
Abstract
Eight highly oxygenated germacranolides (1–8) including four new ones (2–5) were isolated from the whole plant of Carpesium divaricatum. The planar structures and relative configurations of the new compounds were determined by NMR experiment and HRESIMS data. The absolute configuration of 1 was established by circular dichroism (CD) method and X-ray diffraction, and the stereochemistry of the new compounds 2–5 were determined by similar CD spectra with 1. Compound 2 is the first hydroperoxyl germacrane from the genus Carpesium. The 13C NMR data of 1, NMR data of 6–7, and their absolute configurations were reported for the first time. Two new compounds (2 and 4) and two known compounds (6 and 8) exhibited potent cytotoxicity against human cervical cancer (HeLa) cells, superior to that of the positive control doxorubicin.
Similar content being viewed by others
Introduction
Carpesium divaricatum Sieb.et Zucc, belonging to the genus Carpesium (Asteraceae), is widely distributed in China, traditionally used for the treatment of fevers, colds, bruises, and inflammatory diseases1,2,3,4,5. The constituents of this plant have been previously investigated and shown to contain a number of sesquiterpenoids6,7,8,9. Previous investigations indicate that sesquiterpene lactones possessing an α-methylene-γ-lactone moiety in the structure have cytotoxic activity to human cancer cells9,10,11,12,13,14. Recently, Carpesium plants have attracted much attention due to eleven isolated sesquiterpene lactone dimers with novel skeletons displaying significantly cytotoxic activity15,16,17,18. The parent nucleus of the germacranes contains a special ten member ring with different post-modification to produce diverse structural features. A survey of the literature has shown that a large number of germacranolides are isolated from the genus Carpesium, but their absolute configurations have rarely been reported6,7,8,12,13,19,20,21,22,23.
In our ongoing search for new/novel and bioactive products from the medicinal plants in China, four new (2–5) and four known (1 and 6–8) germacranolides were isolated from the whole plant of C. divaricatum. In this paper, the structural elucidation including absolute configuration and bioactive evaluation of these compounds were present.
Results and Discussion
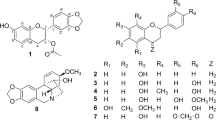
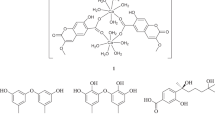
Compound 1 (Fig. 1) was identified as incaspitolide A (1)24, by comparison of its MS, 1H NMR data, as well as optical rotation data with reported data. However, its 13C NMR data have not been reported and absolute configuration has not been determined. The13C NMR data was assigned by 1H-1H COSY and HSQC spectral data. The CD spectrum (Fig. 2) of 1 exhibited two positive Cotton effects at near 252 nm (α-methylene-γ-lactone region) and 294 nm (ketone n, π* region), supporting 6S, 7S configuration12. Fortunately, a suitable crystal was obtained for X-ray diffraction to confirm the absolute configuration. The X-ray crystallographic analysis [flack parameter: −0.02 (10)] established unambiguously the absolute configuration of 1 to be 4S, 5R, 6S, 7S, 8R and 10R (Fig. 3). Herein, the 13C NMR data and absolute configuration of 1 were reported for the first time.

chemical structures of compounds 1–8.

CD spectra of compounds 1–5.

X-ray ORTEP drawing of 1.
Compound 2 was obtained as white needles. The molecular formula was assigned as C24H34O10 on the basis of the positive-ion HRESIMS peak at m/z 505.2036 [M + Na]+, together with its 1H and 13C NMR data (Tables 1 and 2). Its IR spectrum showed hydroxyl (3400 cm−1) and carbonyl (1775 and 1729 cm−1) absorptions. The 1H and 13C NMR spectra of 2 showed an α-methylene-γ-lactone at δH 6.27 (1H, d, J = 2.0 Hz, Ha-13) and 5.96 (1H, d, J = 2.0 Hz, Hb-13), δC 134.7 (C-11), 127.4 (C-13) and 170.6 (C-12); three carbonyl carbons at δC 213.5 (C-9), 175.0 (C-1′) and 167.1 (C-1″); an oxygenated quaternary carbons at δC 73.9 (C-4); five methines including three oxygenated ones at δH 4.75 (1H, d, J = 6.5 Hz, H-5), 4.63 (1H, dd, J = 6.5, 2.0 Hz, H-6), 3.87 (1H, dd, J = 11.5, 2.0 Hz, H-7), 4.92 (1H, d, J = 11.0 Hz, H-8) and 3.27 (1H, m, H-10), δC 79.4 (C-5), 73.3 (C-6), 46.3 (C-7), 79.8 (C-8), and 42.7 (C-10); and two methyl groups at δH 1.01 (3H, d, J = 7.0 Hz, CH3-14), 1.19 (3H, s, CH3-15). These signals (1H and 13C NMR data) of 2 were similar to those of 812 (Table S8.2, Supplementary Information), except for the ester residue at C-5. The singlet signals of 3′-Me and 4′-Me together with chemical shift difference of C-2′ (δ 34.9 → 84.8) implied that C-2′ was an oxygenated quaternary carbon compared with 8. Considering the chemical shift value of C-2′ (δ 84.8) and molecular formula of 2 confirmed that a hydroperoxyl moiety was attached at C-2′ 25,26,27, which was further confirmed by the HRESIMS with fragment peaks at m/z 345.1645 [M + 1-H2O-HOiBu-OOH]+ and 245.1102 [M + 1-H2O-HOiBu-OOH-HOAng]+. The HMBC correlations from both of H3-3′ (δH 1.46, 3H, s) and H3-4′ (δH 1.42, 3H, s) to C-1′ (δc 175.0) and C-2′ (δc 84.8) allowed a reasonable connection of the hydroperoxyl moiety to C-2′. The 1H-1H COSY spectrum (Fig. 4) showed two partial structure sequences for 2: CH2(3)CH2(2)CH2(1)CH(10)CH3(14) and CH(5)CH(6)CH(7)CH(8). The C–C interconnectivity of all fragments was established from the HMBC spectrum (Fig. 4) as correlations of H-15 with C-3 and C-5, H-14 with C-1 and C-9, H-13 with C-7 and C-12, H-8 with C-1″ (ester carbonyl of angeloyloxy group), and H-5 with C-1′ (ester carbonyl of 2′-hydroperoxyl-isobutyryloxy group). On the basis of these data, the planar structure of 2 was established.

Key 1H-1H COSY and HMBC correlations of compounds 2–5.
The relative configuration of 2 was determined by analysis of ROESY data. The key NOE correlations of H-8/H-6, H-7/H-10, H-7/H-5 and H-5/H3-15 indicated that 2 had the same relative configuration as 1. The CD spectrum of 2 showed two positive Cotton effects at near 252 and 294 nm, which closely resembled those of 1. Similar ROESY and CD data of 2 and 1 assigned the absolute configuration of 2 as 4S, 5R, 6S, 7S, 8R and 10R. Thus, the structure of compound 2 was defined as shown, named divarolide A.
Compounds 3–4 possessed molecular formulas of C24H34O9 and C23H32O8, from their HRESIMS at m/z 489.2108 [M + Na]+, and m/z 459.1971 [M + Na]+, respectively. The 1H and 13C NMR data of 3–4 were similar to those of incaspitolide A (1)24, except that the 2′-hydroxy-isobutyryloxy group at C-5 and the angeloyloxy group at C-8 in 3 were observed in place of two isobutyryloxy groups in 1, and an isobutyryloxy group at C-8 in 1 was replaced by the 2-methylacryloyl group in 4, respectively. These observations were confirmed by analyses of relevant 1H-1H COSY, HSQC and HMBC data (Fig. 4). The relative configurations of 3–4 were determined to be the same as that of 1 by comparison of ROESY data for relevant protons. Similar CD data of 3–4 and 1 revealed the same absolute configurations of 3–4 as that of 1. Thus, the structures of compounds 3–4 were established as shown, named divarolide B and divarolide C, respectively.
The molecular formula of compound 5 was assigned as C24H32O8 by HRESIMS (471.1988 [M + Na]+). A comparison of the NMR data of 5 with those of 8 suggested that both of them had the same substituted groups at C-5 and C-8, but that the two mutually coupled methylene units (C-2–C-3) in 8 were oxidized to an olefin moiety in 5. The C-2/C-3 double bond was assigned E-geometry on the basis of the large coupling constant observed for olefinic protons (17.0 Hz). The H-1H COSY, HSQC and HMBC spectra (Fig. 4) of 5 confirmed this observation, leading to the assignment of its planar structure. The relative and absolute configurations of 5 were deduced to be the same as those of 1, on the basis of similar ROESY and CD data. Thus, the structure of compound 5 was elucidated as shown, named divarolide D.
Compounds 6–7 shared the same molecular formula C24H36O8, established from their HRESIMS at m/z 475.2317 [M + Na]+ and m/z 475.2305 [M + Na]+. The 1H and 13C NMR data of 6–7 showed a great similarity with those of 1, except for the ester residues at C-8. The isobutyryloxy group at C-8 in 1 was placed by a 3-methylbutyry- loxy group in 6 and the 2-methylbutyryloxy group of 7, respectively. Compounds 6–7 have been reported as a mixture from Inula cuspidata24. Actually, the exact linkage sites of the substituted groups have not been determined in the previous report and the authors speculate the mixture may contain two pairs of mixtures (incaspitolide B and C). Although the isolation of 6–7 is a huge challenge as they are highly oxygenated and similar, both of them were separated successfully in the present paper. Similarly, their relative and absolute configurations were determined as same as those of 1 by comparison of the ROESY and CD data. Thus, the structures of compounds 6–7 were established as shown, named incaspitolide B1 and incaspitolide B2, respectively.
Compound 8 was a known analogue of 1–7, identified as (4S, 5R, 6S, 7S, 8R, 10R)-8-angeloyloxy-4-hydroxy-5-isobutyryloxy-9-oxo-germacran-7, 12-olide, by comparison of its MS, NMR and optical rotation data with reported data12.
Compounds 1, 2, 4 and 6–8 were obtained in sufficient amounts to be evaluated for their cytotoxic activity against human cervical cancer (HeLa), hepatocellular cancer (Hep G2), stomach cancer (MGC-803), and lung cancer (A549) cell lines. All evaluated compounds exhibited strong cytotoxicity against HeLa (IC50 values of 4.36, 0.83, 1.18, 0.57, 3.58 and 1.70 μM), Hep G2 (IC50 values of 6.41, 8.40, 14.20, 18.10, 9.55 and 8.28 μM), and MGC-803 (IC50 values of 4.63, 4.48, 2.93, 3.49, 4.63 and 2.70 μM) cell lines, but only compounds 2, 4, 6 and 8 had IC50 values of 0.83, 1.18, 0.57 and 1.70 μM against HeLa cell lines, superior to that of the positive control doxorubicin (IC50 value 2.21 μM). Besides, new compound 2 also displayed strong cytotoxicity against A549 with IC50 value of 8.93 μM (the positive control doxorubicin showed IC50 value of 4.18 μM).
In conclusion, eight highly oxygenated germacranolides including four new ones (2–5) were isolated from the whole plant of C. divaricatum. To the best of our knowledge, this is the first report of hydroperoxyl germacrane from the genus Carpesium. New compounds 2 and 4, as well as known compounds 6 and 8, exhibited potent cytotoxicity against HeLa cell lines, superior to that of the positive control doxorubicin. These findings are an important addition to the present knowledge on the structurally diverse and biologically important germacranolide family.
Methods
General Experimental Procedures
Optical rotations were measured on a Perkin-Elmer 241 polarimeter (Perkin-Elmer, Waltham, MA, USA) and UV spectra were recorded on Shimadzu UV-2501 PC (Shimadzu, Kyoto, Japan). IR data were recorded using a Shimadzu FTIR-8400S spectrophotometer (Shimadzu, Kyoto, Japan). 1H and 13C-NMR data were acquired with Bruker 500 instruments (Bruker, Rheinstetten, Germany) using the solvent signals (CD3OD: δH 4.87/δC 49.0 ppm;) as references. HRESIMS data were acquired using Q-TOF analyzer in SYNAPT HDMS system (Waters, Milford, MA, USA). CD spectra were recorded on a JASCO J-815 Spectropolarimeter (Jasco, Tokyo, Japan).
X-ray diffraction data were collected on the Agilent GEMINITME instrument (CrysAlisPro software, Version 1.171.35.11; Agilent, Santa Clara, CA, USA). HPLC was performed using Waters 2535 system (Waters, Milford, MA, USA) with the following components: preparative column, a Daisogel-C18-100A (10 μm, 30 × 250 mm, ChuangXinTongHeng Sci.&Tech., Beijing, China) and a YMC-Pack ODS-A column (5 μm, 10 × 250 mm, YMC, Kyoto, Japan); and detector, Waters 2489 UV. Sephadex LH-20 (40–70 μm, Pharmacia Biotech AB, Uppsala, Sweden), silica gel (60–100, 100–200, and 200–300 mesh) and silica gel GF254 sheets (0.20–0.25 mm) (Qingdao Marine Chemical Plant, Qingdao, China) were used for column chromatography and TLC, respectively. TLC spots were visualized under UV light and by dipping into 5% H2SO4 in EtOH followed by heating.
Plant Material
The whole plant of C. divaricatum were collected from EnShi, Sichuan province of China, in August, 2013. They were identified by Prof. Ben-Gang Zhang of Institute of Medicinal Plant Development. A voucher specimen (No. 20130828) was deposited in the Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences and Peking Union Medical College (CAMS & PUMC), China.
Extraction and Isolation
The air-dried plants (9 kg) were extracted three times (7 days each time) with EtOH–H2O (95:5) at room temperature. The combined extract was concentrated under reduced pressure to furnish a dark brown residue (570 g), which was suspended in H2O and partitioned in turn with petroleum ether (bp 60–90 °C), EtOAc, and n-BuOH. The EtOAc extract (207 g) was separated chromatographically on silica gel column (60–100 mesh, 16 × 20 cm) with a gradient mixture of CH2Cl2–MeOH (100:1, 60:1, 30:1, 15:1, and 6:1) as eluent. Five fractions (fraction A–E) were collected according to thin layer chromatography (TLC) analysis. Fraction A (CH2Cl2–MeOH, 100:1, 140 g) was separated by silica gel column chromatography (CC) (100–200 mesh, 16 × 20 cm) with petroleum ether–aceton (50:1, 25:1, 20:1, 15:1, 12:1, 10:1, 7:1, 5:1, 3:1 and 1:1) as eluent to give fraction A1–A11. Fraction A7 (petroleum ether–aceton, 7:1, 8 g) was separated by Sephadex LH-20 CC (5 × 200 cm, MeOH) to give Fr.A7S1–Fr.A7S3. Fraction A7S2 (MeOH–H2O, 5 g) was purified using preparative HPLC (Daisogel–C18–100A, 10 μm; 250 × 30 mm; 20 mL/min, 70% MeOH in H2O) to yield 8 (3.9 g) and a mixture of 1–7 (800 mg). The mixture of 1–7 (800 mg) was further purified using semipreparative HPLC (60–90% MeOH in H2O for 40 min; 40–80% MeCN in H2O for 40 min) to yield 1 (100 mg), 2 (5 mg), 3 (3.5 mg), 4 (8 mg), 5 (2.6 mg), 6 (30 mg) and 7 (25 mg).
Incaspitolide A (1): white needles (CH3OH),  +57.7 (c 0.25, CHCl3); UV (MeOH) λmax(logε): 207 (4.35) nm, IR (neat) νmax: 3544, 1776, 1746, 1720, 1666 cm−1; CD (MeOH) 256 (Δε +0.09), 310 (Δε +0.22), 207 (Δε −0.44) nm; HRESIMS (pos.): m/z 461.2154 [M + Na]+ (calcd for C23H34O8Na, 461.2151); 1H NMR data see Table 1, 13C NMR data see Table 2.
+57.7 (c 0.25, CHCl3); UV (MeOH) λmax(logε): 207 (4.35) nm, IR (neat) νmax: 3544, 1776, 1746, 1720, 1666 cm−1; CD (MeOH) 256 (Δε +0.09), 310 (Δε +0.22), 207 (Δε −0.44) nm; HRESIMS (pos.): m/z 461.2154 [M + Na]+ (calcd for C23H34O8Na, 461.2151); 1H NMR data see Table 1, 13C NMR data see Table 2.
Divarolide A (2): white needles (CH3OH),  +35.7 (c 0.2, CHCl3); UV (MeOH) λmax(logε): 214 (3.99) nm, IR (neat) νmax: 3400, 1775, 1729, 1645 cm−1; CD (MeOH) 250 (Δε +0.06), 310 (Δε +0.11), 209 (Δε −0.17) nm; HRESIMS (pos.): m/z 505.2036 [M + Na]+ (calcd for C24H34O10Na, 505.2050); 1H NMR data see Table 1, 13C NMR data see Table 2.
+35.7 (c 0.2, CHCl3); UV (MeOH) λmax(logε): 214 (3.99) nm, IR (neat) νmax: 3400, 1775, 1729, 1645 cm−1; CD (MeOH) 250 (Δε +0.06), 310 (Δε +0.11), 209 (Δε −0.17) nm; HRESIMS (pos.): m/z 505.2036 [M + Na]+ (calcd for C24H34O10Na, 505.2050); 1H NMR data see Table 1, 13C NMR data see Table 2.
Divarolide B (3): white needles (CH3OH),  +121.0 (c 0.11, CHCl3); UV (MeOH) λmax(logε): 213 (4.22) nm, IR (neat) νmax: 3500, 1768, 1726, 1643 cm−1; CD (MeOH) 249 (Δε +0.12), 309 (Δε +0.21); HRESIMS (pos.): m/z 489.2108 [M + Na]+ (calcd for C24H34O9Na , 489.2101); 1H NMR data see Table 1, 13C NMR data see Table 2.
+121.0 (c 0.11, CHCl3); UV (MeOH) λmax(logε): 213 (4.22) nm, IR (neat) νmax: 3500, 1768, 1726, 1643 cm−1; CD (MeOH) 249 (Δε +0.12), 309 (Δε +0.21); HRESIMS (pos.): m/z 489.2108 [M + Na]+ (calcd for C24H34O9Na , 489.2101); 1H NMR data see Table 1, 13C NMR data see Table 2.
Divarolide C (4): white needles (CH3OH),  +48.7 (c 0.10, CHCl3); UV (MeOH) λmax(logε): 208 (3.93) nm, IR (neat) νmax: 3508, 1776, 1728, 1665, 1636 cm−1; CD (MeOH) 220 (Δε +0.07), 254 (Δε +0.08), 310 (Δε +0.15) nm; HRESIMS (pos.): m/z 459.1971 [M + Na]+ (calcd for C23H32O8Na, 459.1995); 1H NMR data see Table 1, 13C NMR data see Table 2.
+48.7 (c 0.10, CHCl3); UV (MeOH) λmax(logε): 208 (3.93) nm, IR (neat) νmax: 3508, 1776, 1728, 1665, 1636 cm−1; CD (MeOH) 220 (Δε +0.07), 254 (Δε +0.08), 310 (Δε +0.15) nm; HRESIMS (pos.): m/z 459.1971 [M + Na]+ (calcd for C23H32O8Na, 459.1995); 1H NMR data see Table 1, 13C NMR data see Table 2.
Divarolide D (5): white needles (CH3OH),  +18.9 (c 0.25, CHCl3); UV (MeOH) λmax(logε): 210 (4.03) nm, IR (neat) νmax: 3514, 1771, 1728, 1665, 1646 cm−1; CD (MeOH) 255 (Δε +0.04), 309 (Δε +0.07), 205 (Δε −0.10) nm; HRESIMS (pos.): m/z 471.1988 [M + Na]+ (calcd for C24H32O8Na, 471.1995); 1H NMR data see Table 1, 13C NMR data see Table 2.
+18.9 (c 0.25, CHCl3); UV (MeOH) λmax(logε): 210 (4.03) nm, IR (neat) νmax: 3514, 1771, 1728, 1665, 1646 cm−1; CD (MeOH) 255 (Δε +0.04), 309 (Δε +0.07), 205 (Δε −0.10) nm; HRESIMS (pos.): m/z 471.1988 [M + Na]+ (calcd for C24H32O8Na, 471.1995); 1H NMR data see Table 1, 13C NMR data see Table 2.
Incaspitolide B1 (6): white needles (CH3OH),  +123.1 (c 0.15, CHCl3); UV (MeOH) λmax(logε): 210 (3.83) nm, IR (neat) νmax: 3529, 1777, 1746, 1721, 1665 cm−1; CD (MeOH) 256 (Δε +0.08), 310 (Δε +0.19), 207 (Δε −0.39) nm; HRESIMS (pos.): m/z 475.2317 [M + Na]+ (calcd for C24H36O8Na, 475.2308); 1H NMR data see Table 1, 13C NMR data see Table 2.
+123.1 (c 0.15, CHCl3); UV (MeOH) λmax(logε): 210 (3.83) nm, IR (neat) νmax: 3529, 1777, 1746, 1721, 1665 cm−1; CD (MeOH) 256 (Δε +0.08), 310 (Δε +0.19), 207 (Δε −0.39) nm; HRESIMS (pos.): m/z 475.2317 [M + Na]+ (calcd for C24H36O8Na, 475.2308); 1H NMR data see Table 1, 13C NMR data see Table 2.
Incaspitolide B2 (7): white needles (CH3OH),  +31.2 (c 0.18, CHCl3); UV (MeOH) λmax(logε): 202 (3.77) nm, IR (neat) νmax: 3532, 1781, 1746, 1719, 1667 cm−1; CD (MeOH) 257 (Δε +0.06), 310 (Δε +0.17), 206 (Δε −0.34) nm; HRESIMS (pos.): m/z 475.2305 [M + Na]+ (calcd for C24H36O8Na, 475.2308); 1H NMR data see Table 1, 13C NMR data see Table 2.
+31.2 (c 0.18, CHCl3); UV (MeOH) λmax(logε): 202 (3.77) nm, IR (neat) νmax: 3532, 1781, 1746, 1719, 1667 cm−1; CD (MeOH) 257 (Δε +0.06), 310 (Δε +0.17), 206 (Δε −0.34) nm; HRESIMS (pos.): m/z 475.2305 [M + Na]+ (calcd for C24H36O8Na, 475.2308); 1H NMR data see Table 1, 13C NMR data see Table 2.
X-ray crystal structure analysis
X-ray diffraction data were collected on the Agilent GEMINITME instrument (CrysAlisPro software, Version 1.171.35.11), with enhanced Cu Kα radiation (λ = 1.54184 Å). The structure was solved by direct methods and refined by full-matrix least-squares techniques (SHELXL-97). All non-hydrogen atoms were refined with anisotropic thermal parameters. Hydrogen atoms were located by geometrical calculations and from positions in the electron density maps. Crystallographic data (excluding structure factors) for 1 in this paper has been deposited with the Cambridge Crystallographic Data Centre (deposition number CCDC 1441395). Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44 12 23336033 or e-mail: deposit@ccdc.cam.ac.uk).
A colorless triclinic crystal (0.50 × 0.50 × 0.40 mm) of 1 was obtained from CH2Cl2 –MeOH (3:1). Crystal data: 3C23H34O8·2H2O (M = 459.86), T = 105.5 K, triclinic, space group P1, a = 9.7574(3) Å, b = 10.9450(5) Å, c = 18.3652(8) Å, α = 102.359 (4)°, β = 99.183(3)°, γ = 101.095(3)°, V = 1838.35(13) Å3, Z = 3, ρ = 1.246 mg/mm3, μ(Cu Kα) = 0.781 mm−1, measured reflections = 24294, unique reflections = 12313 (Rint = 0.0202), largest difference peak/hole = 1.010/−0.331 e Å−3, and flack parameter = −0.02(10). The final R indexes [I > 2σ (I)] were R1 = 0.0435, and wR2 = 0.1176. The final R indexes (all data) were R1 = 0.0448, and wR2 = 0.1191. The goodness of fit on F2 was 1.046.
Cytotoxicity assays
The assay was run in triplicate. In a 96-well plate, each well was plated with 2 × 104 cells. After cell attachment overnight, the medium was removed, and each well was treated with 100 μL of medium containing 0.1% DMSO or different concentrations of the test compounds and the positive control doxorubicin. The plate was incubated for 4 days at 37 °C in a humidified, 5% CO2 atmosphere. Cytotoxicity was determined using a modified 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay28. After addition of 10 μL MTT solution (5 mg/mL), cells were incubated at 37 °C for 4 h. After adding 150 μL DMSO, cells were shaken to mix thoroughly. The absorbance of each well was measured at 490 nm in a Multiscan photometer. The IC50 values were calculated by SPSS software and listed in Table 3.
Additional Information
How to cite this article: Zhang, T. et al. New Highly Oxygenated Germacranolides from Carpesium divaricatum and their Cytotoxic Activity. Sci. Rep. 6, 27237; doi: 10.1038/srep27237 (2016).
References
Editorial board of Chinese Materia Medica. Chinese Materia Medica, Vol. 21 (eds Hu, X. M. et al.) 7761 (Shanghai Science & Technology Press, Shanghai (1999).
Kim, E. J. et al. Suppression by a sesquiterpene lactone from Carpesium divaricatum of inducible nitric oxide synthase by inhibiting nuclear factor-κB activation. Biochem. Pharmacol. 61, 903–910 (2001).
Zee, O. P. et al. A new cytotoxic acyclic diterpene from Carpesium divaricatum. Arch . Pharm. Res. 22, 225–227 (1999).
Zee, O. P., Kim, D. K. & Lee, K. R. Thymol derivatives from Carpesium divaricatum. Arch . Pharm. Res. 21, 618–620 (1998).
Chung, I. M. et al. Antiplasmodial activity of isolated compounds from Carpesium divaricatum . Phytother. Res. 24, 451–453 (2010).
Kim, D. K. et al. Four new cytotoxic germacranolides from Carpesium divaricatum . J. Nat. Prod. 60, 1199–1202 (1997).
Maruyama, M. Sesquiterpene lactones from Carpesium divaricatum . Phytochemistry. 29, 547–550 (1990).
Kim, D. K., Lee, K. R. & Zee, O. P. Sesquiterpene lactones from Carpesium divaricatum . Phytochemistry. 46, 1245–1247 (1997).
Xie, W. D. et al. Sesquiterpenoids from Carpesium divaricatum and their cytotoxic activity. Fitoterapia. 83, 1351–1355 (2012).
Zhang, J. P. et al. The genus Carpesium: A review of its ethnopharmacology, phytochemistry and pharmacology. J. Ethnopharmacol. 163, 173–191 (2015).
Yang, C., Yuan, C. S. & Jia, Z. J. Xanthanolides, germacranolides, and other constituents from Carpesium longifolium . J. Nat. Prod. 66, 1554–1557 (2003).
Gao, X., Lin, C. J. & Jia, Z. J. Cytotoxic germacranolides and acyclic diterpenoides from the seeds of Carpesium triste . J. Nat. Prod. 70, 830–834 (2007).
Lin, Y. L. & Ou, J. C. Napalolides A–D, four new sesquiterpene lactones from Carpesium nepalense . J. Nat. Prod. 59, 991–993 (1996).
Li, X. W. et al. Antiproliferative and apoptotic sesquiterpene lactones from Carpesium faberi . Bio. Med. Chem. Lett. 21, 366–372 (2011).
Yang, Y. X. et al. Carpedilactones A–D, four new isomeric sesquiterpene lactone dimers with potent cytotoxicity from Carpesium faberi . Org. Lett. 16, 4216–4219 (2014).
Wu, J. W. et al. Dicarabrones A and B, a pair of new epimers dimerized from sesquiterpene lactones via a [3 + 2] cycloaddition from Carpesium abrotanoides . Org. Lett. 17, 1656–1659 (2015).
Wang, J. F. et al. Dicarabrol, a new dimeric sesquiterpene from Carpesium abrotanoides L. Bioorg. Med. Chem. Lett. 25, 4082–4084 (2015).
Xu, X. K. et al. Four new isomeric sesquiterpene lactone dimers from Carpesium faberi . Tetrahedron Lett. 56, 6381–6384 (2015).
Lee, H. J. et al. A germacranolide sesquiterpene lactone suppressed inducible nitric oxide synthase by downregulating NF-κB activity. Can. J. Physiol. Pharmacol. 89, 232–237 (2011).
Moon, H. I. & Zee, O. Antiproliferative effect from sesquiterpene lactones of Carpesium rosulatum MlQ consumed in South Korea on the five human cancer cell Lines. Rec. Nat. Prod. 4, 3149–3155 (2010).
Moon, H. I. & Zee, O. Sesquiterpene lactones from Carpesium rosulatum with potential cytotoxicity against five human cancer cell lines. Hum. Exp. Toxicol. 30, 1083–1087 (2010).
Kim, M. R. et al. Sesquiterpene lactones from Carpesium triste var. manshuricum. Phytochemistry. 52, 113–115 (1999).
Kim, M. R. et al. Cytotoxic germacranolide sesquiterpene lactones from Carpesium triste var. manshuricum . Arch. Pharm. Res. 30, 556–560 (2007).
Bohlmann, F., Singh, P. & Jakupovic, J. Further ineupatorolide-like germacranolides from Inula cuspidata . Phytochemistry. 21, 157–160 (1982).
Hegazy, M. E. et al. Sesquiterpenes from an Egyptian herbal medicine, Pulicaria undulate, with inhibitory effects on nitric oxide production in RAW264.7 macrophage cells. Chem. Pharm. Bull. 60, 363–370 (2012).
Rustaiyan, A. & Faramarzi, S. Sesquiterpene lactones from Serratula latifolia. Phytochemistry. 27, 479–481 (1988).
Hegazy, M. E. et al. Rare hydroperoxyl guaianolide sesquiterpenes from Pulicaria undulata . Phytochem. Lett. 12, 177–181 (2015).
Wang, B. J., Won, S. J., Yu, Z. R. & Su, C. L. Free radical scavenging and apoptotic effects of Cordyceps sinensis fractionated by supercritical carbon dioxide. Food Chem. Toxicol. 43, 543–552 (2005).
Acknowledgements
This work was financially supported by the Chinese National S&T Special Project on Major New Drug Innovation (2013ZX09508104), the China Postdoctoral Science Foundation (2014M560915), Peking Union Medical College (PUMC) Postdoctoral Science Foundation, and Program for Innovative Research Team in Institute of Medicinal Plant Development (IMPLAD).
Author information
Authors and Affiliations
Contributions
Z.-M.Z. designed the study; T.Z. performed the experiments with the help of J.-G.S., Q.-B.Z. and G.D. The manuscript was prepared by T.Z. and Z.-M.Z. All authors discussed the results and their interpretation and commented on the manuscript at all stages.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhang, T., Si, JG., Zhang, QB. et al. New Highly Oxygenated Germacranolides from Carpesium divaricatum and their Cytotoxic Activity. Sci Rep 6, 27237 (2016). https://doi.org/10.1038/srep27237
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep27237
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
