
Abstract
Adiponectin has been demonstrated to protect the cardiovascular system and bone marrow mesenchymal stem cells (BMSCs). However, it is unclear whether adiponectin can protect BMSCs against flow shear stress (FSS). In this study, our aim was to explore the effects of adiponectin on BMSCs and to explore the role of AMP-activated protein kinase (AMPK) signaling in this process. Shear stress significantly inhibits the survival and increases the apoptosis of BMSCs in an intensity-dependent manner. The expression levels of TGF-β, bFGF, VEGF, PDGF and Bcl2 are simultaneously reduced and the phosphorylation levels of AMPK and ACC, as well as the expression level of Bax, are increased. Supplementation with adiponectin promotes the survival of BMSCs; reverses the changes in the expression levels of TGF-β, bFGF, VEGF, PDGF, Bcl2 and Bax; and further amplifies the phosphorylation of AMPK and ACC. Furthermore, the protective effects of adiponectin can be partially neutralized by AMPK siRNA. In summary, we have demonstrated for the first time that adiponectin can effectively protect BMSCs from FSS and that this effect depends, at least in part, on the activation of AMPK signaling.
Similar content being viewed by others


Introduction
Valvular heart disease (VHD) refers to the structural and functional disorders of the valves and is a common and growing problem in clinics1. In industrialized countries, the prevalence of VHD is approximately 2.5 percent, most cases of which are attributed to aortic stenosis and mitral regurgitation2 and in the United States, VHD accounts for a significantly increasing number of deaths in the aging population3. Furthermore, the conditions are worse in developing countries, where rheumatic heart disease remains the leading cause of VHD2. Artificial heart valve replacement has become the most effective treatment for VHD, which replaces the native valves with mechanical or bioprosthetic valves, thereby prolonging the lifespan of patients with VHD4,5. However, prosthetic valves are not flawless. Mechanical valves are durable but are more prone to thrombosis and patients require lifelong anticoagulant therapy, which in turn increases the risk of hemorrhage. In contrast, bioprosthetic valves have outstanding hemodynamic performance but are degraded and calcified more easily4,5. Additionally, the inability to grow with pediatric patients is an even greater limitation of these prosthetic valves6. As a result, autologous tissue-engineered heart valves (TEHVs) have become the most attractive replacement valves because they can overcome the limitations of mechanical and bioprosthetic valves with their ability to remodel, regenerate and grow6,7. To engineer heart valves, harvested cells are seeded onto decellularized valvular scaffolds to generate a tissue-engineered construct in vitro. They are then implanted into the diseased heart8. The seeded cells used to construct the TEHVs mainly include adipose mesenchymal stem cells, endothelial progenitor cells and bone marrow mesenchymal stem cells (BMSCs)8,9,10. However, the current TEHVs do not adapt well to high shear stress when transplanted in vivo11. Therefore, there is a need to enhance the resistance of seeded cells to flow shear stress.
Adiponectin (APN, also known as adipocyte complement-related protein of 30 kD, adipoQ, apM1 and GBP28) is an adipokine secreted by adipose tissues and other cells, including cardiomyocytes12, whose expression levels are negatively correlated with cerebrovascular, cardiovascular and metabolic diseases, indicating an important role of adiponectin in the cardiovascular system13,14,15,16. Adiponectin exhibits protective effects on various cellular processes, including energy metabolism, inflammation and proliferation, performing anti-hyperglycemic, anti-inflammatory and anti-atherogenic functions17. In particular, adiponectin maintains myocardial cell survival, attenuates ischemia reperfusion injury (IRI) and protects the heart against pressure overload-induced dysfunction, as well as structural and metabolic remodeling18,19,20. Therefore, we speculated that adiponectin has a protective effect on BMSCs, whereby it increases the attachment of BMSCs to decellularized heart valve scaffolds, as well as increases the resistance of TEHVs to flow shear stress.
Adenosine monophosphate (AMP)-activated protein kinase (AMPK) is a serine/threonine protein kinase with high conservation in evolution that is involved in the regulation of cellular energy status21 by regulating the phosphorylation state of its substrates, especially acetyl CoA carboxylase (ACC)22. Its expression exerts a variety of effects on multiple tissues and organs, such as the liver, brain, skeletal muscle and heart23,24,25. The effects of AMPK activation include the metabolic regulation of glucose, cholesterol and fatty acids26, as well as cell growth, apoptosis and autophagy27. Importantly, it has been reported that the endogenous and exogenous activation of AMPK plays a role in heart protection, including the prevention of myocardial ischemic injury28, cardiac fibrosis21 and heart failure29, as well as protection against cardiac pressure overload30,31. Additionally, studies have shown that adiponectin can activate the AMPK-dependent signaling pathway, exerting its anti-IRI and anti-pressure overload actions18,20,32. Therefore, we hypothesized that the effects of adiponectin on maintaining the attachment of BMSCs to decellularized heart valve scaffolds and enhancing the resistance to flow shear stress are mediated by AMPK signaling.
This study was designed to investigate the effects of adiponectin on the activity and function of BMSCs to facilitate their adaption to FSS (FSS). Furthermore, we aimed to explore the underlying mechanism of AMPK signaling in the resistance of BMSCs to flow shear stress induced by adiponectin, contributing to the development of TEHVs against high pressure and flow in vivo.
Results
Characterization of cultured rat BMSCs
BMSCs were isolated and expanded from SD rats. Most cultured adherent cells showed the fibroblastic morphology that is characteristic of MSCs, particularly in the third-generation cells (Fig. 1A). In addition, FACS analysis demonstrated that BMSCs were 99.4% pure for CD90 and 99.6% pure for CD29. The percentages of contaminated populations of hematopoietic stem cells positive for CD34, CD45 and CD106 were 2.2%, 2.6% and 2.2%, respectively (Fig. 1B).

Morphology and phenotype of rat BM-MSCs.
(A) Rat BMSCs showed homogenous, fibroblast-like morphology especially at the third passage. (B) Fluorescence-activated cell sorting (FACS) analysis of immune markers in rat BMSCs. The results confirmed that rat BMSCs were positive for CD29 and CD90 but negative for CD34, CD45 and CD106.
Effect of FSS on the cellular metabolic viability and apoptosis of rat BMSCs
Four different magnitudes of FSS (0, 7.5, 15 and 30 dynes/cm2) were applied to the MSCs for 24 h. After being seeded in 96-well plates for 24 h, the cellular metabolic viability was measured by the CCK-8 assay after being further cultured for 2 h in a humidified atmosphere of 5% CO2 at 37 °C. The data are shown in Fig. 2A. Compared with the control group (without FSS), stimulation of BMSCs for 24 h with 7.5, 15, or 30 dynes/cm2 FSS inhibited cell viability in an intensity-dependent manner (P < 0.05; Fig. 2A, right). Microscopy images (Fig. 2A, left) indicated that the FSS stimulation resulted in significant cell shrinkage and reduced the rate of cellular attachment compared with the control group.

Effect of FSS on the cellular metabolic viability and apoptosis of rat BMSCs.
(A) Representative morphology of rat BMSCs was demonstrated following exposure to different intensities of FSS (0, 7.5, 15, or 30 dynes/cm2) for 24 h. (B) Apoptotic index of rat BMSCs subjected to FSS injury is shown. Apoptotic cells were visualized by green fluorescence. All data are presented as fold changes vs. the control. The results are expressed as the mean ± SD, n = 6. aaP < 0.05 vs. the control group; bbP < 0.05 vs. the 7.5 dynes/cm2 FSS-treated group; ccP < 0.05 vs. the 15 dynes/cm2 FSS-treated group.
The apoptotic index of FSS-treated rat BMSCs was also measured by a TUNEL assay. After stimulation with 7.5, 15, or 30 dynes/cm2 FSS for 24 h, the apoptotic index (Fig. 2B) increased from (1.56 ± 1.60)% to (31.28 ± 3.65)%, (45.76 ± 4.81)% and (59.62 ± 3.14)%, respectively (P < 0.05), showing an intensity-dependent increase in apoptosis induction.
Effects of FSS stimulation on local bFGF, TGF-β, VEGF and PDGF levels in rat BMSCs
To measure the levels of various trophic factors, ELISAs were used to analyze the protein supernatants obtained from the BMSC cultures from each group. As shown in Fig. 3, the levels of bFGF, TGF-β, VEGF and PDGF differed between the groups stimulated with FSS (7.5, 15 and 30 dynes/cm2) for 24 h. Additionally, the bFGF level was reduced in BMSCs from (426 ± 10.61)% to (363 ± 9.88)%, (323 ± 10.42)% and (140 ± 9.89)% following FSS stimulation at 7.5, 15 and 30 dynes/cm2, respectively (P < 0.05, Fig. 3A), while TGF-β levels were significantly decreased from (151 ± 9.57)% to (139 ± 10.62)%, (108 ± 11.51)% and (59.6 ± 5.88)% after FSS stimulation (P < 0.05, Fig. 3B). VEGF and PDGF have been reported to function as the ligands for PDGFR activation in MSCs33. After stimulation with different intensities of FSS, VEGF and PDGF levels changed in a similar manner from VEGF levels of (256 ± 10.44)% to (229 ± 11.29)%, (189 ± 10.26)% and (82.9 ± 8.66)% (P < 0.05, Fig. 3C) and from PDGF levels of (941 ± 16.94)% to (844 ± 15.88)%, (705 ± 16.46)% and (327 ± 15.51)%, respectively (P < 0.05, Fig. 3D).

Effects of FSS stimulation on local production of bFGF, TGF-β, VEGF and PDGF levels in rat BMSCs.
Differences in cytokine levels in BMSC lysates. Productions of these cytokines were measured by an enzyme-linked immunosorbent assay (ELISA). All data are presented as fold changes vs. the control. (A) Levels of bFGF. (B) Levels of TGF-β. (C) Levels of VEGF. (D) Levels of PDGF. The results are expressed as the mean ± SD, n = 6. aaP < 0.05 vs. the control group; bbP < 0.05 vs. the 7.5 dynes/cm2 FSS-treated group; ccP < 0.05 vs. the 15 dynes/cm2 FSS-treated group.
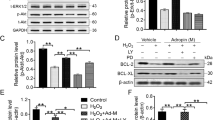
Effects of FSS stimulation on AMPK signaling and the Bcl2 protein family in rat BMSCs
Activation of the AMPK pathway plays an important role in the regulation of BMSCs in many diseases34,35. To investigate the role of AMPK signaling in FSS stimulation-induced BMSC injury, AMPK-related molecules were detected by Western blotting. As shown in Fig. 4, FSS stimulations at different intensities induced a dose-dependent up-regulation of the phosphorylation of AMPK (P < 0.05). As a downstream target of AMPK, ACC phosphorylation was also examined. After being stimulated with FSS, the phosphorylation of ACC was similarly enhanced (P < 0.05). Additionally, FSS stimulation decreased the expression of Bcl2 and increased the expression of Bax (P < 0.05).

Effects of FSS stimulation on AMPK signaling and the Bcl2 protein family in rat BMSCs.
BMSCs were stimulated with different intensities of FSS (0, 7.5, 15, or 30 dynes/cm2) for 24 h. Cell lysates were collected as described in the Materials and Methods section and subjected to Western blotting analysis with antibodies specific to p-AMPK, AMPK, p-ACC, ACC, Bcl2, Bax and β-actin; β-actin served as the control. The statistics are representative of bands for the control and treatment groups. The results are expressed as the mean ± SD, n = 6. aaP < 0.05 vs. the control group; bbP < 0.05 vs. the 7.5 dynes/cm2 FSS-treated group; ccP < 0.05 vs. the 15 dynes/cm2 FSS-treated group.
APN promoted the cellular viability of and reduced apoptosis in FSS-cultured rat BMSCs
To determine the effect of APN on the cellular viability of and apoptosis in rat BMSCs, the cells were exposed to the FSS environment with or without adiponectin pretreatment at different concentrations (10, 20 and 30 μg/mL) for 18 h36. In the experimental groups, BMSCs were chosen to undergo treatment with 15 dynes/cm2 FSS for 24 h because they were too severely damaged following treatment with 30 dynes/cm2 FSS according to our above trial (Fig. 2A). The cellular viability and apoptotic index of rat BMSCs were assessed by CCK-8 analysis and the TUNEL assay. The APN-treated BMSCs exhibited better survival than untreated MSCs under FSS stimulation, especially at the 30-μg/mL concentration (P < 0.05; Fig. 5A, right). Microscopy images (Fig. 5A, left) indicated that the APN treatments significantly ameliorated cell shrinkage and increased the rate of cellular attachment induced by FSS.

APN promoted the cellular viability of and reduced apoptosis in FSS-cultured rat BMSCs.
BMSCs were exposed to the FSS environment (15 dynes/cm2) with or without adiponectin pretreatment at different concentrations (10, 20 and 30 μg/mL) for 18 h. The cellular viability and apoptotic index were assessed as described in the Materials and Methods section. (A) Representative morphology of rat BMSCs. (B) Apoptotic index of rat BMSCs. Apoptotic cells were visualized by green fluorescence. All data are presented as fold changes vs. the control. The results are expressed as the mean ± SD, n = 6. aaP < 0.05 vs. the control group; bbP < 0.05 vs. the 15 dynes/cm2 FSS-treated group; ccP < 0.05 vs. the 15 dynes/cm2 FSS-treated + 10 μg/mL APN group; ddP < 0.05 vs. the 15 dynes/cm2 FSS-treated + 20 μg/mL APN group.
Additionally, the apoptotic index of APN-pretreated BMSCs exposed to the FSS environment was also measured. After pre-treatments with 10, 20 and 30 μg/mL APN for 18 h, the apoptotic index induced by FSS was reduced from (46.21 ± 2.14)% to (44.58 ± 3.27)%, (39.15 ± 3.83)% and (24.66 ± 4.01)%, respectively (P < 0.05, Fig. 5B), demonstrating a dose-dependent decrease in apoptosis induction.
APN restores the local production levels of bFGF, TGF-β, VEGF and PDGF in FSS-cultured rat BMSCs
The above experiments demonstrated the reductions of cytokines in BMSCs mediated by FSS injury. Next, the protective effect of adiponectin on cytokines in BMSCs subjected to FSS injury was examined in a similar manner. As demonstrated in Fig. 6A–D, pre-treatment with APN (10, 20, or 30 μg/mL) for 18 h resulted in a dose-dependent increase in bFGF production following FSS-induced injury, from (321 ± 11.49)% to (325 ± 10.61)%, (362 ± 11.65)% and (394 ± 9.88)%, respectively (P < 0.05, Fig. 6A). The TGF-β level was also significantly increased from (108 ± 6.21)% to (106 ± 6.87)%, (129 ± 5.88)% and (137 ± 7.68)%, respectively, after APN pre-treatment (P < 0.05, Fig. 6B). Furthermore, after APN administration (10, 20 and 30 μg/mL) for 18 h, VEGF and PDGF levels recovered in a similar manner from (186 ± 10.91)% to (186 ± 11.67)%, (201 ± 12.55)% and (236 ± 10.25)% for VEGF (P < 0.05, Fig. 6C) and from (698 ± 17.01)% to (687 ± 15.77)%, (760 ± 15.21)% and (827 ± 16.42)% for PDGF (P < 0.05, Fig. 6D).

APN restored the local production of bFGF, TGF-β, VEGF and PDGF in FSS-cultured rat BMSCs.
BMSCs were exposed to the FSS environment (15 dynes/cm2) with or without adiponectin pretreatment at different concentrations (10, 20 and 30 μg/mL) for 18 h. The cytokines, such as bFGF, TGF-β, VEGF and PDGF, were assessed as described in the Materials and Methods section. All data are presented as fold changes vs. the control. (A) Levels of bFGF. (B) Levels of TGF-β. (C) Levels of VEGF. (D) Levels of PDGF. The results are expressed as the mean ± SD, n = 6. aaP < 0.05 vs. the control group; bbP < 0.05 vs. the 15 dynes/cm2 FSS-treated group; ccP < 0.05 vs. the 15 dynes/cm2 FSS-treated + 10 μg/mL APN group; ddP < 0.05 vs. the 15 dynes/cm2 FSS-treated + 20 μg/mL APN group.
APN pre-treatment modulates AMPK signaling and the Bcl2 protein family in FSS-cultured rat BMSCs
To assess the impact of the APN pre-treatment on signaling pathways activated in FSS-cultured rat BMSCs, the expression and activity of AMPK signaling and the expression of the Bcl2 protein family were compared between the groups. APN pre-treatment further increased the phosphorylation of AMPK and ACC expression, which were altered in FSS-injured BMSCs (Fig. 7). Additionally, APN pre-treatment increased the expression of Bcl2 and decreased the expression of Bax (P < 0.05).

APN pre-treatment modulated AMPK signaling and the Bcl2 protein family in FSS-cultured rat BMSCs.
BMSCs were exposed to the FSS environment (15 dynes/cm2) with or without adiponectin pretreatment at different concentrations (10, 20 and 30 μg/mL) for 18 h. Cell lysates were collected as described in the Materials and Methods section and subjected to Western blotting analysis with antibodies specific to p-AMPK, AMPK, p-ACC, ACC, Bcl2, Bax and β-actin; β-actin served as the control. The statistics are representative of bands corresponding to the control and treatment groups. The results are expressed as the mean ± SD, n = 6. aaP < 0.05 vs. the control group; bbP < 0.05 vs. the 15 dynes/cm2 FSS-treated group; ccP < 0.05 vs. the 15 dynes/cm2 FSS-treated + 10 μg/mL APN group; ddP < 0.05 vs. the 15 dynes/cm2 FSS-treated + 20 μg/mL APN group.
APN protects against FSS-induced changes in cellular viability and cytokines in rat BMSCs via AMPK
To investigate the role of AMPK on the protective effects of APN, AMPK was silenced by siRNA, followed by APN (30 μg/mL) treatment for 18 h and FSS (15 dynes/cm2) for an additional 24 h. AMPK siRNA did not affect the cell viability and cytokines (P > 0.05 compared with the control group, Supplementary Fig. 1). However, AMPK siRNA transfection significantly attenuated the APN-induced up-regulation of AMPK (P < 0.05 compared with the control siRNA + APN + FSS group, Fig. 8). Furthermore, silencing AMPK reversed the protective effects of APN against FSS-induced BMSC death (P < 0.05 compared with the control siRNA + FSS group, Fig. 9A). Inhibition of AMPK by siRNA also reduced the productions of cytokines (P < 0.05 compared with the control siRNA + APN + FSS group, Fig. 9B), such as bFGF, TGF-β, VEGF and PDGF, in rat BMSCs.

AMPK siRNA inhibited the expression of its downstream proteins.
BMSCs were transfected with siRNA targeting AMPK or nonspecific scramble siRNA (control siRNA) for 48 h and then treated with APN (30 μg/mL) for 18 h, followed by exposure to FSS (15 dynes/cm2) for 24 h. Cell lysates were collected as described in the Materials and Methods section and subjected to Western blotting analysis with antibodies specific to p-AMPK, AMPK, p-ACC and ACC; AMPK and ACC served as controls. The statistics are representative of bands corresponding to the control and treatment groups. The results are expressed as the mean ± SD, n = 6. aaP < 0.05 vs. the control siRNA + 15 dynes/cm2 FSS-treated group; bbP < 0.05 vs. the control siRNA+ 30 μg/mL APN + 15 dynes/cm2 FSS-treated group; ccP < 0.05 vs. the AMPK siRNA+ 30 μg/mL APN + 15 dynes/cm2 FSS-treated group.

APN protected against FSS-induced changes in cellular viability and cytokines in rat BMSCs via AMPK.
BMSCs were transfected with siRNA targeting AMPK or nonspecific scramble siRNA (control siRNA) for 48 h and then treated with APN (30 μg/mL) for 18 h, followed by exposure to FSS (15 dynes/cm2) for 24 h. The cellular viability and cytokine levels were assessed as described previously. (A) Cell viability and representative morphology of rat BMSCs. (B) Levels of bFGF. (C) Levels of TGF-β. (D) Levels of VEGF. (E) Levels of PDGF. The results are expressed as the mean ± SD, n = 6. aaP < 0.05 vs. the control siRNA + 15 dynes/cm2 FSS-treated group; bbP < 0.05 vs. the control siRNA + 30 μg/mL APN + 15 dynes/cm2 FSS-treated group; ccP < 0.05 vs. the AMPK siRNA + 30 μg/mL APN + 15 dynes/cm2 FSS-treated group.
Discussion
VHD is a common and growing problem world-wide2,3. Artificial heart valve replacement has become the most effective treatment for VHD to prolong the lifespan and improve the life quality of patients4,5. However, there are problems that limit the utilization of prosthetic and mechanical valves4,5,6. TEHVs have attracted the most attention in valve replacement6,7. Additionally, adipose mesenchymal stem cells, endothelial progenitor cells and BMSCs are often used8,9,10. However, the current TEHVs do not adapt well to high shear stress when transplanted in vivo11. Luo et al.37 found that FSS caused a dose-related reduction of the proliferation rate of MSCs, with the majority of cells being arrested in the G0 or G1 phase in vitro. Using confocal microscopy, Cartmell and colleagues38 also observed that a high flow rate of 1.0 mL/min resulted in substantial cell death throughout the constructs after one week of culture. Other study has also demonstrated that a high flow velocity significantly affected cell morphology, cell-cell interactions, matrix production and matrix composition39. In our study, we also found that the apoptosis of BMSCs increases with the amplification of shear stress, which can be dose-dependently attenuated by APN.
TGF-β, bFGF, VEGF and PDGF are important growth factors involved in healing and are markedly up-regulated following tendon injury40. Furthermore, they are also important in the development of robust TEHVs41,42,43. Kural et al.41 found that isometric cell force, passive retraction and collagen production can be tuned by independently altering boundary stiffness and TGF-β1 concentration. The ability to stimulate matrix production without inducing high active tension will aid in the development of robust TEHVs and other connective tissue replacements, where minimizing tissue shrinkage upon implantation is critical. In particular, the combination of TGF-β1 with EGF and PDGF induces vascular smooth muscle cell proliferation and expression of extracellular matrix constituents found in the aortic valve42. In this study, we found that TGF-β, bFGF, VEGF and PDGF levels can be decreased by shear stress in an intensity-dependent manner. However, these phenomena can be dose-dependently inhibited by APN.
APN is an adipokine secreted by adipose tissues and other cells12 and has demonstrated protective roles in the cardiovascular system13,14,15. The protection afforded by APN from myocardial ischemia-reperfusion injury has been widely demonstrated18,44. APN can also inhibit the structural and metabolic remodeling of the heart. During pressure overload, APN deficiency accelerated the transition from cardiac hypertrophy to heart failure20 and cardiac fibrosis36, but pretreatment with recombinant APN significantly decreased H2O2-induced cardiomyocyte hypertrophy by decreasing total protein levels, protein synthesis and atrial natriuretic factor and brain natriuretic peptide expression36. In doxorubicin-induced cardiomyopathy, APN also improved cardiac function through anti-apoptotic effects45. More importantly, APN infusion ameliorated diabetic mobilopathy of BMSCs and contributed to the mobilization and recruitment of BMSCs to participate in tissue repair and regeneration46. However, whether APN can protect BMSCs in the development of TEHVs is still unclear. In the current study, we observed that APN contributes to the survival of and apoptosis inhibition in BMSCs and neutralizes the influence of FSS on BMSCs in TEHVs.
Previous studies have demonstrated that the activation of AMPK contributes to the protection of the heart against myocardial ischemic injury28, cardiac fibrosis21, heart failure29 and cardiac pressure overload30,31. Additionally, the primary biological effects of APN are induced by the stimulation of AdipoR1 and AdipoR2, which increase the activities of 5′AMP and the peroxisome proliferator-activated receptor, respectively and stimulate AMPK phosphorylation and uncoupling protein 2 up-regulation47,48,49. In particular, numerous studies have shown that through the activation of the AMPK-dependent signaling pathway, APN exerts anti-IRI, anti-pressure overload and anti-apoptotic actions18,20,32,45. ACC is an important substrate of APN and the activation of AMPK can further promote the phosphorylation of ACC22. In this study, we found that both AMPK and ACC phosphorylation are increased under FSS, which can be further amplified by APN. Bcl2 and Bax have been demonstrated to function as an apoptotic activator and apoptotic inhibitor, respectively. The activation of various molecular signals from AMPK can regulate the Bcl2/Bax apoptosis pathway50,51,52,53. In particular, Kim et al.50 reported that triggering the activation of AMPK by various factors resulted in an increase in Bax gene expression and promotion of apoptosis. Other study has also found that the activation of AMPK alpha-1 alleviates endothelial cell apoptosis by increasing the expression of Bcl-253. Our results demonstrated that the activation of AMPK by APN can reverse the increased apoptosis of BMSCs by the upregulation of Bcl2 and downregulation of Bax. Furthermore, the AMPK siRNA can partially reverse the protective effect of APN against BMSCs by decreasing TGF-β, bFGF, VEGF and PDGF expression levels and inhibiting ACC phosphorylation.
As shown in Fig. 10, we have demonstrated for the first time that APN can effectively protect BMSCs from FSS during the development of TEHVs. Additionally, the protective effects of APN are attributed to the activation of AMPK, which can further promote ACC phosphorylation, Bcl2 up-regulation and Bax down-regulation.

Adiponectin enhances bone marrow mesenchymal stem cell resistance to flow shear stress through AMP-activated protein kinase signaling.
APN can effectively protect BMSCs from FSS during the development of TEHVs. Additionally, the protective effects of APN are attributed to the activation of AMPK, which can further promote ACC phosphorylation, Bcl2 up-regulation and Bax down-regulation.
Methods and Materials
Materials
Recombinant globular rat APN, dimethylsulfoxide (DMSO), 4′,6-diamino-2-phenylindole (DAPI), trypsin and protease inhibitor cocktail were purchased from Sigma-Aldrich (St. Louis, MO, USA). Low-glucose Dulbecco’s modified Eagle’s medium (L-DMEM), antibiotic/antimycotic solution and fetal bovine serum (FBS) were obtained from Gibco Laboratories (Life Technologies, Inc., Burlington, ON, Canada). The Cell Counting Kit-8 (CCK8) assay was purchased from Dojindo Molecular Technologies (Kumamoto, Japan). Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) kits were purchased from Roche (Mannheim, Germany). IgG Isotype Control and the FITC-conjugated anti-mouse CD29, CD45 and CD90 were purchased from eBioscience (San Diego, CA, USA) and CD34 and CD106 were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Antibodies against adenosine monophosphate (AMP)-activated protein kinase (AMPK), phospho-AMPK (p-AMPK) and β-actin were obtained from Cell Signaling Technology (Beverly, MA, USA). Antibodies against acetyl CoA carboxylase (ACC), phospho-ACC (p-ACC), Bcl2 and Bax were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit anti-goat, goat anti-rabbit and goat anti-mouse secondary antibodies were purchased from the Zhongshan Company (Beijing, China). The BCA assay kit was purchased from Pierce (Rockford, IL, USA). Scramble and AMPK siRNAs were obtained from Ambion (Austin, TX, USA). INTERFERin siRNA transfection reagent was purchased from Polyplus (New York, NY, USA). A bFGF ELISA kit was purchased from Cusabio Biotech (Wuhan, China). The PDGF ELISA kit was purchased from Quantikine (R&D Systems, Minneapolis, MN, Canada). TGF-b1 and VEGF ELISA kits were purchased from eBioscience (San Diego, CA, USA). Other reagents were all purchased from Sigma, unless otherwise specified.
Animals and ethics
Two-month-old male Sprague-Dawley (S-D) rats, with body weights of 110–130 g, were bought from the Fourth Military Medical University Animal Center in China. Animals were bred in an environmentally controlled room (20–23 °C, 60% humidity, 12-hour light-dark cycle) and fed sterile water and standard laboratory rat chow ad libitum. All the procedures and experimental protocols were approved by the Fourth Military Medical University Animal Care and Use Committee and performed in accordance with the Health Guide for the Care and Use of Laboratory Animals of National Institutes.
BMSC culture and expansion in vitro
The isolation and culture of BMSCs were performed as described previously54. In brief, after administering appropriate anesthesia, the bilateral femurs and tibias of SD rats were aseptically excised after removing connective tissues around the bones. Then, the femurs and tibiae were stored in phosphate-buffered saline (PBS) supplemented with 1% penicillin/streptomycin on ice and the surfaces were further trimmed and cleaned. The bones were broken and the bone marrow was flushed with L-DMEM containing 10% FBS and 1% penicillin/streptomycin. The fluid was collected into the beaker and the dispersed bone marrow cells were centrifuged at 350× g for five minutes. Bone marrow cells were harvested and incubated with the same medium supplemented in a humidified atmosphere of 5% CO2 at 37 °C. The medium was changed for the first time after 48 h, with removal of non-adherent cells and the media was subsequently changed once every three days. Once the cell fusion reached 80–90%, the cells were washed with PBS, trypsinized using 0.25% trypsin at 37 °C and subcultured under the same conditions. Cultured BMSCs at the third passage were used for the following experiments.
Fluorescence-activated cell sorting analysis of BMSCs
Cultured cells were subjected to fluorescence-activated cell sorting (FACS) analysis to assess the expression of various signature antigen markers of BMSCs55. Third-generation cells that exhibited good growth were selected to prepare single-cell suspensions. After being trypsinized with 0.25% trypsin, the cells were collected, centrifuged, rinsed and re-suspended in PBS at a concentration of 105 cells/mL. Re-suspended cells were incubated with 5 mL of fluorescein isothiocyanate-labeled anti-rat IgG Isotype Control, CD29, CD34, CD45, CD90 and CD106 in the dark at 4 °C for 30 minutes. After being washed twice with PBS, the cells were analyzed using a flow cytometer (BD FACSCanto™ II; BD Biosciences). The data were analyzed using CellQuest software (BD Biosciences). IgG Isotype Control and the FITC-conjugated anti-mouse, CD29, CD45 and CD90 antibodies were purchased from eBioscience (San Diego, USA); CD34 and CD106 were purchased from Santa Cruz Biotechnology (Dallas, TX, USA); and the others were purchased from Abcam (Cambridge, MA, USA).
Shear stress stimulation and adiponectin treatments
Shear stress stimulation of BMSCs was carried out in a parallel plate flow chamber with a flow loop apparatus that was designed by Jacobs et al.56 and improved by Bai et al.57 in previous studies. The passage-3 BMSCs at 1.5 × 104 cells/cm2 were trypsinized and cultured on the bottom of the parallel plate flow chamber (10.0 cm in length, 2.5 cm in width and 0.03 cm in height) for one day before exposure to FSS. After the cells had adhered to the slide, the medium was driven by a constant hydrostatic pressure through the channel of the flow chamber and cells were exposed to steady and well-defined FSS58. The FSS intensity (τ, dynes/cm2) was computed according to the formula τ = 6 μQ/wh2, where μ is the viscosity of the medium and Q is the flow volume. FSS stimulations were performed under the same culture conditions as those described above. A peristaltic pump was used to generate shear stress. The duration of FSS was 24 h.
Recombinant globular rat adiponectin was dissolved in L-DMEM containing 0.1% BSA to ensure a concentration of 1.0 mg/mL. This stock solution was stored at 4 °C for one week and diluted with culture medium immediately prior to the experiment. After pretreatment with FSS for 24 h, the cells were subjected to different concentrations of APN (10, 20 and 30 μg/mL) for 18 h. The morphology was then assessed under an inverted/phase contrast microscope and images were obtained using a 600D camera (Canon Company, Japan). Finally, the cells were harvested for further analysis.
Cell proliferation analysis
Cell proliferation analysis was carried out as described in a previous study59. First, BMSCs were cultured with different processing methods. Then, cells from each group were seeded into 96-well plates with 100 μL of cell suspension (8,000 cells/well), with five wells for each group at each time point. After incubation at 37 °C for 24 h, the culture supernatant was removed and a total of 100 μL of serum-free medium and 10 μL of CCK-8 solution were added to each well. After incubation at 37 °C with 5% CO2 for 2 h, the optical density (OD) values were measured at 450 nm using a microplate reader (SpectraMax 190, Molecular Devices, USA) and cell viability was expressed as the OD value.
Cell apoptosis assay
Apoptosis was assessed using the terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL) assay60. BMSCs were first plated onto cover slips at a density of 5 × 104 cells/well in 24-well plates. After different treatments, the cover slips with cells were treated with 3% hydrogen peroxide diluted in methanol for 15 minutes to block endogenous peroxidase activity and then fixed in 4% paraformaldehyde for 30 minutes at 4 °C. The cells were then incubated in 0.1% Triton X-100 for 20 minutes on ice. After washing with PBS, the cells were covered with 50 μL of the TUNEL reaction mixture and two negative controls using 50 μL of label solution per control. Next, all of the cell samples were incubated in this solution for 60 minutes at 37 °C in a dark humidified chamber. Finally, the cells were stained with DAPI. The apoptotic cells were visually identified in ten selected fields and photographed at a magnification of ×200 under an Olympus FV1000 (Olympus, Japan) confocal microscope.
Enzyme-linked immunosorbent assay
The bFGF, TGF-b, VEGF and PDGF concentrations were measured using an enzyme-linked immunosorbent assay (ELISA). For this assay, the treated BMSCs were collected and washed with ice-cold PBS. After 48 h, culture supernatants were divided into 200-μL samples in triplicate. Finally, these samples were homogenized in pre-chilled lysis buffer (20 mM HEPES, 0.5 mM EGTA, 1 mM DTT and 0.32 M sucrose; PH 7.4) containing protease inhibitors cocktail and then incubated at 4 °C for 30 minutes. The samples were centrifuged at 15, 000 rpm in a pre-chilled centrifuge at 4 °C for ten minutes. The supernatant from the cell lysate was collected and assayed for protein concentration using the Pierce™ BCA Protein Assay reagent kit (Thermo Fisher Scientific, Waltham, MA, USA). Samples containing equal amounts of total proteins were analyzed by ELISA kits for bFGF, TGF-β, VEGF and PDGF according to the manufacturers’ protocols. The sensitivity of these kits was less than 1 ng/mL, the intra-assay coefficient of variation was 10% and the inter-assay coefficient of variation was 10%. All the reported results are based on three independent experiments on separate batches of cells.
Western blot analysis
Samples were lysed in detergent lysis buffer (20 mM Tris-HCl, pH 7.5; 150 mM NaCl; 1 mM EDTA; 1 mM EGTA; 1% Triton; 2.5 mM sodium pyrophosphate; 1 mM β-glycerophosphate; 1 mM Na3VO4; and 1 μg/mL leupeptin). The lysates were centrifuged at 12, 000 rpm for 25 minutes at 4 °C and protein concentrations were measured using the Pierce™ BCA Protein Assay reagent kit (Thermo Fisher Scientific). Ten micrograms of protein were separated by 7.5–10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (Bio-Rad, Hercules, CA, USA) and separated proteins were electrophoretically transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA). Membranes were blocked with Tween-20 Tris-buffered saline (10 mM Tris-HCl, pH 7.5; 100 mM NaCl; and 0.1% Tween 20) containing 5% nonfat dry milk before incubation with rabbit monoclonal anti-adenosine monophosphate (AMP)-activated protein kinase (AMPK) antibody (1:1,000), rabbit monoclonal anti-phospho-AMPK (p-AMPK) antibody (1:1,000), goat polyclonal anti-acetyl CoA carboxylase (ACC) antibody (1:500), rabbit polyclonal anti-phospho-ACC (p-ACC) antibody (1:500), goat polyclonal anti-Bcl2 antibody (1:500), rabbit polyclonal anti-Bax antibody (1:500) and rabbit monoclonal anti-β-actin antibody (1:1,000). Antigen detection was performed using enhanced chemiluminescence Western blotting detection reagents (Amersham Pharmacia Biotech, Piscataway, NJ, USA) with horseradish peroxidase-conjugated secondary antibodies. Images were obtained and densities were determined using Image J. The protein expression was corrected by the β-actin density and the expression in the control group was arbitrarily set at 100%.
Small interfering RNA transfection
Utilizing specific AMPK RNA, interference was performed against the AMPK target sequence (5′-AAGAGAAGCAGAAGCACGACG-3′)61. A nonspecific scramble siRNA was used as the control. Cells were seeded into six-well plates. After the cells reached 70–80% confluence, they were transfected with AMPK siRNA or NC siRNA. Transfections were performed using Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions with 100 nM of each siRNA62. The efficiency of siRNA-mediated AMPK knockdown was verified by Western blot analysis 48 h after transfection. Twenty-four hours after transfection, the cells were treated with APN (30 μg/mL) for 18 h and FSS (15 dynes/cm2) for 24 h. Finally, the cells were collected and processed for subsequent use in further experiments.
Statistical analysis
All of the values are presented as the mean ± standard deviation (SD). Group comparisons were carried out using ANOVA (SPSS 15.0, SPSS Inc., Chicago, IL, USA). All of the groups were analyzed simultaneously using one-way ANOVA with the Bonferroni correction for multiple comparisons. A P value less than 0.05 was regarded as a statistically significant difference.
Additional Information
How to cite this article: Zhao, L. et al. Adiponectin enhances bone marrow mesenchymal stem cell resistance to flow shear stress through AMP-activated protein kinase signaling. Sci. Rep. 6, 28752; doi: 10.1038/srep28752 (2016).
References
Schoen, F. J. Evolving concepts of cardiac valve dynamics: the continuum of development, functional structure, pathobiology and tissue engineering. Circulation 118, 1864–1880 (2008).
Iung, B. & Vahanian, A. Epidemiology of acquired valvular heart disease. Can J Cardiol 30, 962–970 (2014).
Coffey, S., Cox, B. & Williams, M. J. Lack of progress in valvular heart disease in the pre-transcatheter aortic valve replacement era: increasing deaths and minimal change in mortality rate over the past three decades. Am Heart J 167, 562–567 e562 (2014).
Sacks, M. S., Schoen, F. J. & Mayer, J. E. Bioengineering challenges for heart valve tissue engineering. Annu Rev Biomed Eng 11, 289–313 (2009).
Schoen, F. J. Mechanisms of function and disease of natural and replacement heart valves. Annu Rev Pathol 7, 161–183 (2012).
Masoumi, N. et al. Tri-layered elastomeric scaffolds for engineering heart valve leaflets. Biomaterials 35, 7774–7785 (2014).
Schmidt, D. et al. Living autologous heart valves engineered from human prenatally harvested progenitors. Circulation 114, I125–131 (2006).
Jana, S., Tranquillo, R. T. & Lerman, A. Cells for tissue engineering of cardiac valves. J Tissue Eng Regen Med, doi: 10.1002/term.2010 (2015).
Engelmayr, G. C. Jr., Sales, V. L., Mayer, J. E. Jr. & Sacks, M. S. Cyclic flexure and laminar flow synergistically accelerate mesenchymal stem cell-mediated engineered tissue formation: Implications for engineered heart valve tissues. Biomaterials 27, 6083–6095 (2006).
Bin, F. et al. Construction of tissue-engineered homograft bioprosthetic heart valves in in vitro. ASAIO J 52, 303–309 (2006).
Ye, X., Zhao, Q., Sun, X. & Li, H. Enhancement of mesenchymal stem cell attachment to decellularized porcine aortic valve scaffold by in vitro coating with antibody against CD90: a preliminary study on antibody-modified tissue-engineered heart valve. Tissue Eng Part A 15, 1–11 (2009).
Caselli, C. et al. Back to the heart: the protective role of adiponectin. Pharmacol Res 82, 9–20 (2014).
Ghantous, C. M., Azrak, Z., Hanache, S., Abou-Kheir, W. & Zeidan, A. Differential Role of Leptin and Adiponectin in Cardiovascular System. Int J Endocrinol 2015, 534320 (2015).
Renaldi, O. et al. Hypoadiponectinemia: a risk factor for metabolic syndrome. Acta Med Indones 41, 20–24 (2009).
Li, Q. et al. Plasma adiponectin levels in relation to prognosis in patients with angiographic coronary artery disease. Metabolism 61, 1803–1808 (2012).
Yang, Y. et al. The emerging role of adiponectin in cerebrovascular and neurodegenerative diseases. Biochim Biophys Acta 1852, 1887–1894 (2015).
Nigro, E. et al. New insight into adiponectin role in obesity and obesity-related diseases. Biomed Res Int 2014, 658913 (2014).
Shibata, R. et al. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med 11, 1096–1103 (2005).
Parker-Duffen, J. L. & Walsh, K. Cardiometabolic effects of adiponectin. Best Pract Res Clin Endocrinol Metab 28, 81–91 (2014).
Shimano, M. et al. Adiponectin deficiency exacerbates cardiac dysfunction following pressure overload through disruption of an AMPK-dependent angiogenic response. J Mol Cell Cardiol 49, 210–220 (2010).
Ma, X. et al. Cardiac Fibrosis Alleviated by Exercise Training Is AMPK-Dependent. PLos One 10, e0129971 (2015).
Ding, G. et al. Adiponectin and its receptors are expressed in adult ventricular cardiomyocytes and upregulated by activation of peroxisome proliferator-activated receptor gamma. J Mol Cell Cardiol 43, 73–84 (2007).
Kuwabara, Y. et al. MicroRNA-451 exacerbates lipotoxicity in cardiac myocytes and high-fat diet-induced cardiac hypertrophy in mice through suppression of the LKB1/AMPK pathway. Circ Res 116, 279–288 (2015).
Kim, M. et al. Mutation in the gamma2-subunit of AMP-activated protein kinase stimulates cardiomyocyte proliferation and hypertrophy independent of glycogen storage. Circ Res 114, 966–975 (2014).
Baskin, K. K. & Taegtmeyer, H. AMP-activated protein kinase regulates E3 ligases in rodent heart. Circ Res 109, 1153–1161 (2011).
Young, L. H. AMP-activated protein kinase conducts the ischemic stress response orchestra. Circulation 117, 832–840 (2008).
Zou, M. H. & Xie, Z. Regulation of interplay between autophagy and apoptosis in the diabetic heart: new role of AMPK. Autophagy 9, 624–625 (2013).
Kataoka, Y. et al. Omentin prevents myocardial ischemic injury through AMP-activated protein kinase- and Akt-dependent mechanisms. J Am Coll Cardiol 63, 2722–2733 (2014).
Sasaki, H. et al. Metformin prevents progression of heart failure in dogs: role of AMP-activated protein kinase. Circulation 119, 2568–2577 (2009).
Schisler, J. C. et al. CHIP protects against cardiac pressure overload through regulation of AMPK. J Clin Invest 123, 3588–3599 (2013).
Zaha, V. G. & Young, L. H. AMP-activated protein kinase regulation and biological actions in the heart. Circ Res 111, 800–814 (2012).
Wang, Y. et al. AMP-activated protein kinase deficiency enhances myocardial ischemia/reperfusion injury but has minimal effect on the antioxidant/antinitrative protection of adiponectin. Circulation 119, 835–844 (2009).
Ding, W. et al. Platelet-derived growth factor (PDGF)-PDGF receptor interaction activates bone marrow-derived mesenchymal stromal cells derived from chronic lymphocytic leukemia: implications for an angiogenic switch. Blood 116, 2984–2993 (2010).
Dong, Q., Yang, Y., Song, L., Qian, H. & Xu, Z. Atorvastatin prevents mesenchymal stem cells from hypoxia and serum-free injury through activating AMP-activated protein kinase. Int J Cardiol 153, 311–316 (2011).
de Meester, C. et al. Role of AMP-activated protein kinase in regulating hypoxic survival and proliferation of mesenchymal stem cells. Cardiovasc Res 101, 20–29 (2014).
Essick, E. E. et al. Adiponectin mediates cardioprotection in oxidative stress-induced cardiac myocyte remodeling. Am J Physiol Heart Circ Physiol 301, H984–993 (2011).
Chakrabarti, P. et al. SIRT1 controls lipolysis in adipocytes via FOXO1-mediated expression of ATGL. J Lipid Res 52, 1693–1701 (2011).
Cartmell, S. H., Porter, B. D., Garcia, A. J. & Guldberg, R. E. Effects of medium perfusion rate on cell-seeded three-dimensional bone constructs in vitro. Tissue Eng 9, 1197–1203 (2003).
Grayson, W. L. et al. Optimizing the medium perfusion rate in bone tissue engineering bioreactors. Biotechnol Bioeng 108, 1159–1170 (2011).
Molloy, T., Wang, Y. & Murrell, G. The roles of growth factors in tendon and ligament healing. Sports Med 33, 381–394 (2003).
Kural, M. H. & Billiar, K. L. Mechanoregulation of valvular interstitial cell phenotype in the third dimension. Biomaterials 35, 1128–1137 (2014).
Appleton, A. J., Appleton, C. T., Boughner, D. R. & Rogers, K. A. Vascular smooth muscle cells as a valvular interstitial cell surrogate in heart valve tissue engineering. Tissue Eng Part A 15, 3889–3897 (2009).
Colazzo, F. et al. Shear stress and VEGF enhance endothelial differentiation of human adipose-derived stem cells. Growth Factors 32, 139–149 (2014).
Tao, L. et al. Adiponectin cardioprotection after myocardial ischemia/reperfusion involves the reduction of oxidative/nitrative stress. Circulation 115, 1408–1416 (2007).
Konishi, M. et al. Adiponectin protects against doxorubicin-induced cardiomyopathy by anti-apoptotic effects through AMPK up-regulation. Cardiovasc Res 89, 309–319 (2011).
Yu, L. et al. Adiponectin regulates bone marrow mesenchymal stem cell niche through a unique signal transduction pathway: an approach for treating bone disease in diabetes. Stem Cells 33, 240–252 (2015).
Hug, C. et al. T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc Natl Acad Sci USA 101, 10308–10313 (2004).
Tanabe, H. et al. Crystal structures of the human adiponectin receptors. Nature 520, 312–316 (2015).
Yamauchi, T. et al. Globular adiponectin protected ob/ob mice from diabetes and ApoE-deficient mice from atherosclerosis. J Biol Chem 278, 2461–2468 (2003).
Kim, J. H. et al. AMP-activated protein kinase phosphorylates CtBP1 and down-regulates its activity. Biochem Biophys Res Commun 431, 8–13 (2013).
Jin, J. et al. AMPK inhibitor Compound C stimulates ceramide production and promotes Bax redistribution and apoptosis in MCF7 breast carcinoma cells. J Lipid Res 50, 2389–2397 (2009).
Tacar, O., Sriamornsak, P. & Dass, C. R. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol 65, 157–170 (2013).
Liu, C., Liang, B., Wang, Q., Wu, J. & Zou, M. H. Activation of AMP-activated protein kinase alpha1 alleviates endothelial cell apoptosis by increasing the expression of anti-apoptotic proteins Bcl-2 and survivin. J Biol Chem 285, 15346–15355 (2010).
Lennon, D. P. & Caplan, A. I. Isolation of rat marrow-derived mesenchymal stem cells. Exp Hematol 34, 1606–1607 (2006).
Sun, H., Wu, Y., Fu, D., Liu, Y. & Huang, C. SIRT6 regulates osteogenic differentiation of rat bone marrow mesenchymal stem cells partially via suppressing the nuclear factor-kappaB signaling pathway. Stem Cells 32, 1943–1955 (2014).
Jacobs, C. R. et al. Differential effect of steady versus oscillating flow on bone cells. J Biomech 31, 969–976 (1998).
Bai, K., Huang, Y., Jia, X., Fan, Y. & Wang, W. Endothelium oriented differentiation of bone marrow mesenchymal stem cells under chemical and mechanical stimulations. J Biomech 43, 1176–1181 (2010).
Daculsi, R. et al. Unusual transduction response of progenitor-derived and mature endothelial cells exposed to laminar pulsatile shear stress. J Biomech 41, 2781–2785 (2008).
Zhao, L., Liu, L., Wu, Z., Zhang, Y. & Chu, P. K. Effects of micropitted/nanotubular titania topographies on bone mesenchymal stem cell osteogenic differentiation. Biomaterials 33, 2629–2641 (2012).
Tang, Y. L. et al. Improved graft mesenchymal stem cell survival in ischemic heart with a hypoxia-regulated heme oxygenase-1 vector. J Am Coll Cardiol 46, 1339–1350 (2005).
Aguilar, V. et al. S6 kinase deletion suppresses muscle growth adaptations to nutrient availability by activating AMP kinase. Cell Metab 5, 476–487 (2007).
Hung, B. P. et al. Platelet-Derived Growth Factor BB Enhances Osteogenesis of Adipose-Derived But Not Bone Marrow-Derived Mesenchymal Stromal/Stem Cells. Stem Cells 33, 2773–2784 (2015).
Acknowledgements
This work was supported by National Natural Science Foundation of China (81422004, 81500263, 81470480, 81470477, 81100137, 81100081), China Postdoctoral Science Foundation (2015M572681), National High-tech R&D Program of China (2014AA020514, 2015AA020919), the grants from the Excellent Doctoral Support Project of the Fourth Military Medical University (2015D02) and Technological New Star Program of Shaanxi Province (2014KJXX-56).
Author information
Authors and Affiliations
Contributions
W.Y. and Y.S. designed the research, L.Z., C.X.F., Y.Y. and Y.Z. performed the experiments, D.J.W., C.D., W.H., Z.Q.M., S.J., S.Y.D. and J.J.L. analyzed the data and C.X.F. and Y.Y. wrote the main text and prepared all of the figures. The authors reviewed and approved this manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhao, L., Fan, C., Zhang, Y. et al. Adiponectin enhances bone marrow mesenchymal stem cell resistance to flow shear stress through AMP-activated protein kinase signaling. Sci Rep 6, 28752 (2016). https://doi.org/10.1038/srep28752
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep28752
This article is cited by
-
Adipokines at the crossroads of obesity and mesenchymal stem cell therapy
Experimental & Molecular Medicine (2023)
-
Adiponectin overexpression promotes fracture healing through regulating the osteogenesis and adipogenesis balance in osteoporotic mice
Journal of Bone and Mineral Metabolism (2023)
-
Exosomes secreted from sonic hedgehog-modified bone mesenchymal stem cells facilitate the repair of rat spinal cord injuries
Acta Neurochirurgica (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
