
Abstract
The interaction between endothelial cells (ECs) and smooth muscle cells (SMCs) plays a critical role in the maintenance of vessel wall homeostasis. The X-box binding protein 1 (XBP1) plays an important role in EC and SMC cellular functions. However, whether XBP1 is involved in EC-SMC interaction remains unclear. In this study, In vivo experiments with hindlimb ischemia models revealed that XBP1 deficiency in SMCs significantly attenuated angiogenesis in ischemic tissues, therefore retarded the foot blood perfusion recovery. In vitro studies indicated that either overexpression of the spliced XBP1 or treatment with platelet derived growth factor-BB up-regulated miR-150 expression and secretion via extracellular vesicles (EVs). The XBP1 splicing-mediated up-regulation of miR-150 might be due to increased stability. The SMC-derived EVs could trigger EC migration, which was abolished by miR-150 knockdown in SMCs, suggesting miR-150 is responsible for SMC-stimulated EC migration. The SMC-derived miR-150-containing EVs or premiR-150 transfection increased vascular endothelial growth factor (VEGF)-A mRNA and secretion in ECs. Both inhibitors SU5416 and LY294002 attenuated EVs-induced EC migration. This study demonstrates that XBP1 splicing in SMCs can control EC migration via SMC derived EVs-mediated miR-150 transfer and miR-150-driven VEGF-A/VEGFR/PI3K/Akt pathway activation, thereby modulating the maintenance of vessel wall homeostasis.
Similar content being viewed by others


Introduction
Endothelial cells (ECs) and smooth muscle cells (SMCs) are two major cellular components within the vessel wall. ECs line the lumen forming an intact mono-layered structure that provides a semi-selective barrier between blood and the underlying tissues and modulating vessel tone via nitric oxide production. SMCs are responsible for the contractility of the vessel wall. The liaison between ECs and SMCs plays an essential role in vessel maturation under physiological conditions, vascular injury repair and disease development such as atherosclerosis1,2.
MicroRNAs (miRNAs, miRs) are a set of small non-coding RNAs that participate in multiple cellular processes through regulating gene transcription, mRNA stability and protein translation3. Recent studies have revealed that miRNAs can be released from cells, delivered by extracellular vesicles(EVs) and act on recipient cells in a paracrine way, modulating intercellular communications4,5,6,7,8. The miR-150 family is located in human chromosome 19. MiR-150 has been shown to participate in multiple physiological and pathological processes, including hematopoietic cell differentiation9,10 and suppresses tumour cells proliferation11,12. In cardiovascular system, miR-150 is reported to promote endothelial progenitor cells homing13 and monocyte-EC interaction14, while it may also be involved in the pathogenesis of pulmonary arterial hypertension15,16. In our previous study, we noticed that series of microRNA levels including miR-150 in SMCs were up-regulated during the neointimal formation after vascular injury17. However, the potential role of miR-150 in EC-SMC communication remains unclear.
The X-box binding protein 1 (XBP1) was originally identified as a stress-inducible transcription factor essential for cell survival under stress conditions18,19,20. Under endoplasmic reticulum (ER) stress conditions, XBP1 mRNA undergoes unconventional splicing through an ER-resident kinase that possesses ribonuclease activity, the inositol requiring enzyme 1 alpha (IRE1α)21. Our recent studies and reports from other groups showed that physiological stimuli like vascular endothelial cell growth factor (VEGF) could trigger XBP1 splicing in an ER stress response independent manner22,23,24,25. In ECs, XBP1 splicing plays diverse roles including cell proliferation23, autophagy response26 and apoptosis27. In SMCs, BMP-2 was reported to activate XBP1 splicing28. Our study revealed that XBP1 splicing was activated in vascular SMCs in response to vascular injury in vivo and platelet derived growth factor-BB (PDGF-BB) stimulation in vitro and that XBP1 deficiency in SMCs attenuated neointima formation in mouse model of femoral artery injury17. In this study, we demonstrated that XBP1 splicing in SMC regulated EC migration via EVs-mediated miR-150 transfer.
Results
XBP1 deficiency in SMCs attenuated the foot blood perfusion recovery in mouse hindlimb ischemia model
To test whether XBP1 was involved in EC-SMC interaction, we performed a hindlimb ischemia model in SM22-Cre/XBP1loxP/loxP (XBP1smcko) mice and observed foot blood perfusion recovery. Compared to XBP1loxP/loxP mice, the foot blood perfusion recovery in the injured limb was significantly attenuated in mice at 2 weeks post-surgery (Fig. 1A) as revealed by Doppler scanning analysis. Immunofluorescence staining with anti-CD31 and anti-αSMA antibodies revealed that the capillary vessel density was significantly decreased in the injured skeletal muscle in XBP1smcko mice as compared to XBP1loxP/loxP mice (Fig. 1B). These results suggest XBP1 deficiency in SMCs may affect angiogenesis, therefore causing the retardation of the foot blood perfusion recovery.

Disruption of XBP1 gene in SMCs decreased angiogenesis in ischemic tissue.
The right femoral arteries were banded at two adjacent sites with internal part cut to totally block foot blood supply in XBP1loxP/loxP(WT, n = 7) or SM22-Cre+/XBP1loxP/loxP(XBP1smcko, n = 21) mice. The blood flow (A) at both sides was measured by Doppler Laser Scanner at 30 minutes, 7 days and 14 days post-surgery. After blood flow measurement, the mice were humanely sacrificed and the skeletal muscle tissues around the banding site were harvested and subjected to cryo-sectioning and double immunostaining with anti-CD31 (red) and anti-SMA (green) antibodies (B). (A) Left panel shows the representative images from one mouse of each group at 14 days post-surgery. The white open box indicates the blood flow data collecting area. The right panel shows the average blood flow perfusion ratio of the right to the left foot from all mice in each group. (mean ± SEM, *P < 0.05; ANOVA, Dunnett post-test). (B) Left panel shows the representative images of blood vessels in the skeletal muscle tissues from one mouse of each group at 14 days post-surgery. Nucleus was conterstained with DAPI. Scale bar: 100 μm. Right panel shows the average vessel density (all vessels including capillary vessels) in the tissues collected from all mice in each group. (mean ± SEM, *P < 0.05; Student T-test).
Overexpression of XBP1s in SMCs triggered EC migration via paracrine mechanisms
During the angiogenesis process, the initial step is the migration of ECs out of the intact endothelium, followed by EC proliferation. As described above, XBP1 deficiency in SMC affected angiogenesis in ischemic tissue, indicating that XBP1 in SMC may be involved in EC migration and proliferation via a paracrine mechanism. To test this, the conditioned medium was collected from SMCs infected with Ad-XBP1u and Ad-XBP1s viruses respectively and applied to HUVECs to assess cell proliferation and migration. Cell number calculation assays showed that there was no difference among the conditioned media from SMCs infected with either of the two viruses or the control virus (Ad-null) (Fig. 2A), suggesting that XBP1 in SMC has no effect on EC proliferation. There was a decrease of cell proliferation under conditioned media as compared to fresh medium, which might be due to prior nutrient consumption by SMCs in the conditioned media. However, the EC monolayer wound healing assay revealed that the conditioned medium from SMCs infected with Ad-XBP1s, but not those from cells infected with Ad-XBP1u-infected, had significantly increased EC migration (Fig. 2B). This was further confirmed by transwell-based migration assay (Fig. 2C), suggesting that XBP1 splicing in SMCs may regulate EC migration through some secreted factors.

XBP1 splicing in SMCs increased EC migration via secreted factors.
HSMCs were infected with Ad-null (null), Ad-XBP1s (1s), or Ad-XBP1u (1u) at 10 MOI for 24 hr. The medium was then replaced by M199 medium supplemented with 0.5% FBS and incubated for 8 hr. The conditioned medium was then applied to HUVECs for proliferation assays via cell number counting (A) 24 hr post incubation) and cell migration assessments via wound healing assays (B) 6 hr post incubation, scale bar: 200 μm) and transwell migration assays (C) 6 hr post incubation, scale bar: 25 μm). Data presented are representative images or mean ± SEM of three independent experiments. (*P < 0.05; ANOVA, Dunnett post-test).
MiR-150 was mediated by overexpression of XBP1s in SMCs-induced EC migration
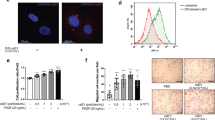
To analyse the potential responsible factors in the conditioned medium, EVs was isolated by ultracentrifugation. The effect of the whole conditioned medium, EVs and EVs-depleted medium on EC migration was performed with the transwell migration assay. As shown in Fig. 3A, both EVs and EVs-depleted medium from Ad-XBP1s-infected SMCs significantly increased EC migration, suggesting that stimulating factors exist in both EVs and medium. In our previous study, we found that activation of XBP1 splicing modulated multiple microRNA levels in SMCs, of which miR-150 was up-regulated17. It has been reported that miR-150 was involved in monocyte-regulated EC migration14,29. Therefore, we wondered whether miR-150 contributed to EVs-mediated pro-migratory effect. Quantitative RT-PCR analysis confirmed that overexpression of XBP1s in SMCs increased cellular and EVs miR-150 levels (Fig. 3B). Transfection of SMCs with anti-miR-150 oligos abolished overexpression of XBP1s-mediated upregulation of miR-150 in both whole cell lysates and EVs (Fig. 3C). As expected, the anti-miR-150 transfection in SMCs abolished the EC pro-migratory effect of the EVs isolated from Ad-XBP1s-infected SMCs as compared to control anti-mir RNA oligos (Fig. 3D). Tube formation is another criterion to evaluate EC functions in angiogenesis. Our studies revealed that EVs isolated from Ad-XBP1s-infected HSMCs significantly increased HUVEC capillary structure formation on Matrigel, which was ablated by anti-miR-150 transfection in SMCs (Fig. 3E). These results suggest that miR-150 plays an essential role in EVs-mediated EC migration and angiogenesis.

miR-150 was mediated by XBP1 splicing in SMC-induced EC migration.
(A) Effect of the whole conditioned medium (W), EVs and supernatant (S) on HUVEC migration in transwell assay. (B) Over-expression of XBP1s in SMC increased miR-150 expression and secretion in EVs. (C) anti-miR-150 abolished XBP1s-induced miR-150 expression and secretion. (D) anti-miR-150 abolished XBP1s/SMC-derived EVs-induced HUVEC migration. Data presented are mean ± SEM of three independent experiments. (E) anti-miR-150 abolished XBP1s/SMC-derived EVs-induced HUVEC tube formation. Left panel shows the representative images. Right panel shows the average tubes/10x view from 3 views/well of 3 wells (*P < 0.05; ANOVA, Dunnett post-test).
PDGF upregulated miR-150 in SMCs in XBP1 splicing dependent manner
To test whether miR-150 could be upregulated in SMCs under physiological or pathological conditions, the effect of PDGF has been examined. Our previous studies have revealed that PDGF treatment triggered XBP1 splicing via IRE1α phosphorylation17. Hence SMCs were infected with IRE1α or XBP1 shRNA lentivirus and treated with PDGF, followed by analysis of miR-150 in whole cell lysate and EVs. As shown in Fig. 4A, PDGF treatment increased miR-150 in both the cell lysate and EVs from non-target shRNA (NTsh) infected SMCs. However, the increase was abolished by either IRE1α shRNA (IRE1αsh) or XBP1 shRNA (XBP1sh). These results suggest that IRE1α-mediated XBP1 splicing is essential for PDGF-induced miR-150 upregulation and secretion via EVs. The EVs isolated from PDGF-treated SMCs increased EC migration, which was abolished by anti-miR-150 transfection (Fig. 4B), suggesting that miR-150 is involved in PDGF-mediated EC/SMC interaction.

PDGF up-regulated miR-150 expression and secretion in SMC in IRE1α/XBP1 dependent manner.
(A) PDGF increased miR-150 expression and secretion in SMC in an XBP1 splicing dependent manner. HSMCs were infected with non-target (NTsh), IRE1α (Ish) or XBP1 (Xsh) shRNA lentivirus at 100 IU for 48 hr and then treated with DMEM supplemented with 0.5% FBS for 24 hr, followed by 20 ng/ml PDGF-BB treatment for 4 hr. The cellular and EV miR-150 levels were assessed by quantitative RT-PCR of three independent experiments. (mean ± SEM, *P < 0.05; ANOVA, Dunnett post-test) (B) The anti-miR-150 abolished PDGF/SMC-mediated EC migration. HSMCs was transfected with anti-miR-150 RNA (anti) and incubated in complete growth medium for 48 hrs and then treated with 0.5% FBS medium for 24 hr, followed by 20 ng/ml PDGF-BB treatment for 4 hr. The effect of isolated EVs on HUVEC migration was assessed by transwell migration assays. The left panel shows the representative images, while the right panel shows the statistical analysis of migrated cells per 10x view. Control anti-miR RNA (ctl) was included. Scale bar: 25 μm. Data presented are representative images or mean ± SEM of three independent experiments. (*P < 0.05; ANOVA, Dunnett post-test).
XBP1s increased miR-150 stabilization
XBP1 is a zinc leucine zip transcription factor. Our previous study has shown that XBP1 splicing in SMCs increased mir-1274B via direct transcriptional regulation17. We wondered whether a similar mechanism exists in miR-150 upregulation. To test this, we cloned a 1.1 kb fragment corresponding to the promoter region of the miR-150 gene into pSI-Check2 vector to replace the SV40 early enhancer promoter. Co-transfection of this plasmid with XBP1s overexpressing plasmid (pShuttle2-XBP1s (pXBP1s)) was performed in SMCs, followed by luciferase activity assays. Surprisingly, overexpression of XBP1s did not increase the reporter gene expression, but instead decreased the reporter gene expression (Fig. 5A). Chromatin-immunoprecipitation assay did not show a direct binding of XBP1s to the promoter region(data not shown). Therefore, we assumed that XBP1s increased miR-150 stability, leading to feedback inhibitory effect on its transcription. Indeed, when the reporter plasmid was co-transfected with miR-150 pre-mir mimic, the reporter gene expression was significantly decreased (Fig. 5B), suggesting a feedback inhibitory effect exists.

XBP1s suppressed miR-150 transcription.
(A) Over-expression of XBP1s suppressed miR-150 promoter reporter gene expression. 0.1 μg/well pSi-CHECK2-miR-150 (pSI-150) was co-transfected with 0.1 μg/well pShuttle2-XBP1s (pXBP1s) into HSMCs in triplicate. Renilla and Firefly luciferase activities were assessed 48 hr post-transfection. pSi-CHECK2 (pSI) and pShuttle2 (Mock) were included as control. (mean ± SEM, *P < 0.05; ANOVA, Dunnett post-test) (B) Exogenous premiR-150 suppressed miR-150 promoter reporter gene expression. 0.1 μg/well pSi-CHECK2-miR-150 (pSI-150) was co-transfected with 10 pmol/well premiR-150 into HSMCs in triplicate. Renilla and Firefly luciferase activities were assessed 48 hr post-transfection. pSi-CHECK2 (pSI) and premiR control (ctl) were included as control. Data presented are mean of three independent experiments. (mean ± SEM, *P < 0.05; ANOVA, Dunnett post-test).
MiR-150 activated VEGF-A autocrine/paracrine-mediated VEGF receptor/PI3K-Akt signalling pathway
It was reported that microvesicle-derived miR-150 could promote tumorigenesis via up-regulating VEGF30. VEGF is a well-known EC activator, modulating multiple EC functions including migration. Thus, we wondered whether there was a crosstalk between miR-150 and VEGF in our system. We firstly assessed the effect of Ad-XBP1s/HSMC-derived EVs on the VEGF-A mRNA in HUVECs and found that the EVs transiently upregulated VEGF-A mRNA level (Fig. 6A). Western blot analysis revealed that Ad-XBP1s/HSMC-derived EVs exerted different effect on two VEGF/VEGF receptor downstream signal pathways, ErK and Akt pathways. The EVs increased Akt phosphorylation but decreased ErK phosphorylation (Fig. 6B). Knockdown of miR-150 by anti-miR-150 transfection in HSMCs abolished XBP1s/HSMC-derived EVs-induced VEGF-A mRNA upregulation (Fig. 6C) and Akt phosphorylation (Fig. 6D), suggesting that EVs-mediated VEGF-A upregulation and Akt phosphorylation is miR-150 dependent. VEGF receptor inhibitor SU5416 abolished EV-induced Akt phosphorylation in HUVECs (Fig. 6E), suggesting that VEGF-A may function as an autocrine or paracrine factor for Akt activation. Further experiments demonstrated that SU5416 and PI3K inhibitor LY294002 significantly decreased Ad-XBP1s/HSMC-derived EVs-induced EC migration as revealed by transwell migration assays (Fig. 6F). These results suggest that miR-150-mediated VEGF-A upregulation and VEGFR/Akt activation are responsible for EV-induced EC migration.

VEGF-PI3K/Akt pathway was responsible for Ad-XBP1s/HSMC-derived EVs-mediated EC migration.
(A,B) Ad-XBP1s/HSMC-derived EVs increased VEGF-A mRNA and Akt phosphorylation in HUVECs. EVs were isolated from Ad-XBP1s/HSMC condition medium (CM), reconstituted in M199 medium supplemented with 5 μg/ml insulin and 5 μg/ml transferrin and applied to HUVECs for time indicated, followed by quantitative RT-PCR analysis of VEGF-A mRNA (A) or western blot analysis of ErK and Akt (S473) phosphorylation (B). (C,D) Anti-miR-150 abolished XBP1s/HSMC-derived EVs-mediated VEGF-A expression and Akt. EVs were isolated from control (ctl) or anti-miR-150 transfected with Ad-XBP1s-infected HSMCs CM, reconstituted and applied to HUVECs for 2 h, followed by VEGF-A mRNA (C) and Akt (S473) phosphorylation assays (D). (E) SU5416 ablated Ad-XBP1s/HSMC-derived EVs-induced Akt phosphorylation. HUVECs were treated with EVs isolated from Ad-XBP1s in the presence of 10 μM SU5416 for 2 h, followed by Akt phosphorylation assay. (F) SU5416 and LY294002 attenuated Ad-XBP1s/HSMC-derived EVs-induced HUVEC migration. Transwell migration assays were performed with EVs isolated from Ad-XBP1s-infected HSMC CM in M199 medium supplemented with insulin and transferrin in the presence of SU5416 (SU) or 10 10 μM LY294002 (LY) for 8 h. Left panel shows the representative images of migrated cells while right panel shows average migrated cells per 10x view. Ad-null and DMSO (DM) were included as control. Data presented are representative images or mean of three independent experiments. (mean ± SEM, *P < 0.05, **P < 0.01; ANOVA, Dunnett post-test).
To further confirm the involvement of miR-150 in VEGF-A mRNA regulation and Akt activation in ECs, we transfected HUVECs directly with premiR-150 or anti-miR-150 followed by VEGF-A mRNA, VEGF-A secretion in medium and VEGF-A expression and Akt action in the cells within 3 hr post transfection. As shown in Fig. 7A, premiR-150 transfection significantly increased VEGF-A mRNA while anti-miR-150 transfection had no effect. ELISA analysis of VEGF-A in cell culture medium revealed a similar trend (Fig. 7B). Western blot analysis indicated that premiR-150 transfection increased both ErK and Akt phosphorylation, which was ablated by the presence of SU5416 (Fig. 7C). A significant increase was detected in VEGF band in the premiR-150 transfection sample, while elevated amount of VEGF could be detected in both control and premiR-150 transfection samples under SU5416 treatment (Fig. 7C). The results suggest that miR-150 can upregulate VEGF-A mRNA, secretion and its receptor-mediated Akt phosphorylation in ECs.

Over-expression of premiR-150 increased VEGF-A expression and VEGF receptor dependent Akt phosphorylation in HUVECs.
(A,B) Over-expression of premiR-150 increased VEGF-A expression and secretion. HUVECs were transfected with premiR-150 or anti-miR-150 in serum free M199 medium for 5 h, then treated with M199 medium supplemented with 5 μg/ml insulin and 5 μg/ml transferrin for 3 h, followed by quantitative RT-PCR analysis of VEGF-A mRNA (A) or ELISA assay to detect VEGF-A in cell culture medium (B) premiR and anti-miR control RNA fragments were included as controls. (C) SU5416 ablated premiR-150-induced Akt and ErK phosphorylation. HUVECs were transfected with premiR-150 or anti-miR-150 in serum free M199 medium for 5 h, then treated with M199 medium supplemented with 5 μg/ml insulin and 5 μg/ml transferrin in the presence of 10 μM SU5416 for 3 h, followed by Western blot analysis of Akt and ErK phosphorylation and VEGF expression. premiR RNA fragments DMSO were included as controls. Data presented are representative images or mean of three independent experiments. (mean ± SEM, *P < 0.05; ANOVA, Dunnett post-test).
Discussion
The communication between ECs and SMCs is critical for the maintenance of vessel wall homeostasis and remodelling. Under physiological conditions, ECs and SMCs liaise with each other via paracrine and/or juxtacrine manner to control vessel tone31. The co-ordinations between ECs and SMCs on their proliferation and migration are essential for arteriogenesis32. Under pathological conditions, risk factors-induced endothelial dysfunction can trigger medial SMC proliferation and/or apoptosis, eventually leading to vascular disease development33,34. Multiple genes and factors have been found to be involved in the liaison of EC-SMC interaction. XBP1 plays diverse roles in ECs and SMCs. In ECs, under oxidative or ER stress, XBP1 splicing contributes to EC autophagic and apoptosis response26,27,35,36,37; while under growth factor stimulation, XBP1 splicing contributes to EC proliferation23,24,38. In SMCs, XBP1s contributes to calponin h1 decrease, leading to SMC proliferation and neointima formation17,39. In this study, we demonstrate that XBP1 is also involved in the liaison between ECs and SMCs.
The compensative angiogenesis in ischemic tissue is essential for cell survival and tissue or organ function restoration. This is a multiple cells and factors-involved multi-step process, including existing ECs-mediated angiogenesis and stem/progenitor cell-mediated neovascularization. VEGF, PDGF and hypoxia inducible factor 1 (HIF-1) are the main modulators of angiogenesis.
Several studies have shown that XBP1 promotes angiogenesis in cardiac hypertrophy via upregulating VEGF expression in ECs25,40,41,42,43. Our previous study revealed that XBP1 is a downstream of VEGF signalling pathway mediator23. In this study, we found that XBP1 splicing in SMCs could upregulate VEGF-A expression and VEGF receptor-dependent Akt phosphorylation in ECs. The activation of Akt phosphorylation may be modulated through VEGF via autocrine or paracrine mechanisms. The overall effect is to induce EC migration and angiogenesis. The key mediator for the XBP1/VEGF crosstalk-mediated EC-SMC liaison seems to be miR-150. MiRNAs are a set of small non-coding RNAs participating in multiple cellular processes including cell motility. Secretion and delivery of miRNAs via EVs contribute to intercellular communication44. It has been reported that miR-150 can be secreted from monocytes via EVs and increase EC migration and angiogenesis14,29,30. Our study demonstrated that miR-150 can be secreted from SMCs via EVs and increase VEGF-A expression and Akt phosphorylation in ECs. Very importantly, direct transfection of miR-150 into HUVECs can increase VEGF-A mRNA, VEGF secretion and Akt phosphorylation. These results suggest a positive role of miR-150 in VEGF-A signalling pathways and EC migration and angiogenesis. Different from miR-150 transfection-induced ErK phosphorylation, SMC-derived EVs suppressed ErK phosphorylation in EC, which may be due to other inhibitory components within the EVs. This may also explain why EVs have no effect on EC proliferation. However, several reports have suggested that endothelial intrinsic miR-150 seems to play a negative role in EC migration and/or angiogenesis45,46,47. The discrepancy among these reports may be derived from different protocols. In our study, we assessed VEGF-A expression, secretion and VEGF-downstream signalling pathway activation within 3 hours post transfection. A negative feedback may exist in a long term period.
PDGF is another important growth factor for angiogenesis under ischemia. It can be induced from ECs in response to ischemia or other stimulus48,49,50, which in turn activates medial SMCs proliferation and migration, leading to vessel wall remodelling51,52. During this process, XBP1 splicing plays an important role17. In the present study, we found that PDGF stimulated SMCs to secret miR-150-containing EVs in XBP1 splicing dependent manner, leading to EC migration. Therefore, we may speculate that PDGF functions as a liaison mediator from ECs to SMCs.
HIF-1 is a critical transcription factor, essential for cell survival and angiogenesis under ischemia. In response to hypoxia under ischemia conditions, HIF-1 upregulates the transcription of a set of genes that are involved in cell proliferation in both ECs and SMCs, contributing to revascularization in the ischemic tissues53. It has been reported that XBP1 can form a complex with HIF-1 in regulating gene transcription in cancer cells54. HIF-1 was reported to negatively regulate miR-150 expression during liver regeneration. In this study, we found that XBP1 suppressed miR-150 transcription, in which HIF-1 might be involved. XBP1/HIF-1 complex may be responsible for the miR-150 downregulation in pulmonary airway SMCs under hypertension16. Further detailed investigation will be required to address the crosstalk between XBP1 and HIF-1 in both ECs and SMCs following ischemia.
Taken together, we may speculate that under ischemia the activated ECs secrete PDGF, which in turn activates XBP1 splicing in SMCs. The spliced XBP1 stabilizes miR-150 and increases miR-150 secretion via EVs. The miR-150-containing EVs are taken by ECs. The miR-150 upregulates VEGF-A mRNA and protein expression and secretion. VEGF activates Akt phosphorylation via an autocrine or paracrine mechanism, leading to ECs migration and angiogenesis (Fig. 8). Thus, PDGF, XBP1 splicing, miR-150 and VEGF form an important signaling pathway to liaise EC-SMC interaction, in which XBP1 splicing plays a central role and HIF-1 may be also involved through interaction with XBP1.

A schematic illustration of PDGF-IRE1α-XBP1-miR-150 pathway in EC-SMC interaction.
Under ischemia, the activated ECs may secret PDGF, which in turn activates IRE1α phosphorylation, leading to XBP1 mRNA splicing to remove 26-nucleotide intron in SMCs. The spliced XBP1 stabilizes miR-150 and increases its secretion via extracellular vesicles (EVs). The miR-150-containing EVs are taken by ECs, increasing VEGF-A mRNA transcription or stability, leading to VEGF-A protein production and secretion. VEGF-A triggers the ErK and Akt phosphorylation via binding to VEGF receptor in an autocrine or paracrine manner. The activated Akt contributes to EC migration, leading to angiogenesis.
So far, we have demonstrated that XBP1 splicing is involved in EC and SMC proliferation and their interaction, similar to many other important molecules, like PDGF, HIF-1. This will require us to make critical thinking when we intend to develop therapeutic strategies to intervene human diseases. For example, if we want to target XBP1 splicing in SMCs to suppress airway SMC proliferation under pulmonary hypertension, it may affect the EC-SMC interaction and EC proliferation as well. So, a cell specific targeting strategy will be required.
Methods
Chemicals and Reagents
All cell culture serum and media were purchased from Thermo Fisher Scientific, while cell culture supplements were purchased from Sigma. Rat anti-CD31 (553369) was purchased from Pharmingen while antibodies against alpha SMA (ab7817), phosphor-ErK (ab50011) and ErK1/2 (ab17942) were from Abcam. The antibodies against phospho-Akt (sc-7985R), Akt1 (sc-1619) and GAPDH (sc-25778) were from Santa CruZ Biotech. The antibody against VEGF (SAB1402390) was from Sigma. All secondary antibodies were from Dakocytomation. All microRNA reagents were purchased from Thermo Fisher Scientific. PDGF, insulin, holo-transferrin, SU5416, LY294002, DMSO, DAPI and Giemsa were purchased from Sigma.
Cell culture
Human aortic smooth muscle cells (HSMC, ATCC-PCS-100-012) were purchased from ATCC and cultured on 0.04% gelatin-coated flasks in DMEM medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin and streptomycin in humidified incubator supplemented with 5% CO2. The cells were split every three days at a ratio of 1:3. Human umbilical vein ECs (HUVECs, ATCC-PCS-100-010) were cultured on 0.04% gelatin-coated flasks in M199 medium supplemented with 1 ng/ml β-EC growth factor, 3 μg/ml EC Growth Supplement from bovine neural tissue, 10 μ/ml heparin, 1.25 μg/ml thymidine, 10% FBS, 100 μ/ml penicillin and streptomycin in humidified incubator supplemented with 5% CO2. The cells were split every three days at a ratio of 1:5. Both HSMCs and HUVECs up to passage 10 were used in this study. HEK293 and HEK293T cells were maintained in DMEM supplemented with 10% FBS and penicillin/streptomycin and were split every three days at a ratio of 1:4.
Adenoviral and shRNA lentiviral infection
For adenoviral infection, HSMCs were incubated with Ad-null, or Ad-XBP1u(unspliced XBP1) or Ad-XBP1s(spliced XBP1) virus at 10 MOI for 6 hr and then cultured in fresh complete growth medium for time duration indicated in figure legends. For shRNA lentiviral infection, HSMCs were incubated with 100 transfection unit/cell of non-target shRNA or XBP1 shRNA or IRE1α shRNA lentivirus in the presence of 10 mg/mL polybrene for 16 hr, followed by culture in fresh complete growth medium for 72 hr and subjected to further treatments.
Cell counting
HUVECs were seeded in complete growth medium at a 1 × 105 cells/well in 6-well plates for 24 hr. The medium was changed to M199 supplemented with 0.5% FBS (control)-based conditioned medium from the Ad-XBP1s infected SMCs and incubated for 24 hr. Upon harvest, the cells were trypsinized to single cell suspension and subjected to cell counting using a multisizer 3 coulter counter (Beckman Coulter), according to the manufacturer’s instructions.
Wound healing migration assay
Wound was created by scratching confluent HUVECs in 6-well plates. After removal of the cell debris and medium, fresh conditioned medium was added and incubated for 6 hr. Three scratching lines were created in each well and each conditioned medium was performed in three wells. Images were taken with at three different sites on each scratching line at 0 hr and 6 hr. The average migrated cells were calculated from 3 sites/line x 3 lines/well x3 wells.
Transwell migration assay
The HUVECs were detached by using trypsin and resuspended in M199 containing 0.5% FBS. A 100 μl of 5 × 105 cells/ml cell suspension was added into the insert and 600 μl of M199 containing 0.5% FBS or conditioned medium or reconstituted EVs was added into the holder of the transwell (8 μm pore size), followed by incubation for 6 hr. The cells were then fixed with methanol/acetic acid (3:1) for 15 minutes and stained with Giemsa solution (Sigma) for 15 minutes. After removal of the cells inside the insert, the migrated cells were observed under Nikon Eclipse TS100 microscope and images were taken by Nikon Digital Sight system and processed with Adobe Photoshop software. Cells were calculated from 3 10x view/well x 3 wells.
Tube formation assay
The effect of EVs on EC angiogenesis was assessed by tube formation assay as described previously55. Briefly, 50 μl/well of growth factor-reduced Matrigel solution (Millipore) was added to 96-well plate and solidified at 37 °C. EVs were isolated from anti-miR-150 or control miR-transfected Ad-null or Ad-XBP1s-infected HSMCs and reconstituted in M199 medium supplemented with 5 μg/ml insulin (Sigma) and 5 μg/ml transferrin (Sigma). 3000 HUVECs were resuspended in 100 ul EV-reconstituted medium and added to matrigel containing wells in triplicate and incubated in 37 °C humidified incubator supplemented with 5% CO2 for 4 h. Tube formation was observed under Nikon Eclipse TS100 microscope and images were taken by Nikon Digital Sight system and processed with Adobe Photoshop software. Cells were calculated from 3 10x view/well x 3 wells.
EVs isolation
The EVs were isolated from SMCs culture supernatants by differential centrifugations as previously described56. Briefly, the conditioned medium was collected from Ad-XBP1s-infected or PDGF-BB treated SMCs and centrifuged at 500 g (Sorvall Legend RT,75006445 swing-out rotor) at 4 °C for 5 min to remove cell debris. The supernatant was collected and centrifuged again at 4500 g at 4 °C for 30 min to remove smaller debris. The supernatant was then transferred to Ultra-Clear™ tube (Beckman coulter, Item No: 344059) and ultracentrifuged at 120000 g (Hitachi CP100NX,TH-641 rotor) at 4 °C for 60 min. Pellets were resuspended and washed in 1 ml of cold PBS and ultracentrifuged again at the same condition. The EVs pellets were subjected to miRNAs isolation and quantitative RT-PCR analysis for miR-150 or reconstituted into the original volume with M199 medium containing 0.5% FBS (Figs 2, 3, 4 except Fig. 3E) or M199 medium supplemented with 5 μg/ml insulin (Sigma) and 5 μg/ml transferrin (Sigma) (Figs 3E, 6 and 7) for cellular function assays.
MicroRNA analysis
The cellular total RNA was isolated with mirVana™ miRNA isolation kit according to the manufacturer’s instruction. The reverse transcription of miRNAs was performed with the Applied Biosystems® TaqMan® MicroRNA Reverse Transcription kit and Applied Biosystems® 5x RT primer. The quantitative PCR amplifications of samples were done via Applied Biosystems® TaqMan® Universal PCR Master Mix, No AmpErase® UNG and 20x TaqMan small RNA Assay with protocol provided.
VEGF-A mRNA analysis
The cellular total RNA was isolated with Qiagen RNeasy kit with protocol provided. One microgram of RNA was reverse transcribed into cDNA using Impro-RT transcription system (Promega) and 20 ng cDNA (relative to RNA amount) was amplified by a real time PCR SYBR master mix (Applied Biosystems) with a primer set of 5′> ctg tct tgg gtg cat tgg agc <3′ and 5′> ctg cat ggt gat gtt gga ctc <3′ for VEGF-A. The 18s rRNA primer set of 5′> ata cat gcc gac ggg cgc tg <3′ and 5′> gga gag ggg ctg acc ggg tt <3′ was included as internal control. The fold of induction was defined as ct for VEGF-A minus ct for 18s rRNA with that of control group set as 1.0.
Plasmid construction and transient transfection
The 1.1 kb fragment of miR-150 promoter region was amplified by PCR using a primer set of 5′> gcg cag atc tat tca ctt aat taa aga caa aga g <3′ and 5′> tat atc tag agc cgc cgc tgc cgc tgc tgc ctc g <3′ and inserted into the BglII/NheI sites of pSi-CHECK2 to replace the SV40 early enhancer promoter, designated as pSI-150. The plasmid was verified by DNA sequencing. For transient transfection assay, HSMCs were seeded in 12-well plates at 5 × 104 cells/well in triplicate 24 hr prior to transfection. Medium was changed to serum and antibiotics free DMEM. 0.1 μg/well reporter plasmid DNA (pSi-CHECK2 or pSI-150) and 0.1 μg/well pShuttle2-XBP1s or 10 pmol premiR-150 were transfected into HSMCs with 1 μl lipofectamine RNAiMax (Thermo Fisher Scientific) according to the procedure provided. Five hours later, the transfection solution was removed and fresh complete growth medium was added and the cells were incubated for 24 hr, followed by luciferase activity assay with the dual reporter system (Promega) according to the protocols provided. The relative luciferase activity was defined as the ratio of readout for Renilla luciferase to that for Firefly luciferase with that of control group set as 1.0. For the knockdown of endogenous miR-150, HSMC were transfected by 50 pmol/25 ml flask anti-miR-150 (hsa-miR-150-5p inhibitor, Sigma) with lipofectamine RNAiMax (Thermo Fisher Scientific) according to the procedure provided.
ELISA analysis of VEGF
HUVECs were transfected with premiR control, premiR-150, anit-miR control or anti-miR-150 in serum free M199 medium for 5 h, followed by incubation with M199 medium supplemented with 5 μg/ml of insulin and 5 μg/ml of transferrin for 3 h. The medium was collected and cell debris was removed by centrifugation at 2000 rpm at 4 °C for 5 min. The medium was then concentrated 20 fold using Ultracel-10 K centrifuge unit (Millipore) and subjected to VEGF concentration assessment using VEGF human ELISA Kit (ab100663, Abcam) with protocol provided. The fold of induction was defined as the relative A450 nm absorbance with that of premiR control group set as 1.0.
Western blot analysis
Western blot analysis was performed according to standard procedures described elsewhere. Briefly, the cells were lysed with IP-A buffer [10 mM Tris-HCl, pH7.5, 120 mM NaCl, 1 mML EDTA, 1% Triton X-100 plus protease inhibitor cocktail tablets (one tablet for 50 mL, Roche)] and the protein concentration was measured by BioRad protein assay (BioRad)). 50 μg lysate was applied to SDS-PAGE, PVDF membrane (Amersham) transfer, blocking with 5% milk powder in TBST buffer (10 mM Tris-HCl, pH7.5, 120 mM NaCl, 1 mM EDTA, 0.05% Tween-20), primary and secondary antibodies incubation, ECL development and X-ray film exposure (Amersham). The images were processed by Adobe Photoshop software.
Hindlimb ischemia
The XBP1LoxP/LoxP(regarded as wild type, WT) and SM22Cre/XBP1loxP/loxP (SMC conditional knockout, XBP1smcko) mice were created as described previously17 and were anaesthetized using a combination of Hypnorm (25 mg/kg; Veta Pharma, UK) and Hypnovel (25 mg/kg; Roche). The right femoral artery was ligated permanently, i.e. the femoral artery was ligated at two adjacent sites with the middle part cut23. The foot blood flow was measured with LDI Doppler laser scanner (Moor Instruments) 30 minutes post-surgery, 1 week and 2 weeks post-surgery. The reperfusion percentage was defined as the ratio of mean measurement in the foot area of ligated hindlimb to that of contralateral unligated hindlimb. The data was processed with Microsoft Excel software. The flow image was processed with Adobe Photoshop software. The mice were sacrificed humanely after flow measurement at 2 weeks post-surgery. Adductor muscle tissues of ligated side were harvested and cryo-sectioned, followed by immunofluorescent staining with anti-CD31 and smooth muscle actin antibodies. All animal experiments in this study were performed according to protocols approved by the Institutional Committee for Use and Care of Laboratory Animals.
Indirect immunofluorescence staining
Immunofluorescence staining was performed according to standard procedure57. Briefly, the mouse skeletal muscle cryo-sections were air dried at room temperature and fixed with cold methanol (4 °C) for 15 min. The fixed sections were permeabilized with 0.1% Triton X-100 at RT for 15 min, followed by blocking with diluted swine serum (1:20) for 1 hr, incubation with diluted primary antibodies for 1 hr and with secondary antibodies for 45 min. Nucleus was counterstained with DAPI. Images were taken by using SP5 confocal microscope (Leica, Germany) and were processed by Adobe Photoshop software. Magnification was indicated in figure legends as scale bars.
Statistical analysis
Data were expressed as the mean ± SEM and analyzed using GraphPad Prism 5 software with t-test for pair-wise comparisons or analysis of variance (ANOVA) and significance was depicted by asterisks, *p < 0.05, **p < 0.01.
Additional Information
How to cite this article: Zhao, Y. et al. XBP1 splicing triggers miR-150 transfer from smooth muscle cells to endothelial cells via extracellular vesicles. Sci. Rep. 6, 28627; doi: 10.1038/srep28627 (2016).
References
Jain, R. K. Molecular regulation of vessel maturation. Nat Med 9, 685–693 (2003).
Lao, K. H., Zeng, L. & Xu, Q. Endothelial and smooth muscle cell transformation in atherosclerosis. Curr Opin Lipidol 26, 449–456 (2015).
Ambros, V. microRNAs: tiny regulators with great potential. Cell 107, 823–826 (2001).
Yeh, Y. Y. et al. Characterization of CLL exosomes reveals a distinct microRNA signature and enhanced secretion by activation of BCR signaling. Blood 125, 3297–3305 (2015).
Climent, M. et al. TGFbeta Triggers miR-143/145 Transfer From Smooth Muscle Cells to Endothelial Cells, Thereby Modulating Vessel Stabilization. Circ Res 116, 1753–1764 (2015).
Das, S. & Halushka, M. K. Extracellular vesicle microRNA transfer in cardiovascular disease. Cardiovasc Pathol 24, 199–206 (2015).
Fabbri, M., Paone, A., Calore, F., Galli, R. & Croce, C. M. A new role for microRNAs, as ligands of Toll-like receptors. RNA Biol 10, 169–174 (2013).
Tetta, C., Ghigo, E., Silengo, L., Deregibus, M. C. & Camussi, G. Extracellular vesicles as an emerging mechanism of cell-to-cell communication. Endocrine 44, 11–19 (2013).
Zhou, B., Wang, S., Mayr, C., Bartel, D. P. & Lodish, H. F. miR-150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. Proc Natl Acad Sci USA 104, 7080–7085 (2007).
Garzon, R. & Croce, C. M. MicroRNAs in normal and malignant hematopoiesis. Curr Opin Hematol 15, 352–358 (2008).
Watanabe, A. et al. The role of microRNA-150 as a tumor suppressor in malignant lymphoma. Leukemia 25, 1324–1334 (2011).
Ito, M. et al. MicroRNA-150 inhibits tumor invasion and metastasis by targeting the chemokine receptor CCR6, in advanced cutaneous T-cell lymphoma. Blood 123, 1499–1511 (2014).
Wang, W. et al. MiR-150 enhances the motility of EPCs in vitro and promotes EPCs homing and thrombus resolving in vivo. Thromb Res 133, 590–598 (2014).
Zhang, Y. et al. Secreted monocytic miR-150 enhances targeted endothelial cell migration. Mol Cell 39, 133–144 (2010).
Bienertova-Vasku, J., Novak, J. & Vasku, A. MicroRNAs in pulmonary arterial hypertension: pathogenesis, diagnosis and treatment. J Am Soc Hypertens 9, 221–234 (2015).
Perry, M. M. et al. Role of non-coding RNAs in maintaining primary airway smooth muscle cells. Respir Res 15, 58 (2014).
Zeng, L. et al. XBP 1-Deficiency Abrogates Neointimal Lesion of Injured Vessels Via Cross Talk With the PDGF Signaling. Arterioscler Thromb Vasc Biol 35, 2134–2144 (2015).
Shen, X. et al. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 107, 893–903 (2001).
Romero-Ramirez, L. et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res 64, 5943–5947 (2004).
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T. & Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 (2001).
Ron, D. & Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8, 519–529 (2007).
Zhou, Y. et al. Regulation of glucose homeostasis through a XBP-1-FoxO1 interaction. Nat Med 17, 356–365 (2011).
Zeng, L. et al. Vascular endothelial cell growth-activated XBP1 splicing in endothelial cells is crucial for angiogenesis. Circulation 127, 1712–1722 (2013).
Liu, L. et al. Targeting the IRE1alpha/XBP1 and ATF6 arms of the unfolded protein response enhances VEGF blockade to prevent retinal and choroidal neovascularization. Am J Pathol 182, 1412–1424 (2013).
Silvestre, J. S. Vascular endothelial growth factor and angiogenesis: the XBP1 games. Circulation 127, 1644–1646 (2013).
Margariti, A. et al. XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J Biol Chem 288, 859–872 (2013).
Zeng, L. et al. Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proc Natl Acad Sci USA 106, 8326–8331 (2009).
Liberman, M., Johnson, R. C., Handy, D. E., Loscalzo, J. & Leopold, J. A. Bone morphogenetic protein-2 activates NADPH oxidase to increase endoplasmic reticulum stress and human coronary artery smooth muscle cell calcification. Biochem Biophys Res Commun 413, 436–441 (2011).
Li, J. et al. Microvesicle-mediated transfer of microRNA-150 from monocytes to endothelial cells promotes angiogenesis. J Biol Chem 288, 23586–23596 (2013).
Liu, Y. et al. Microvesicle-delivery miR-150 promotes tumorigenesis by up-regulating VEGF and the neutralization of miR-150 attenuate tumor development. Protein & cell 4, 932–941 (2013).
Kerr, P. M. et al. Endothelial feedback and the myoendothelial projection. Microcirculation 19, 416–422 (2012).
Simons, M. & Eichmann, A. Molecular controls of arterial morphogenesis. Circ Res 116, 1712–1724 (2015).
Ross, R. Atherosclerosis–an inflammatory disease. N Engl J Med 340, 115–126 (1999).
Wang, P. et al. Vascular smooth muscle cell apoptosis is an early trigger for hypothyroid atherosclerosis. Cardiovasc Res 102, 448–459 (2014).
Gargalovic, P. S. et al. Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proc Natl Acad Sci USA 103, 12741–12746 (2006).
Placido, A. I., Oliveira, C. R., Moreira, P. I. & Pereira, C. M. Enhanced amyloidogenic processing of amyloid precursor protein and cell death under prolonged endoplasmic reticulum stress in brain endothelial cells. Mol Neurobiol 51, 571–590 (2015).
Li, J., Wang, J. J. & Zhang, S. X. Preconditioning with endoplasmic reticulum stress mitigates retinal endothelial inflammation via activation of X-box binding protein 1. J Biol Chem 286, 4912–4921 (2011).
Duan, Q. et al. MicroRNA-214 Is Upregulated in Heart Failure Patients and Suppresses XBP1-Mediated Endothelial Cells Angiogenesis. J Cell Physiol 230, 1964–1973 (2015).
Wan, X. J. et al. Involvement of BK channel in differentiation of vascular smooth muscle cells induced by mechanical stretch. Int J Biochem Cell Biol 59, 21–29 (2015).
Duan, Q. et al. Deregulation of XBP1 expression contributes to myocardial vascular endothelial growth factor-A expression and angiogenesis during cardiac hypertrophy in vivo. Aging Cell (2016).
Ghosh, R. et al. Transcriptional regulation of VEGF-A by the unfolded protein response pathway. PLoS One 5, e9575 (2010).
Pereira, E. R., Liao, N., Neale, G. A. & Hendershot, L. M. Transcriptional and post-transcriptional regulation of proangiogenic factors by the unfolded protein response. PLoS One 5, e12521 (2010).
Miyagi, H. et al. Transcriptional regulation of VEGFA by the endoplasmic reticulum stress transducer OASIS in ARPE-19 cells. PLoS One 8, e55155 (2013).
Guduric-Fuchs, J. et al. Selective extracellular vesicle-mediated export of an overlapping set of microRNAs from multiple cell types. BMC genomics 13, 357 (2012).
Liu, C. H. et al. Endothelial microRNA-150 is an intrinsic suppressor of pathologic ocular neovascularization. Proc Natl Acad Sci USA 112, 12163–12168 (2015).
Narasaraju, T. et al. Role of microRNA-150 and glycoprotein nonmetastatic melanoma protein B in angiogenesis during hyperoxia-induced neonatal lung injury. Am J Respir Cell Mol Biol 52, 253–261 (2015).
Yu, Z. Y., Bai, Y. N., Luo, L. X., Wu, H. & Zeng, Y. Expression of microRNA-150 targeting vascular endothelial growth factor-A is downregulated under hypoxia during liver regeneration. Mol Med Rep 8, 287–293 (2013).
Kaiser, M., Weyand, C. M., Bjornsson, J. & Goronzy, J. J. Platelet-derived growth factor, intimal hyperplasia and ischemic complications in giant cell arteritis. Arthritis Rheum 41, 623–633 (1998).
Machens, H. G. et al. Platelet-derived growth factor-AA-mediated functional angiogenesis in the rat epigastric island flap after genetic modification of fibroblasts is ischemia dependent. Surgery 131, 393–400 (2002).
Lin, C. M. et al. Ferulic acid augments angiogenesis via VEGF, PDGF and HIF-1 alpha. J Nutr Biochem 21, 627–633 (2010).
Drusbosky, L. et al. Endothelial cell derived angiocrine support of acute myeloid leukemia targeted by receptor tyrosine kinase inhibition. Leuk Res 39, 984–989 (2015).
Myllarniemi, M., Calderon, L., Lemstrom, K., Buchdunger, E. & Hayry, P. Inhibition of platelet-derived growth factor receptor tyrosine kinase inhibits vascular smooth muscle cell migration and proliferation. FASEB J 11, 1119–1126 (1997).
Fraisl, P., Mazzone, M., Schmidt, T. & Carmeliet, P. Regulation of angiogenesis by oxygen and metabolism. Dev Cell 16, 167–179 (2009).
Chen, X. et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature 508, 103–107 (2014).
Zeng, L. et al. HDAC3 is crucial in shear- and VEGF-induced stem cell differentiation toward endothelial cells. J Cell Biol 174, 1059–1069 (2006).
Thery, C., Amigorena, S., Raposo, G. & Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Current protocols in cell biology/editorial board, Juan S. Bonifacino … [et al.] Chapter 3, Unit 3.22 (2006).
Margariti, A. et al. Histone deacetylase 7 controls endothelial cell growth through modulation of beta-catenin. Circ Res 106, 1202–1211 (2010).
Acknowledgements
This work is supported by British Heart Foundation grant [PG12-11-29408 and PG13-63-30419 to L.Z.].
Author information
Authors and Affiliations
Contributions
Y.Z. and Y.L. contributed to experimental design, performance, data collection and analysis and manuscript writing. P.L., Y.G., J.Y., K.-H.L., G.W. and Y.H. contributed to experimental performance and data analysis. G.C. contributed to HUVEC isolation and manuscript revision. T.L., Q.X. and L.Z. contributed to experiment design, data analysis and manuscript revision.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhao, Y., Li, Y., Luo, P. et al. XBP1 splicing triggers miR-150 transfer from smooth muscle cells to endothelial cells via extracellular vesicles. Sci Rep 6, 28627 (2016). https://doi.org/10.1038/srep28627
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep28627
This article is cited by
-
Exosomes in cardiovascular diseases: a blessing or a sin for the mankind
Molecular and Cellular Biochemistry (2022)
-
The Role of Extracellular Non-coding RNAs in Atherosclerosis
Journal of Cardiovascular Translational Research (2022)
-
Cardiovascular Exosomes and MicroRNAs in Cardiovascular Physiology and Pathophysiology
Journal of Cardiovascular Translational Research (2021)
-
The Exosome-Derived Biomarker in Atherosclerosis and Its Clinical Application
Journal of Cardiovascular Translational Research (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
