
Abstract
The present study applied the PacBio single molecule, real-time sequencing technology (SMRT) in evaluating the quality of silage production. Specifically, we produced four types of Medicago sativa silages by using four different lactic acid bacteria-based additives (AD-I, AD-II, AD-III and AD-IV). We monitored the changes in pH, organic acids (including butyric acid, the ratio of acetic acid/lactic acid, γ-aminobutyric acid, 4-hyroxy benzoic acid and phenyl lactic acid), mycotoxins, and bacterial microbiota during silage fermentation. Our results showed that the use of the additives was beneficial to the silage fermentation by enhancing a general pH and mycotoxin reduction, while increasing the organic acids content. By SMRT analysis of the microbial composition in eight silage samples, we found that the bacterial species number and relative abundances shifted apparently after fermentation. Such changes were specific to the LAB species in the additives. Particularly, Bacillus megaterium was the initial dominant species in the raw materials; and after the fermentation process, Pediococcus acidilactici and Lactobacillus plantarum became the most prevalent species, both of which were intrinsically present in the LAB additives. Our data have demonstrated that the SMRT sequencing platform is applicable in assessing the quality of silage.
Similar content being viewed by others


Introduction
Medicago sativa is one of the most important forage crops that are widely used in feeding dairy cows1,2. It contains essential nutrient ingredients for cattle, including proteins, vitamins and minerals3. In China, most planting areas of M. sativa are located in the monsoon regions. The frequent rains in these regions do not only reduce the nutritional value of the crops, but also increase the risk of proliferation of undesirable microbes. To make the best use of M. sativa, there is a growing interest in improving the ensilage technology. Most published researches address the agronomical, biochemical, microbiological, nutritional and engineering aspects of the process4. Among them, recent successes have been achieved by incorporating various additives to aid the fermentation process, which has become a common practice5.
The assessment of silage quality is typically based on determining the fermentation qualities and changes in microbial compositions6. During the past few years, the evaluation of silage quality relied on a comprehensive range of chemical analysis7. Common silage quality indicators include the silage dry matter weight and content, water-soluble carbohydrate concentration, and target bacterial counts8. Although these indicators may partially reflect the silage quality, the assessment process is time-consuming and the results are sometimes inaccurate, especially because of the limited information obtained regarding the silage microbial composition. Recently, real-time polymerase chain reaction and in-depth 16S rRNA gene libraries sequence analysis have been designed to quantify the bacterial microbiota of silages prepared with or without commercial inoculants9,10. These studies have demonstrated the applicability of using such molecular techniques in quantifying certain silage-specific species under a wide variety of conditions. However, the silage bacterial microbiota profiles described by the previous studies are restricted to the genus precision due to the relatively low taxonomical resolution of the traditional DNA sequencing technique that could only determine the partial sequence of the 16S rRNA gene.
The Pacific Biosciences (PacBio) single molecule, real-time sequencing technology (SMRT) is able to depict the bacterial profile of target samples to species level precision because of its power in generating long sequence reads11,12. In the present study, apart from analyzing the silage quality using conventional indicators like pH, organic acid contents, and mycotoxin formation, we specifically focused on detecting and comparing the bacterial microbiota composition of M. sativa silages produced by adding four different lactic acid bacteria (LAB) additives using the PacBio SMRT method. Our data have demonstrated that the SMRT sequencing platform is suitable for assessing the quality of silage.
Results
Fermentative changes in silage pH and organic acids content
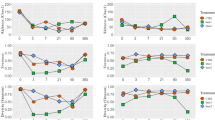
The silage fermentative changes in pH and various organic acids including butyric acid, the ratio of acetic acid/lactic acid, γ-aminobutyric acid, 4-hyroxy benzoic acid and phenyl lactic acid are shown in Fig. 1 and Table 1. Generally, the addition of any of the four LAB additives (AD-I, AD-II, AD-III and AD-IV) resulted in varying degree of fermentative changes in pH and organic acids content. After fermentation, the silage inoculated with AD-IV had both elevated butyric acid concentration and acetic acid/lactic acid ratio. The highest concentration of γ-aminobutyric acid was found in the silage fermented with AD-I, while the lowest one was that fermented with AD-IV. The silage with AD-II had a relatively higher content of both 4-hyroxy benzoic acid and phenyl lactic acid.

The changes in silage pH before and after fermentation.
Changes in silage mycotoxin content after fermentation
The changes in the silage aflatoxin B1, vomitoxin and zearalonone concentrations are given in Table 2. The silage concentrations of the three investigated toxins generally decreased upon fermentation. However, some exceptions were found in the silages fermented with AD-III and AD-IV. The former had a slight increase in aflatoxin B1 concentration, while the latter had mild elevation in both aflatoxin B1 and zearalonone levels.
Changes in microbial composition after silage fermentation
SMRT sequencing of the full length 16S rRNA gene was performed to obtain accurate bacterial profiles of the silage samples at species level. A total of 67,199 raw reads were generated from 8 silage samples, with an average of 8,399 reads for each sample. The Shannon-Wiener diversity curves showed that the sequence depth was adequate for all samples (Fig. 2). Results from the Shannon index, Simpson index, Chao1 index and number of observed species (Table 3) indicated that most samples had a high bacterial biodiversity.

Shannon diversity index curves showing the diversity of taxa present in the dairy products.
Using the RDP classifier, more than 960 species were identified from all samples; twelve of them had a relative abundance of >1%, namely Pediococcus acidilactici, Lactobacillus plantarum, Lactobacillus pobuzihii, Bacillus megaterium, Salmonella enterica, Enterobacter cloacae, Pantoea agglomerans, Ochrobactrum lupini, Weissella cibaria, Enterococcus durans, Bacillus cereus and Bacillus marisflavi. In fact, great variations were found among the species number (Fig. 3) and relative abundances of detected bacteria before and after fermentation (Fig. 4). Before fermentation, Bacillus megaterium was the most abundant species in the silage samples. After fermentation, P. acidilactici (41.18%) and L. plantarum (20.15%) appeared to be the dominant species in the silages (Fig. 4). Clearly, the prevalent species existed in the fermented silages were highly dependent on the original bacterial composition in the LAB additives. By PCoA analysis based on the weighted and unweighted UniFrac distances (Fig. 5), distinct clusters were identified in relation to the silage fermentation status.

Each dot represents a specific sample.

Relative abundances of the silage bacterial species before and after fermentation.

UniFrac weighted and unweighted principle coordinate analysis scores plot based on PC1 and PC2.
Discussion
Silages made from M. sativa are widely used in feeding dairy cows. To improve the fermentation, preservation and nutritional value of silages, LAB-containing additives are often added to aid the fermentative process. Good quality silages also maximize the feed efficiency and thus profitability. To ensure the quality of silages and its production process, it is therefore crucial to perform proper quality control. Traditionally, such evaluation is largely based on determining the microbial composition using biochemical methods, which is unsatisfactory, as the results may sometimes be difficult to interpret and ambiguous. In the present study, the full-length 16S rRNA gene-SMRT sequencing method was applied to monitor the quality of silage production.
Typically, the fermentation of M. sativa silage with LAB additives results in a decrease in pH due to the production of organic acids during the process. Commonly the drop in pH values was mainly caused by lactic acid production during fermentation; and a low pH is favorable, as the silages are better preserved and are more stable. In particular, acetic acid and butyric acid were found to increase the stability of the silage under aerobic conditions13. On the other hand, some compounds, e.g. butyric acid, were implicated as being responsible for reducing silage intake in a study published in 196314. However, only moderate correlation was found between these fermentation acids and silage intake from a large number of animal trials15. Thus, it is still not entirely clear whether a high acidic content in the silage would affect the feed palatability; and whether the high concentration of butyric acid in the AD-IV-treated silage would have any negative effect on feeding remains to be further determined.
In addition to the aforementioned organic acids, 4-hyroxy benzoic acid and phenyl lactic acid have been shown to play a role in maintaining the quality of the silage by inhibiting the production of fungal mycotoxins16. Mycotoxins can cause nervous system disorders, vomiting, loss of appetite, immune function reduction, abortion, stillbirth and monster in cows17,18. Moreover, they can diminish dairy cattle feed intake and milk yield, and even lead to death. There is also an increased health risk for human after consuming milk derived from cows that have ingested contaminated feeds19. It is therefore interesting to observe that the silages treated with AD-I and AD-II have reduced levels of aflatoxin B1 and zearalonone, which is a highly desirable property for future applications. Specifically, these two mycotoxins were negatively correlated to rumen motility and infertility, while their acceptable levels in livestock feeds were below 20 μg/kg and 0.56 mg/kg, respectively20.
The microbiota profile is another indicator that reflects silage quality. Particularly, good quality silage should not contain any pathogenic bacteria. Consistent with previous studies21, the additives used here significantly reduced the populations of pathogenic bacteria that were likely present in the silage raw materials. Some major bacterial species, including B. megaterium, B. marisflavi and S. enteric, may have cause diseases22,23. The minor bacterial species, E. durans, is considered undesirable, even though no report has yet indicated any pathogenic effect of this bacterium on dairy cows. Previous studies have however shown its correlation with antibiotic gene transfer24. One possible mechanism of LAB additives in shaping the silage microbiota during the fermentation process was their ability in releasing antagonistic materials25. The silage predominant species, P. acidilactici and L. plantarum, are both capable of producing antibacterial substances that can inhibit the growth of B. marisflavi, S. enteric and E. durans26,27. Especially, L. plantarum was also reported to be able to produce a broad spectrum of antifungal compounds28, which could explain the decreases in the silage mycotoxin concentrations after fermentation.
To conclude, the four additive LAB inoculants mostly have positive effects on the fermentation of M. sativa silage. Apart from regulating the organic acid and toxin contents, the LAB additives also modulated the bacterial compositions in the fermented silages. Although only eight of the samples were analyzed with the SMRT sequencing technology, our data have shown that this is a prospective method for silage quality assessment.
Methods
Silage production
In this study, M. sativa planted in the Wuhe city of Anhui province was used. M. sativa which was in the budding stage was harvested with a precision chop harvester equipped with an applicator for liquid additives. To reduce the moisture content of the M. sativa, the grass was tedded in the sun. As the water content of the M. sativa reached 45–50%, the grass was chopped to 1–2 cm. During the cutting process, each of the four different commercial LAB-containing additives (Table 4) were sprayed separately and evenly to the respective chopped M. sativa at the concentration of 105 cfu/g. In particular, the first two additives consisted of the same strains, namely L. plantarum Ps-8 and L. plantarum Ps-9, that were obtained from the Key Laboratory of Dairy Biotechnology and Engineering, Ministry of Education, Inner Mongolia Agricultural University, China. The treated M. sativa were then quickly transported with a special truck to the packing site. After unloading from the truck, M. sativa was baled and compacted by a strapping machine. Each group consisted of 5 × 600 kg of silage. The fermentation process lasted for 35 days. Samples from the silage, before and after the fermentation, were collected with sterilized containers and were kept in ice boxes during transportation. For the sequencing analysis, protectant was added into the samples to avoid DNA degradation.
Organic acid analysis and pH measurement
Sample preparation
About 225 g deionized water was added into 25 g of silage sample, followed by mixing at 150 rpm for 30 min in a homogenizer. The mixture was acidified with 1 mol/L hydrochloric acid (1:3, w/w) in a centrifuge tube. Then, the homogenate was centrifuged at 9000 rpm for 10 min. Ten milliliter supernatant was filtered through a 0.22 μm pore size membrane filter before chromatographic analysis. For γ-aminobutyric acid analysis, the homogenate was centrifuged at 1 × 104 rpm/min for 5 min.
Determination of 4-hyroxy benzoic acid and phenyl lactic acid
Separations with high-performance liquid chromatography (HPLC) were performed on an Agilent 1100 Series LC system. A preparative BEHC18 column (1.7 μm, 2.1 × 100 mm, Waters, America) was used. Solvent A was formic acid diluted in deionized water (1:999), and solvent B was formic acid diluted in acetonitrile (1:999) solution. Elution was performed with a linear gradient as follows: solvent B 20–50% in 2 min, 50–95% in 2.1–3 min, 95–5% in 3–3.1 min. Analytical column temperature was 30 °C, and the flow rate was 0.4 mL/min. The operation conditions for the MS analysis of aflatoxin B1, vomitoxin and zearalonone in positive ionization mode (ESI+) were as follows: capillary voltage, 2.5 kV; cone voltage, 40 V; desolvation gas, 0 L/hr; cone gas, 600 L/hr; source temperature, 100 °C; desolvation temperature, 600 °C.
Determination of butyric, acetic and lactic acids
A preparative ZORBAX Elipse AAA C18 column (3.5 μm, 4.6 × 150 mm) was used. Solvent A was phosphate buffer solution (pH 2.5), and solvent B was methanol solution. Elution was performed with a gradient of 97:3. Analytical column temperature was 300 °C, and the flow rate was 1 mL/min. Absorbance was detected at 210 nm.
Determination of γ-aminobutyric acid
The o-phthalaldehyde (OPA) derivative reagent was prepared as described previously29. Briefly, 10 mg OPA (99%, Sigma) was dissolved in 0.5 mL methanol, then 30 μL 2- mercaptoethanol and 2 mL 0.4 mol/L borate buffer (HPLC grade) (pH 9.4) were added. Before injecting into the machine, 10 μL of sample solution was mixed with 90 μL OPA derivative reagent, reacting for 1 min. A preparative ZORBAX Elipse AAA C18 column (3.5 μm, 4.6 × 150 mm) was used. Solvent A was sodium hydrogen phosphate buffer solution (pH 7.8), and solvent B is the mixture of methanol, acetonitrile and deionized water (45:45:10). Elution was performed with a gradient of 97:3. Analytical column temperature was 35 °C, and the flow rate was 2.0 mL/min. Fluorescence detector was employed for detection with the excitation and emission wavelengths of 340 nm and 450 nm, respectively.
pH measurement
25g silage sample was dissolved in 225 mL of deionized water. After vortex mixing for 30 min, a pH meter was used for the measurement.
Mycotoxin analysis
Sample preparation
About 250 g sample was dried to constant weight at 60 °C. After crushing by high speed rotating mill, 25 g crushed sample was transferred to a beaker, followed by adding the mixture of acetonitrile : water (4:6, v/v) in the ratio of 1:8 and leaching for 12 h at room temperature. The mixture was homogenized for 20–30 min in a high speed homogenizer for mycotoxin extraction. Then, it was filtered through a qualitative filter paper. The filtrate was further centrifuged. The resultant supernatant was mixed with acetonitrile and deionized water. After 24 h, the mixture was centrifuged again and the supernatant was collected, enriched by an immunoaffinity chromatography column (BIOTEZ, America) before being analysed with a ultraperformance liquid chromatography–electrospray ionization–quadrupole time-of-flight mass spectrometry (UPLC-ESI-QTOFMS) system (Waters, Milford, MA).
UPLC conditions for determination of aflatoxin B1
A preparative ZORBAX Elipse AAA C18 column (3.5 μm, 4.6 × 150 mm) was used. The mobile phase was the mixture of methanol, acetonitrile and deionized water (5:1:1). Analytical column temperature was 30 °C, and the flow rate was 2.0 mL/min. Fluorescence detector was employed with the excitation and emission wavelengths at 235 nm and 460 nm, respectively.
UPLC conditions for determination of vomitoxin
A preparative ZORBAX Elipse AAA C18 column (3.5 μm, 4.6 × 150 mm) was used. Solvent A was deionized water, and solvent B was the mixture of methanol and acetonitrile (36:64). Elution was performed with a gradient of 97:3. Analytical column temperature was 35 °C, and the flow rate was 2.0 mL/min. Fluorescence detector was employed with the excitation and emission wavelengths at 235 nm and 460 nm, respectively.
UPLC conditions for determination of zearalonone
A preparative C18 column (5 μm, 4.6 × 100 mm) was used. Solvent A was the mixture of methanol, acetonitrile and deionized water (96:2:2), and solvent B was the acetonitrile. Analytical column temperature was 35 °C, and the flow rate was 0.5 mL/min. Absorbance was detected at 220 nm.
QTOFMS conditions for determination of mycotoxins
The operation conditions for the MS analysis of aflatoxin B1, vomitoxin and zearalonone in electrospray ionization mode (ESI+) were as follows: capillary voltage, 2.5 kV; cone voltage, 2.5 kV; desolvation gas, 0 L/hr; cone gas, 0 L/hr; source temperature, 100 °C; desolvation temperature, 100 °C.
SMRT analysis of microbial composition
A total of eight samples, including four each before and after fermentation, were collected, respectively. Sample and sequence information are tabulated (Table 4). DNA was extracted using the OMEGA DNA isolation kit (Omega, D5625-01, USA) following the manufacturer’s instructions. The quality of extracted DNA was checked by 1% agarose gel electrophoresis and spectrophotometry (optical density at 260 nm/280 nm ratio). All extracted DNA samples were stored at −20 °C for further analysis.
The bacterial 16S rRNA was amplified by PCR for barcoded SMRT sequencing with the forward 27F (5′-GAGAGTTTGATCCTGGCTCAG-3′) and the reverse 1541R (5′- AAGGAGGTGATCCAGCCGCA-3′) primers. These primers contained a set of 16-nucleotide barcodes. PCR amplifications of the 16S rRNA regions were performed as described previously30. The PCR program was as follows: 95 °C for 2 min; 30 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s with a final extension of 72 °C for 5 min. The amplicons were sequenced using P6-C4 chemistry on a PacBio RS II instrument (Pacific Biosciences). The quality control for PCR amplifications and sequence preprocessing was performed as described previously31.
Raw data processing was carried out using the protocol RS_ReadsOfinsert.1 available in SMRT Portal version 2.7 as described previously12. The extraction of high-quality sequences was firstly performed with the Quantitative Insights Into Microbial Ecology (QIIME) package (version 1.7). Then, PyNAST32 and UCLUST33 were applied to align the extracted high-quality sequences under 100% clustering of sequence identity to obtain representative sequences. The unique sequence set was classified into operational taxonomic units (OTUs) under the threshold of 98.6% identity using UCLUST after the selection of the representative sequences34. The taxonomy of each OTU representative sequence was assigned using the Ribosomal Database Project (RDP) II database that classified at a minimum bootstrap threshold of 80%35. A de novo taxonomic tree was constructed employing a representative OTU set in FastTree for downstream analysis36, including the beta diversity calculation. The Shannon-Wiener, Simpson’s diversity, Chao1 and rarefaction estimators were calculated to evaluate the alpha diversity. The UniFrac distance was calculated based on the phylogenetic tree34. Both weighted and unweighted calculations were performed for the principal coordinate analysis (PCoA). The graph presentations were generated by the R package version 3.1.2 and the Origin software version 8.5. The sequence data reported in this study have been deposited in the MG-RAST database (accession No. 4678995.3–4679002.3).
Statistical analysis
Experimental data were analyzed by the SAS software (SAS version 9.0, SAS Institute Inc. Cary, NC), and the statistical significance was tested by ANOVA. The chemical composition of each sample was tested three times, and the results were expressed as mean ± standard deviation.
Additional Information
How to cite this article: Bao, W. et al. Assessing quality of Medicago sativa silage by monitoring bacterial composition with single molecule, real-time sequencing technology and various physiological parameters. Sci. Rep. 6, 28358; doi: 10.1038/srep28358 (2016).
References
Mohammed, R., Stevenson, D. M., Beauchemin, K. A., Muck, R. E. & Weimer, P. J. Changes in ruminal bacterial community composition following feeding of alfalfa ensiled with a lactic acid bacterial inoculant. J. Dairy Sci. 95, 328–339, doi: 10.3168/jds.2011-4492 (2012).
Hassanat, F., Gervais, R., Masse, D. I., Petit, H. V. & Benchaar, C. Methane production, nutrient digestion, ruminal fermentation, N balance, and milk production of cows fed timothy silage- or alfalfa silage-based diets. J. Dairy Sci. 97, 6463–6474 (2014).
Arndt, C., Powell, J. M., Aguerre, M. J. & Wattiaux, M. A. Performance, digestion, nitrogen balance, and emission of manure ammonia, enteric methane, and carbon dioxide in lactating cows fed diets with varying alfalfa silage-to-corn silage ratios. J. Dairy Sci. 98, 418–430 (2015).
Fernandes, B. T. & Martins, C. F. H. Technological innovations in silage production and utilization. Rev. Bras. Saúde Prod. Anim., Salvador 13, 629–641 (2012).
Hashemzadeh-Cigari, F. et al. Interactive effects of molasses by homofermentative and heterofermentative inoculants on fermentation quality, nitrogen fractionation, nutritive value and aerobic stability of wilted alfalfa (Medicago sativa L) silage. J Anim Physiol Anim Nutr (Berl). 98, 290–299 (2014).
Weinberg, Z. G., Ashbell, G., Hen, Y. R. & Azrieli, A. The effect of applying lactic acid bacteria at ensiling on the aerobic stability of silages. J. Appl. Bacteriol. 75, 512–518 (1993).
Slottner, D. & Bertilsson, J. Effect of ensiling technology on protein degradation during ensilage. Anim. Feed Sci. Technol. 127, 101–111 (2006).
Sheperd, A. C., Maslanka, M., Quinn, D. & Kung, L., Jr. Additives containing bacteria and enzymes for alfalfa silage. J. Dairy Sci. 78, 565–572 (1995).
Stevenson, D. M., Muck, R. E., Shinners, K. J. & Weimer, P. J. Use of real time PCR to determine population profiles of individual species of lactic acid bacteria in alfalfa silage and stored corn stover. Appl. Microbiol. Biotechnol. 71, 329–338 (2006).
McGarvey, J. A. et al. Bacterial population dynamics during the ensiling of Medicago sativa (alfalfa) and subsequent exposure to air. J. Appl. Microbiol. 114, 1661–1670, (2013).
Zhang, W., Sun, Z., Menghe, B. & Zhang, H. Short communication: Single molecule, real-time sequencing technology revealed species- and strain-specific methylation patterns of 2 Lactobacillus strains. J. Dairy Sci. 98, 3020–3024 (2015).
Hou, Q. et al. Evaluation of bacterial contamination in raw milk, ultra-high temperature milk and infant formula using single molecule, real-time sequencing technology. J. Dairy Sci. 98, 8464–8472 (2015).
Danner, H., Holzer, M., Mayrhuber, E. & Braun, R. Acetic acid increases stability of silage under aerobic conditions. Appl. Environ. Microbiol. 69, 562–567 (2003).
Harris, C. E. & Raymond, W. F. The effect of ensiling on crop digestibility. J. Br. Grassl. Soc. 18, 204–212 (1963).
Wright, D. A. J. G. F., Steen, R. W. J. & Patterson, D. C. Factors influencing the response in intake of silage and animal performance after wilting of grass before ensiling: a review. Grass Forage Sci. 55, 1–13 (2000).
Broberg, A., Jacobsson, K., Strom, K. & Schnurer, J. Metabolite profiles of lactic acid bacteria in grass silage. Appl. Environ. Microbiol. 73, 5547–5552 (2007).
Martins, H. M., Marques, M., Almeida, I., Guerra, M. M. & Bernardo, F. Mycotoxins in feedstuffs in Portugal: an overview. Mycotoxin Res. 24, 19–23 (2008).
Price, W. D., Lovell, R. A. & McChesney, D. G. Naturally occurring toxins in feedstuffs: center for veterinary medicine perspective. J. Anim. Sci. 71, 2556–2562 (1993).
Sugiyama, K., Hiraoka, H. & Sugita-Konishi, Y. Aflatoxin M1 contamination in raw bulk milk and the presence of aflatoxin B1 in corn supplied to dairy cattle in Japan. Shokuhin Eiseigaku Zasshi. 49, 352–355 (2008).
Adams, R., Kephart, K., Ishler, V., Hutchnson, L. & Roth G. Mold and mycotoxin problems in livestock feeding. Available online: http://www.das.psu.edu/dairynutrition/documents/mold.pdf (1993).
Santos, A. O., Avila, C. L. & Schwan, R. F. Selection of tropical lactic acid bacteria for enhancing the quality of maize silage. J. Dairy Sci. 96, 7777–7789 (2013).
López, A. C., Minnaard, J., Pérez, P. F. & Alippi, A. M. In vitro interaction between Bacillus megaterium strains and Caco-2 cells. Int J Microbiol. 16, 27–33 (2013).
Singla, N., Bansal, N., Gupta, V. & Chander, J. Outbreak of Salmonella Typhi enteric fever in sub-urban area of North India: a public health perspective. Asian Pac J Trop Med. 6, 167–168 (2013).
Jahne, M. A., Rogers, S. W., Ramler, I. P., Holder, E. & Hayes, G. Hierarchal clustering yields insight into multidrug-resistant bacteria isolated from a cattle feedlot wastewater treatment system. Environ Monit Assess. 187, 4168 (2015).
Hu, X. et al. Fermentation characteristics and lactic acid bacteria succession of total mixed ration silages formulated with peach pomace. Asian-australas. J. Anim. Sci. 28, 502–510 (2015).
Cintas, L. M. et al. Isolation and characterization of pediocin L50, a new bacteriocin from Pediococcus acidilactici with a broad inhibitory spectrum. Appl. Environ. Microbiol. 61, 2643–2648 (1995).
Hernandez, D., Cardell, E. & Zarate, V. Antimicrobial activity of lactic acid bacteria isolated from Tenerife cheese: initial characterization of plantaricin TF711, a bacteriocin-like substance produced by Lactobacillus plantarum TF711. J. Appl. Microbiol. 99, 77–84 (2005).
Wang, H. K. et al. Production and characterization of antifungal compounds producted by Lactobacillus plantarum IMAU10014. Plos One. 7, e29452 (2012).
Tcherkas, Y. V., Kartsova, L. A. & Krasnova, I. N. Analysis of amino acids in human serum by isocratic reversed-phase high-performance liquid chromatography with electrochemical detection. J Chromatogr A. 913, 303–308 (2001).
Liu, W. J. et al. High-throughput sequencing for the detection of the bacterial and fungal diversity in Mongolian naturally fermented cow’s milk in Russia. BMC Microbiol. 15, 45 (2015).
Mosher, J. J., Bernberg, E. L., Shevchenko, O., Kan, J. & Kaplan, L. A. Efficacy of a 3rd generation high-throughput sequencing platform for analyses of 16S rRNA genes from environmental samples. J. Microbiol. Methods. 95, 175–181 (2013).
Caporaso, J. G. et al. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267 (2010).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Lozupone, C. & Knight, R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235 (2005).
Cole, J. R. et al. The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res. 35, D169–172 (2007).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 26, 1641–1650 (2009).
Acknowledgements
This research was supported by the China Agriculture Research System (Grant No. CARS-37).
Author information
Authors and Affiliations
Contributions
W.Y.Z., L.Y.K. and H.P.Z. designed the study and wrote the manuscript. W.C.B. and Z.H.M. designed and performed the experiments. H.Y.X. and Y.Z. analyzed the data. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Bao, W., Mi, Z., Xu, H. et al. Assessing quality of Medicago sativa silage by monitoring bacterial composition with single molecule, real-time sequencing technology and various physiological parameters. Sci Rep 6, 28358 (2016). https://doi.org/10.1038/srep28358
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep28358
This article is cited by
-
Enzyme additives influence bacterial communities of Medicago sativa silage as determined by Illumina sequencing
AMB Express (2021)
-
Isolation, identification and utilization of lactic acid bacteria from silage in a warm and humid climate area
Scientific Reports (2021)
-
Changes in physico-chemical characteristics and viable bacterial communities during fermentation of alfalfa silages inoculated with Lactobacillus plantarum
World Journal of Microbiology and Biotechnology (2021)
-
Fermentative profile and lactic acid bacterial dynamics in non-wilted and wilted alfalfa silage in tropical conditions
Molecular Biology Reports (2019)
-
Assessment of the physicochemical properties and bacterial composition of Lactobacillus plantarum and Enterococcus faecium-fermented Astragalus membranaceus using single molecule, real-time sequencing technology
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
