs for important economic traits cannot be accurately detected. In this study, we identified 2,510 markers, including 2,467 SLAF markers and 43 SSR markers and grouped them into seven linkage groups for construction of an integrated map. This genetic map, with an average inter-marker distance of 0.37 cM, represents, to our knowledge, the most extensive orchardgrass genetic map to date. This map also has a much higher marker density than most genetic maps constructed with SLAF markers23,34,38,39,40,41,42. HighMap software and collinearity with the genome sequence32 were used to evalute the quality of previous genetic maps. The marker order and map distances of our map were validated with HighMap21. Since the genome of orchardgrass has not been sequenced, the collinearity could not be analyzed. We therefore used SSR markers selected from the first tetraploid orchardgrass genetic map to evaluate the quality of our map. SSR MarkerOG365N1 and MarkerOG365N3, located in LG2, were grouped in A-H2 and MarkerOG402N2, located in LG6, was found in L-H6. These results validate the reliability of our map. However, our genetic map can be further saturated to cover the whole genome with recently enriched orchardgrass-specific markers developed through different methods2,17,18.
Flowering time is an important trait of forage grass cultivars and is strongly affected by energy density and dry matter digestibility43. Heading date mainly determines adaptability to regional and environmental conditions and has been a major target in breeding programs44. Here, we measured FT and HD in four environments (two locations over two years) and analyzed the phenotypic variation and frequency distribution (Fig. 2). Previous studies showed that FT and HD are complex traits controlled by a network of regulatory genes involved in development and signal transduction43. Specifically, vernalization45,46 and photoperiodicity47,48 have been reported to affect the genetic and physiological mechamisms of grass FT and HD. One crucial and challenging factor in the genetic analysis of complex traits is the degree of phenotypic variation49. This study demonstrated a wide range of phenotypic variation in the FT and HD of orchardgrass in a segregating population in four environments. The shortest average FT and HD were 99 days and 53 days, respectively, in E2 and the longest were 118 days and 69, respectively, in E1 (Table 3). This indicates that the two traits respond similarly to different environments. The range of FT and HD variation showed that E1 had the smallest influence on the traits, as there was a narrower range of variation than in the other three environments. The values of skewness were all nearly 0 for FT and HD at E1–E4, while the values of kurtosis were all around 3. The spearman correlation coefficients was analyzed by SAS which showed that these results indicated accurate phenotyping for QTL analysis (Table 4).
The mapping of QTLs in grasses is challenging, especially for autotetraploid and self-incompatible grasses with high heterozygosity and large genome sizes, such as orchardgrass35. Therefore, it is difficult to generate a permanent population and to develop sufficient numbers of molecular markers. The first HD QTL analysis in orchardgrass was reported using the F1 population of a ‘asch621’ and ‘him271’ cross. Xie et al.15 conducted a QTL study for HD using SSR and AFLP markers; the sequence region harboring the two flanking SSR markers exhibited homology to physically mapped rice genes and explained 12–24% of the phenotypic variation. With advances in sequencing technology and its application in marker development, significant progress has been made in the analysis of FT and HD QTLs in key crops such as rice50, wheat51, barley52 and maize53. In the present study, 11 QTLs associated with HD and FT were found to be distributed across three LGs (LG1, LG3 and LG5). The results showed that the FT QTLs that were detected explained 8.20–27.00% of the phenotypic variance, while the HD QTLs explained 10.10–23.60% (Table 5). In previous research, many major and minor QTLs related to HD were detected in barley at different locations52. Similarly, in rice, several different QTLs associated with HD were located across different chromosomes and Hd1 was identified as a candidate gene by means of a map-based cloning strategy50. Five QTLs for vernalization response, measured as days to heading, were identified and mapped to four linkage groups in perennial ryegrass54, implicating the gene VRN1. These results showed that both HD and FT QTLs could be detected and identified by high-densty map constrution and QTL analysis. In our study, we detected several QTLs associated with HD and FT. After annotation, an HD-related QTL was annotated as belonging to the hd1 gene and an FT-related QTL was annotated as belonging to the VRN1 (Lp_74D14_1 and Lp_7D23_1) genes. The fact that these genes are associated with FT and HD is unsurprising, since vernalization has been reported to be associated with flowering and development in previous HD QTL analyses. Markers surrounding these QTLs could be listed as candidate genes for further screening and verification and applied to association analysis for fine mapping. One of the most significant results of this study was the mapping of an HD and FT QTL on LG3 that was consistently found across two different years. Future studies involving the genotyping of this region on LG3 with a handful of existing markers (Marker167780 and Marker139469) would be useful. Marker167780 and Marker139469, detected in all four environments, were not annotated with any related genes. These consistent QTLs with stable and significant effects on the phenotypes of FT and HD may be valuable resources for candidate gene exploration in the future.
Materials and Methods
Plant materials and DNA isolation
An F1 population of 213 individuals derived from the cross between two Chinese orchardgrass cultivars–‘Kaimo’ (early flowering time) and ‘01436’ (late flowering time)–was used for map construction. Plants were grown in a research field of the Grassland Science Department, Sichuan Agricultural University in Ya’an, Sichuan, China. Each individual plant in the F1 population was clonally divided in the Baoxing Research Field in the summer of 2013. Two replicates of each of the 213 genotypes in the mapping population and the two parental lines were planted in October of 2013. The first few young of each genotype were collected for DNA isolation. Samples were lyophilized and stored at −20 °C until use. Total genomic DNA was isolated using the Plant Genomics DNA Kit (TIANGEN Biotech) according to the manufacturer’s instructions. The concentration and quality of genomic DNA were tested by agarose gel electrophoresis and ND-1000 Spectrophotometer (NanoDrop).
SLAF and SSR data analysis and genotyping
Genomic DNA samples of the two parents and 213 F1 progeny individuals were subjected to SLAF-seq with some modifications21. Briefly, SLAFs were selected for paired-end sequencing on an Illumina HiSeq 2500 sequencing platform, performed by the Beijing Biomarker Technologies Corporation. The sequencing data obtained in our study has been deposited in the NCBI-short read archive (SRA) database under BioProject accession number (PRJNA321844). All paired-end reads (200 bp per read) generated from SLAF-seq raw reads were clustered according to sequence similarity. Identical reads were merged to avoid repeat computing requirements and sequences with over 90% identity were grouped into one SLAF locus, as described21. Differences in high-depth fragments were defined as SNPs or indels. In order to construct a high-quality genetic map, we filtered the SLAFs using five criteria: 1) removal of SLAFs from parents where the sequencing depth was less than 10X; 2) removal of SLAFs with more than three SNPs; 3) removal of SLAFs with the aa × bb segregation pattern; 4) removal of SLAFs with missing in more than 30% of offspring; 5) removal of segregation-distorted markers (p < 0.01). SLAFs that passed the five-step filtering process were used to construct a high-density genetic map. In this study, SLAFs followed the 1:1 Mendelian segregation pattern were used for genetic map construction. The generated SLAF markers were compared with the whole-genome sequences of perennial ryegrass (Lolium perenne L.), barley (Hordeum vulgare L.), rice (Oryza sativa L.) and Brachypodium distachyon by BLAST using an E-value cutoff of 1e-10 and an identity cutoff of 90% and requiring two paired matches.
SSR primers (Supplementary Table 7) were selected from several previous articles14,15. PCR reactions were performed according to Xie et al., with slight modifications14,15. Following are the conditions used: 5 min denaturation at 94 °C; 35 cycles of 1 min at 94 °C, 30 s at 52–58 °C and 1 min at 72 °C; and then a final extension of 10 min at 72 °C and storage at 4 °C. The segregation data for SSR markers in the same population were detected and 43 of them were used for constructing the genetic map.
Map construction and QTL analysis
HighMap software, which consists of four modules–linkage grouping, marker ordering, error genotyping correction and map evaluation22–was used for map construction. The single-linkage clustering algorithm was used to cluster the markers into linkage groups. The SLAF and SSR markers were ordered in the linkage groups and genotyping errors were corrected using HighMap software. Based on the integrated map, significant loci associated with HD and FT were identified based on LOD scores larger than the 5% cutoff value determined through 1,000 permutation tests using the CIM method from “qtl” package of R. MapQTL6.055 was used to conduct logarithm of odds and percentage of phenotypic variance explained analysis and interval mapping (IM)56 was used to detect QTLs for target traits. HD and FT were investigated in 2014 and 2015 in Baoxing and Ya’an during the spring and summer seasons. HD and FT were recorded as the number of days from January 1st to the date when the first inflorescence fully emerged from the flag leaf and to the date of the first open floret on the plant, respectively15. The spearman correlation coefficient was analyzed by SAS.
Conclusions
In conclusion, a high-density genetic linkage map was constructed with 2,467 SLAF markers and 43 SSR markers in orchardgrass. This map will serve as an invaluable tool for MAS in orchardgrass breeding programs. Comparative genomic analysis provided an overview of the relationship between orchardgrass and four related grass species. QTL analysis revealed stable and significant effects on flowering time and heading date phenotypes and provided valuable avenues for candidate gene exploration in the future. These data provide useful genomic resources for molecular dissection of complex traits, genome sequence assembly and genetic improvement of the forage grass orchardgrass.
Additional Information
How to cite this article: Zhao, X. et al. Construction of high-density genetic linkage map and identification of flowering-time QTLs in orchardgrass using SSRs and SLAF-seq. Sci. Rep. 6, 29345; doi: 10.1038/srep29345 (2016).
References
Stewart, A. & Ellison, N. Dactylis. In Wild Crop Relatives: Genomic and Breeding Resources, Springer Berlin Heidelberg, 73–87 (2011).
Hirata, M., Yuyama, N. & Cai, H. Isolation and characterization of simple sequence repeat markers for the tetraploid forage grass Dactylis glomerata. Plant breeding 130, 503–506 (2011).
Hannaway, D. et al. Orchardgrass (dactylis glomerata L.). ([Corvallis, Or.]: Oregon State University Extension Service;[Olympia, Wash.]: Washington State University Cooperative Extension;[Moscow, Idaho]: University of Idaho Cooperative Extension System;[Washington, DC]: US Dept. of Agriculture, 1999).
Bushman, B. S., Robins, J. G. & Jensen, K. B. Dry matter yield, heading date and plant mortality of orchardgrass subspecies in a semiarid environment. Crop Science 52, 745–751 (2012).
Ream, T. S. et al. Interaction of photoperiod and vernalization determines flowering time of Brachypodium distachyon. Plant physiology 164, 694–709 (2014).
Casler, M. et al. Sparse-flowering orchardgrass represents an improvement in forage quality during reproductive growth. Crop Science 54, 421–429 (2014).
Van Santen, E. & Sleper, D. A. Orchardgrass. Cool-season forage grasses, 503–534 (1996).
Casler, M. et al. Genetic progress from 40 years of orchardgrass breeding in North America measured under hay management. Crop Science 40, 1019–1025 (2000).
Lowry, D. B. et al. QTLs for Biomass and Developmental Traits in Switchgrass (Panicum virgatum). BioEnergy Research 8, 1856–1867 (2015).
Liu, S. et al. High density genetic map of Miscanthus sinensis reveals inheritance of zebra stripe. GCB Bioenergy (2015).
Hegarty, M. et al. Genotyping by RAD sequencing enables mapping of fatty acid composition traits in perennial ryegrass (Lolium perenne (L.)). Plant biotechnology journal 11, 572–581 (2013).
Wang, F. et al. Sequence-tagged high-density genetic maps of Zoysia japonica provide insights into genome evolution in Chloridoideae. The Plant Journal (2015).
Xie, W. et al. Genetic maps of SSR and SRAP markers in diploid orchardgrass (Dactylis glomerata L.) using the pseudo-testcross strategy. Genome 54 (2011).
Song, Y. et al. Construction of a simple sequence repeat marker-based genetic linkage map in the autotetraploid forage grass Dactylis glomerata L. Grassland science 57, 158–167 (2011).
Xie, W., Robins, J. G., Bushman, B. S. & Gustafson, J. A genetic linkage map of tetraploid orchardgrass (Dactylis glomerata L.) and quantitative trait loci for heading date. Genome 55, 360–369 (2012).
Marques, C., Brondani, R., Grattapaglia, D. & Sederoff, R. Conservation and synteny of SSR loci and QTLs for vegetative propagation in four Eucalyptus species. Theoretical and Applied Genetics 105, 474–478 (2002).
Bushman, B. S. et al. Orchardgrass (Dactylis glomerata L.) EST and SSR marker development, annotation and transferability. Theoretical and applied genetics 123, 119–129 (2011).
Huang, L. et al. Identifying differentially expressed genes under heat stress and developing molecular markers in orchardgrass (Dactylis glomerata L.) through transcriptome analysis. Molecular ecology resources (2015).
Davey, J. W. et al. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nature Reviews Genetics 12, 499–510 (2011).
Baird, N. A. et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PloS one 3 (2008).
Sun, X. et al. SLAF-seq: an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PloS one 8, e58700 (2013).
Qi, Z. et al. A high-density genetic map for soybean based on specific length amplified fragment sequencing PLoS One 9.8, e104871 (2014).
Zhang, Y. et al. Construction of a high-density genetic map for sesame based on large scale marker development by specific length amplified fragment (SLAF) sequencing. BMC plant biology 13, 141 (2013).
Xu, X. et al. QTL mapping of cucumber fruit flesh thickness by SLAF-seq. Scientific reports 5 (2015).
Zhu, Y. et al. Construction of a high-density genetic map using specific length amplified fragment markers and identification of a quantitative trait locus for anthracnose resistance in walnut (Juglans regia L.). BMC genomics 16, 1–13, doi: 10.1186/s12864-015-1822-8 (2015).
Mao, D. et al. Multiple cold resistance loci confer the high cold tolerance adaptation of Dongxiang wild rice (Oryza rufipogon) to its high-latitude habitat. Theoretical and Applied Genetics, 1–13 (2015).
Zhao, X. et al. Loci and candidate gene identification for resistance to Sclerotinia sclerotiorum in soybean (Glycine max L. Merr.) via association and linkage maps. The Plant Journal 82, 245–255 (2015).
Byrne, S. L. et al. A synteny-based draft genome sequence of the forage grass Lolium perenne. The Plant Journal 84, 816–826 (2015).
Huang, S. et al. Draft genome of the kiwifruit Actinidia chinensis. Nature communications 4 (2013).
Chen, S. et al. The development of 7E chromosome-specific molecular markers for Thinopyrum elongatum based on SLAF-seq technology. PLoS One 8.6, e65122 (2013).
Xu, X. et al. A high-density genetic map of cucumber derived from Specific Length Amplified Fragment sequencing (SLAF-seq). Frontiers in plant science 5 (2014).
Guo, Y. et al. Using specific length amplified fragment sequencing to construct the high-density genetic map for Vitis (Vitis vinifera L. × Vitis amurensis Rupr.). Name: Frontiers in Plant Science 6, 393 (2015).
Zhang, J. et al. High-density genetic map construction and identification of a locus controlling weeping trait in an ornamental woody plant (Prunus mume Sieb. et Zucc). DNA Research, dsv003 (2015).
Wang, L. et al. Construction of a high-density linkage map and fine mapping of QTL for growth in Asian seabass. Scientific reports 5 (2015).
Unamba, C. I., Nag, A. & Sharma, R. K. Next Generation Sequencing technologies: The doorway to the unexplored genomics of non-model plants. Frontiers in Plant Science 6, doi: 10.3389/fpls.2015.01074 (2015).
Wang, X. et al. Telomere-centric genome repatterning determines recurring chromosome number reductions during the evolution of eukaryotes. New Phytologist 205, 378–389 (2015).
Higgins, J. A., Bailey, P. C. & Laurie, D. A. Comparative genomics of flowering time pathways using Brachypodium distachyon as a model for the temperate grasses. PloS one 5, e10065 (2010).
Li, B. et al. Construction of a high-density genetic map based on large-scale markers developed by specific length amplified fragment sequencing (SLAF-seq) and its application to QTL analysis for isoflavone content in Glycine max. BMC genomics 15, 1086 (2014).
Wei, Q. et al. An SNP-based saturated genetic map and QTL analysis of fruit-related traits in cucumber using specific-length amplified fragment (SLAF) sequencing. BMC genomics 15, 1158 (2014).
Wang, J. et al. Construction of Commercial Sweet Cherry Linkage Maps and QTL Analysis for Trunk Diameter. PloS one 10, e0141261 (2015).
Ma, J.-Q. et al. Large-Scale SNP Discovery and Genotyping for Constructing a High-Density Genetic Map of Tea Plant Using Specific-Locus Amplified Fragment Sequencing (SLAF-seq). PloS one 10, e0128798 (2015).
Cai, C., Cheng, F., Wu, J., Zhong, Y. & Liu, G. The First High-Density Genetic Map Construction in Tree Peony (Paeonia Sect. Moutan) using Genotyping by Specific-Locus Amplified Fragment Sequencing. PloS one 10, e0128584–e0128584 (2014).
Jung, C. & Müller, A. E. Flowering time control and applications in plant breeding. Trends in plant science 14, 563–573 (2009).
Fujino, K., Yamanouchi, U. & Yano, M. Roles of the Hd5 gene controlling heading date for adaptation to the northern limits of rice cultivation. Theoretical and applied genetics 126, 611–618 (2013).
Dubcovsky, J. et al. Effect of photoperiod on the regulation of wheat vernalization genes VRN1 and VRN2. Plant molecular biology 60, 469–480 (2006).
Trevaskis, B., Hemming, M. N., Peacock, W. J. & Dennis, E. S. HvVRN2 responds to daylength, whereas HvVRN1 is regulated by vernalization and developmental status. Plant Physiology 140, 1397–1405 (2006).
Serrano, G. et al. Chlamydomonas CONSTANS and the evolution of plant photoperiodic signaling. Current Biology 19, 359–368 (2009).
Jackson, S. D. Plant responses to photoperiod. New Phytologist 181, 517–531 (2009).
Xu, L. et al. Genome-wide association study reveals the genetic architecture of flowering time in rapeseed (Brassica napus L.). DNA Research, dsv035 (2015).
Yano, M. et al. Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. The Plant Cell 12, 2473–2483 (2000).
Chen, A. et al. PHYTOCHROME C plays a major role in the acceleration of wheat flowering under long-day photoperiod. Proceedings of the National Academy of Sciences 111, 10037–10044 (2014).
Cuesta-Marcos, A. et al. Heading date QTL in a spring × winter barley cross evaluated in Mediterranean environments. Molecular breeding 21, 455–471 (2008).
Buckler, E. S. et al. The genetic architecture of maize flowering time. Science 325, 714–718 (2009).
Jensen, L. B. et al. QTL mapping of vernalization response in perennial ryegrass (Lolium perenne L.) reveals co-location with an orthologue of wheat VRN1. Theoretical and Applied Genetics 110, 527–536, doi: 10.1007/s00122-004-1865-8 (2004).
Van Ooijen, J. MapQTL® 5, Software for the mapping of quantitative trait loci in experimental populations. Kyazma BV, Wageningen 63 (2004).
Jansen, R. C. Interval mapping of multiple quantitative trait loci. Genetics 135, 205–211 (1993).
Acknowledgements
This research work was funded by the National Basic Research Program (973 program) in China (No. 2014CB138705), the earmarked fund for Modern Agro-Industry Technology Research System (No. CARS-35-05) and the National Natural Science Foundation of China (NSFC 31372363). Finally, we would like to thank Shaun Bushman worked at United States Department of Agriculture Agricultural Research Service, Forage and Range Research Lab for helpful excellent technical assistance.
Author information
Authors and Affiliations
Contributions
L.H. and X. Zhang designed and conceived the experiments. X. Zhao, L.H., D.Y., J.L. and L.T. performed the experiments. X. Zhao, L.H., X.L. and T.S. analyzed the data. X. Zhao, L.H. and J.W. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhao, X., Huang, L., Zhang, X. et al. Construction of high-density genetic linkage map and identification of flowering-time QTLs in orchardgrass using SSRs and SLAF-seq. Sci Rep 6, 29345 (2016). https://doi.org/10.1038/srep29345
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep29345
This article is cited by
-
QTL mapping for growth-related traits by constructing the first genetic linkage map in Simao pine
BMC Plant Biology (2022)
-
Investigation of genetic relationships within three Miscanthus species using SNP markers identified with SLAF-seq
BMC Genomics (2022)
-
Genome-wide association study identifying genetic variants associated with carcass backfat thickness, lean percentage and fat percentage in a four-way crossbred pig population using SLAF-seq technology
BMC Genomics (2022)
-
Comparative Transcript Profiling Suggests Distinct Flowering Response of Early- and Late-Flowering Phenotypes in Forage Grass Dactylis glomerata L
Journal of Plant Growth Regulation (2021)
-
Identification of fruit size associated quantitative trait loci featuring SLAF based high-density linkage map of goji berry (Lycium spp.)
BMC Plant Biology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
