
Abstract
The fungus Humicola insolens is one of the most powerful decomposers of crystalline cellulose. However, studies on the β-glucosidases from this fungus remain insufficient, especially on glycosyl hydrolase family 3 enzymes. In the present study, we analyzed the functional diversity of three distant family 3 β-glucosidases from Humicola insolens strain Y1, which belonged to different evolutionary clades, by heterogeneous expression in Pichia pastoris strain GS115. The recombinant enzymes shared similar enzymatic properties including thermophilic and neutral optima (50–60 °C and pH 5.5–6.0) and high glucose tolerance, but differed in substrate specificities and kinetics. HiBgl3B was solely active towards aryl β-glucosides while HiBgl3A and HiBgl3C showed broad substrate specificities including both disaccharides and aryl β-glucosides. Of the three enzymes, HiBgl3C exhibited the highest specific activity (158.8 U/mg on pNPG and 56.4 U/mg on cellobiose) and catalytic efficiency and had the capacity to promote cellulose degradation. Substitutions of three key residues Ile48, Ile278 and Thr484 of HiBgl3B to the corresponding residues of HiBgl3A conferred the enzyme activity towards sophorose and vice versa. This study reveals the functional diversity of GH3 β-glucosidases as well as the key residues in recognizing +1 subsite of different substrates.
Similar content being viewed by others


Introduction
The depletion of fossil fuel at enhanced rate and accompanied adverse effects on the global economic and environment has accelerated the research on its alternatives. Cellulosic materials like agricultural wastes and crop by-products (corn stover, wheat straw, bagasse, etc) are the most abundant polysaccharides in nature and represent the most valuable source of renewable energy1. Thus efficient utilization of cellulose biomass has been attracting attentions worldwide for the sustainable development and eco-efficiency2,3,4. Cost-effective process of enzymatic hydrolysis requires low production cost and highly active enzymes with great inhibitor tolerance and synergistic actions. In nature, complete hydrolysis of cellulose needs the synergistic action of a whole cellulolytic enzyme system, which includes endo-β-glucanase (EC 3.2.1.4), cellobiohydrolase (EC 3.2.1.91) and β-glucosidase (EC 3.2.1.21)1,5,6. β-Glucosidase can accelerate the decomposition of cellulose and improve the glucose yield by catalyzing the rate-limiting step of cellobiose hydrolysis7,8. The hyperproducing mutant strains of Trichoderma reesei are commercial producers of highly active cellulase (i.e. Celluclast 1.5 L, Novo Nodisk A/S, Bagsvaerd, Danmark), but have low β-glucosidase activities. Thus the Celluclast 1.5 L alone is inefficient in biomass degradation. To alleviate this limitation, it is a common practice to supplement other fungal β-glucosidase to avoid cellobiose inhibition and increase saccharification efficiency8. Thus, the discovery and biochemical characterization of novel fungal β-glucosidases are of great importance.
In general, β-glucosidases are hydrolases that acts upon β-bonds linking two glucose or glucose-substituted molecules (i.e., the disaccharide cellobiose or isoflavone aglycone). Based on amino acid sequences, the β-glucosidases are grouped into six families of glycoside hydrolase (GH), i.e. GH1, 3, 5, 9, 30 and 116 (http://www.cazy.org/)9,10. Besides biomass conversion, β-glucosidases are applied in other biological processes, such as biogenesis of various functional molecules (e.g., terpenols, flavonoids, phytohormones) from glycoside precursors11,12,13. Considering the great differences among homologous GH3 β-glucosidases14, an insight into the substrate specificity of β-glucosidases is beneficial for better utilization of this multifunctional biocatalyst. With the development of crystal determination, there have been several resolved GH3 β-glucosidase structures15,16,17. And several conserved substrate bind sites were verified in single protein by crystallisation of inhibitor complex or experimental determination, such as Arg156 and Tyr511 of GH3 β-glucosidase AaBGL1 from Aspergillus aculeatus (PDB: 4IIB)15 and Trp49 of β-glucosidase from Aspergillus niger18. But few researches were conducted to investigate the functional diversity and substrate specificity of multiple β-glucosidases from the same species.
Thermophilic filamentous fungi are excellent microbial sources of highly-active, thermostable β-glucosidases for industrial purposes19. To date, filamentous fungi including Trichoderma, Aspergillus and Penicillium are the main microbial sources of industrial cellulases20,21. Thermophilic Humicola spp. are also reported to have the ability to produce various cellulolytic enzymes22,23,24,25,26,27,28. However, the studies on β-glucosidases from this fungus appeared rather scanty. Up to now, an intracellular glucose- and xylose-stimulated β-glucosidase of GH1 (BglHi1) and a purified extracellular β-glucosidase of GH3 (BglHi2) from H. insolens have been biochemically characterized24,25. In this study, three GH3 β-glucosidase encoding genes (Hibgl3A–C) were cloned from H. insolens strain Y1 and successfully expressed in Pichia pastoris strain GS115. The enzymes were all most active under neutral and mesophilic conditions, but showed distinguished substrate specificity, catalytic efficiency and glucose tolerance. Further site-directed mutagenesis revealed the vital role of three residues in the substrate specificity of GH3 β-glucosidases.
Results
Gene cloning and sequence analysis
Three β-glucosidase genes of GH3, i.e. Hibgl3A, Hibgl3B and Hibgl3C, were cloned from the cDNA of H. insolens Y1. Their sequence information is summarized in Table 1. Although deduced HiBgl3A and HiBgl3B exhibit the highest identities with the GH3 protein from Myceliophthora thermophila ATCC 42464 (XP_003663420, 77% and 73%, respectively), they are only 39.9% identical to each other based on the analysis of AlignX from Vector NTI (Invitrogen). Deduced HiBgl3C is most similar to the GH3 β-glucosidase from Trichoderma viride (AAQ76093, 79% identity) and shared 35.4% and 37.3% sequence identity with HiBgl3A and HiBgl3B, respectively. Multiple sequence alignments of these three β-glucosidases with other GH3 proteins (see Fig. 1) indicated the conserved catalytic residues, Asp299 and Glu527 for HiBgl3A, Asp286 and Glu 515 for HiBgl3B and Asp261 and Glu 463 for HiBgl3C, respectively.

Sequence alignment of HiBgl3A, HiBgl3B and HiBgl3C with other GH3 β-glucosidases.
The source and PDB codes or genebank accession numbers of these β-glucosidases are Aspergillus aculeatus (4IIB), Hypocrea jecorina (3ZYZ), Myceliophthora thermophila ATCC 42464 (XP_003663420), Thielavia terrestris NRRL 8126 (XP_003655388), Neurospora crassa OR74A (XP_956104), Trichoderma viride (AAQ76093) and Talaromyces stipitatus ATCC 10500 (XP_002485128). Identical and similar amino acids are indicated by black and gray shades, respectively. The putative catalytic residues were marked with asterisks. The three unique residues existed in HiBgl3B were framed by red rectangle.
Expression and purification of recombinant β-glucosidases
The cDNA fragments of Hibgl3A, Hibgl3B and Hibgl3C without the signal peptide-coding sequences were successfully expressed in P. pastoris GS115 with methanol induction in laboratory flasks. The β-glucosidase activities in the cultural supernatants of the recombinant strains harboring pIC9-Hibgl3A, pIC9-Hibgl3B and pIC9-Hibgl3C were 0.42, 0.76 and 3.54 U/ml, respectively. The crude enzymes were then concentrated, desalted and purified by anion exchange chromatography. SDS-PAGE analyses (see Supplementary Fig. S1) indicated the apparent molecular weights of purified HiBgl3A, HiBgl3B and HiBgl3C were about 120, 127 and 80 kDa, respectively. After deglycosylation with endo-β-N-acetylglucosaminidase H (Endo H), all enzymes showed reductions in the molecular masses, which were in agreement with their calculated values.
Enzymatic properties
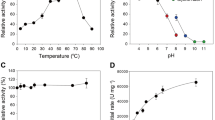
The enzymatic properties of H. insolens β-glucosidases were determined by using 4-nitrophenyl β-d-glucopyranoside (pNPG) as the substrate. All enzymes showed optimal activities at neutral pH, i.e. pH 5.5 for HiBgl3A and HiBgl3C and pH 6.0 for HiBgl3B (Fig. 2A). HiBgl3A and HiBgl3C retained stable (more than 80% activity) over the pH range of 5.0–10.0 at 37 °C for 1 h, while HiBgl3B was stable in a narrower range (pH 5.0–9.0) (Fig. 2B). When assayed under the pH optimum of each enzyme, HiBgl3A, HiBgl3B and HiBgl3C exhibited maximum activities at 60, 50–55 and 60 °C, respectively (Fig. 2C). All enzymes were highly stable at 50 °C (Fig. 2D). The H. insolens β-glucosidases showed similar performance in the presence of 5 mM metal ions or chemical reagents tested, being activated by Mn2+, highly resistant to most metal ions, EDTA and β-mercaptoethanol and sensitive to Ag+ and SDS (see Supplementary Table 1). Moreover, HiBgl3A and HiBgl3C retained more activities than HiBgl3B did in the presence of chemicals.

Enzymatic properties of the purified recombinant β-glucosidases using pNPG as the substrate.
The relative activities of HiBgl3A (red triangle), HiBgl3B (green square) and HiBgl3C (blue circle) were plotted in the line chart. (A) Effect of pH on activities. (B) pH stability. (C) Effect of temperature on activities. (D) Thermostability at different temperatures. Each value in the panel represents the means ± SD (n = 3).
Substrate specificity, kinetic parameters and inhibition constants
The substrate specificities of the three H. insolens β-glucosidases are shown in Table 2. When using disaccharides of different linkages as the substrate, the enzymes showed different preference, gentiobiose (β-1,6 linkage) > sophorose (β-1,2 linkage) > cellobiose (β-1,4 linkage) for HiBgl3A, sophorose > cellobiose > gentiobiose for HiBgl3C, respectively and no HiBgl3B activity against all tested disaccharides. Aryl β-glycoside substrates (4-nitrophenyl compounds and soy isoflavones) that have a phenyl at subsite +1 were also tested. All enzymes showed much higher activities towards pNPG (over 8 fold) than against pNPC, pNPX, pNPGal and pNPAf, but varied in the hydrolysis of soy isoflavones. The activities of HiBgl3B and HiBgl3C against soy isoflavones followed the order of daidzin > genistin > glycitin, while HiBgl3A had relatively low but similar activities against the three tested soy isoflavone substrates. All enzymes had no observable activity on polysaccharides (barley β-glucan, sodium carboxymethylcellulose, Avicel, laminarin and lichenin).
The kinetics of H. insolens β-glucosidases on substrates pNPG and cellobiose are shown in Table 3. In comparison with the other two counterparts, HiBgl3C exhibited much higher substrate affinity (the lowest Km) and catalytic efficiency (kcat/Km). The glucose inhibition was also evaluated using pNPG as the substrate. HiBgl3B and HiBgl3C exhibited relatively high tolerance to glucose than that of HiBgl3A.
Design, construction and specific activities determination of mutants
Three isoenzymes exhibited distinct features in terms of substrate specificity. To investigate the evolutionary relationship of GH3 β-glucosidases, a phylogenetic analysis on the amino acid sequences of H. insolens β-glucosidases and counterparts obtained from the NCBI database using the Neighbor-Joining (NJ) method (shown in Fig. 3) indicated that HiBgl3A, HiBgl3B and HiBgl3C belonged to different evolutionarily related clades. Those belonging to clade II and III have similar length, about 860 amino acids, with different substrate specificities. Sequence alignment suggested that three distinct unique residues (framed by red rectangle in Fig. 1) existed in HiBgl3B and the homologous protein XP_003661483, i.e. Ile48, Ile278 and Thr484, which were generally Trp, Phe and Tyr, respectively, in most GH3 β-glucosidases. These three residues are all located at the entrance of the enzyme’s catalytic pocket and may relate to substrate specificity. Thus we conducted site-directed mutagenesis on HiBgl3A and HiBgl3B of similar lengths to verify the impact of these three sites on recognizing +1 subsite of different substrates. The specific activities and kinetic values of mutants towards aryl β-glycoside pNPG and three disaccharides were measured (shown in Table 4). Although the single and combined mutants I48W, I278F, T484Y and I48W/I278F/T484Y of HiBgl3B showed decreased activities towards pNPG, all the mutants conferred obvious hydrolysis activities on sophorose, which were much higher than the activities on pNPG of their own. However, no activity on the other two disaccharides was detected. In contrast, negative mutants of HiBgl3A all lost hydrolysis activities towards disaccharides and the mutants W69I and F304I even became completely inactivated towards pNPG (data not shown). The turnover numbers (kcat) of HiBgl3B mutants all decreased substantially while no significant change was found in the Km values, leading to declines in catalytic efficiency. However, introduction of mutation Y509T caused the increase of the Km value, but did not affect the kcat value.

The phylogenetic tree generated from the analysis of H. insolens β-glucosidases and other closely related β-glucosidases amino acid sequences in the NCBI database using the Neighbor-Joining method.
The numbers on nodes correspond to the percentage bootstrap values for 1,000 replicates. The accession number of each SOD in GenBank is labelled prior to the species name.
Enzymatic saccharification of cellulose materials
To investigate the application potential of the most efficient H. insolens β-glucosidase HiBgl3C in biomass conversion, the sacchrification efficiencies of different enzyme combinations using corn stover and Avicel as the cellulosic materials were compared as shown in Fig. 4. Over 96 h incubation at pH 5.5 and 50 °C, supplementation of the commercial T. reesei cellulase Celluclast 1.5 L at the dosage of 5 FPU per gram dry materials (DM) released 15.95 mM and 17.09 mM of reducing sugars from corn stover and Avicel, respectively, in which glucose accounted for 3.46 (21%) and 4.85 mM (28%), respectively. The culture supernatants of H. insolens Y1 showed considerable ability to decompose cellulose materials with the activities of 4 FPU/ml and 30 BGU/ml. When supplemented the crude supernatants of H. insolens Y1 (at the dosage of 11 BGU/g DM of β-glucosidase) into commercial Celluclast 1.5 L, the amounts of reducing sugar and yields of fermentable sugars were increased substantially. Their synergistic action resulted in the increased release of reducing sugars from corn stover and Avicel (31.43 and 40.28 mM, respectively) and increased glucose conversion rate (percentage of glucose amount to that of reducing sugars, 99% for corn stover and 77% for Avicel, respectively). When substituted the culture supernatants of H. insolens Y1 with commercial Novozyme 188 (Sigma-Aldrich) at the same dosage of β-glucosidase (11 BGU/g DM), two-fold increase of saccharification efficiency was achieved. As results, 32.91 mM and 36.76 mM of reducing sugars were released from corn stover and Avicel, respectively and the glucose conversion rates were higher than 90%. HiBgl3C showed comparable performance to Novozyme 188 in saccharification, releasing more than 80% of the reducing sugars of Novozyme 188 (27.67 mM and 28.83 mM, respectively) and converting 84% and 93% of the reducing sugars into glucose for corn stover and Avicel, respectively.

Enzymatic saccharifications of pretreated cellulose materials.
The concentrations (mM) of reducing sugar and glucose in the hydrolyzates of corn stover and Avicel by different enzyme combinations (cellulase at 5 FPU/g and β-glucosidase at 11 BGU/g, resepctivley) were shown in (A–D), respectively. Circle: Celluclast 1.5 L only; square: Celluclast 1.5 L plus crude enzyme of H. insolens Y1; upper triangle: Celluclast 1.5 L plus Novozyme 188; lower triangle: Celluclast 1.5 L plus HiBgl3C.
Discussion
The fungus H. insolens is one of the most powerful decomposers of crystalline cellulose and represents a potential industrial producer of cellulases19,23,27. However, the studies on H. insolens β-glucosidases are much limited to only two reports24,25 and the inner peptide sequences of the purified GH3 β-glucosidase BglHi225 showed 100% sequence identity to HiBgl3A of the present work. Although heterologous expression of cellulolytic enzymes is becoming the research focus29, none of the β-glucosidase genes has been cloned from H. insolens Y1 or heterologously expressed as well as functionally characterized. In this study, three GH3 β-glucosidase genes were identified based on the partial genome sequence of H. insolens Y1 and their functions and saccharification performance were revealed after heterologous expressions in P. pastoris GS115. Although the three protein sequences were discrepant and grouped into three different evolutionary clades, their biochemical properties had some similarities. For example, HiBgl3A, HiBgl3B and HiBgl3C exhibited maximum activities at 50–60 °C and pH 5.0–6.0. In contrast, most β-glucosidases from filamentous fungi, i.e. Trichoderma and Aspergillus, have acidic pH optimal in the range of pH 4.0–5.0 (Table 2)30,31,32,33,34,35,36.
The three β-glucosidases from H. insolens Y1 exhibited distinct difference in the substrate specificity towards disaccharides and aryl β-glycosides. HiBgl3A and HiBgl3C are typical GH3 β-glucosidases with strong affinities to a wide range of substrates including both disaccharides and aryl-glycosides although the performances are not the same. It might due to the distinct structures of these two β-glucosidases with inhomogeneous protein sequences, which were grouped into different evolutionary clades. The activities of these two enzymes on cellobiose are higher than that of many fungal counterparts41,42,43 including that from T. reesei (Hypocrea jecorina, 21 U/mg)33. However, HiBgl3B has no activity on all tested disaccharides and only showed considerable hydrolysis ability on pNPG. It’s similar to the BglF from Aspergillus oryzae that has no activity on oligosaccharides (<0.01 U/mg) and a very low specific activity on pNPG (0.6 U/mg)14. This kind of β-glucosidase is defined as aryl β-glucosidase and widely exists in microorganisms44. A previous study has reported that GH3 β-glucosidases have a strict stereochemical requirement to accommodate β-D-glucopyranose at subsite −1, while subsites + seem insignificant in both substrate binding and hydrolysis10. This relative plasticity at subsite +1 might account for the broad substrate specificity of GH3 β-glucosidases towards different aglycon structures. Besides, β-glucosidases of different sequence clades may vary in conformation at the subsite +1, consequently leading to variations in the activities on substrates with different aglycons at this site. Based on the multiple sequence alignment and structure analysis, three conserved substrate recognizing residues for the subsite +1 of cellobiose were identified in the β-glucosidases capable of hydrolyzing cellobiose15,17, for instance, Trp68, Phe305 and Tyr511 of AaBGL1 from Aspergillus aculeatus (PDB: 4IIB), were substituted by Ile48, Ile278 and Thr484 of HiBgl3B, respectively (see Fig. 1)15. The lack of ability to interact with the second sugar ring of disaccharides might be responsible for its unique substrate specificity. It could be speculated that this sort of β-glucosidases does not contribute to the deconstruction of cellulose, though the natural function is unclear yet. This difference indicates their functional diversity.
Activity changes between wild type and mutant enzymes suggested that Ile48, Ile278 and Thr484 have effects on the substrate recognition of enzymes. To investigate the interactions between I48, I278 and T484 and sophorose, a molecular docking was performed and the results were shown in Fig. 5A. The interactions were analyzed by software Ligplot+ and shown in the planar graph Fig. 5B. W48, F278 and Y484 are supposed to interact with ligand sophorose at the subsite +1 by hydrophobic and polar interactions, which are absent in wild type HiBgl3B. And compared to other two disaccharides β-1-4-cellobiose and β-1-6-gentiobiose, the hydroxyl at the C6 site of +1 subunit of β-1-2-sophorose was outward, thus requiring smaller spatial position to locate into the binding pocket. It may explain why HiBgl3B mutants had no activity on cellobiose and gentiobiose. In contrast, wild type HiBgl3B is able to combine the highly hydrophobic aromatic ring at its +1 binding site. However, the binding affinity seems to vary significantly among different sequences and structures because the same case did not apply to the negative mutants of HiBgl3A. For example, the single mutants of HiBgl3A had no activity towards disaccharides and no accumulated effect was found in the combination mutant of HiBgl3B. We conjecture that these three residues in HiBgl3A must have an integral role in the binding with disaccharide substrates. Without Trp69 and Phe304, the enzyme lost hydrolysis activities on both aryl aglycones and disaccharides, indicating the indispensable role played by these two residues. On the other hand, HiBgl3A and HiBgl3B share only 39.9% sequence identity, which may accounts for the huge differences in protein structure, especially the dramatic difference at substrates recognition sites. The disordered conformations of HiBgl3B mutants or abnormal binding to pNPG is also supported by the sharp decline of kcat values. The changes in the Km values of wild type enzymes and mutants also provided proofs of quite distinct effect of these three sites in different clade of β-glucosidases. As for the current job, they all contributed to the binding of disaccharide substrates for HiBgl3A and HiBgl3B. The only difference is that they had no effect on the recognition of pNPG for HiBgl3B while affected the recognition of HiBgl3A.

The stereoview of the catalytic pocket of docked model of mutant I48W/I278F/T484Y combining with sophorose (A) and schematic plan of interaction analyses between the ligand and residues (B). Sophorose (green) and several key residues (red for catalyst and blue for W48, F278 and Y484) are depicted as sticks. Ligand–protein interactions were obtained using Ligplot+.
Moreover, H. insolens β-glucosidases also exhibited diversity in kinetics and glucose tolerance. Most of the microbial β-glucosidases that have the ability to hydrolyze cellobiose are very sensitive to glucose (with the Ki value in the range of 0.35–10 mM glucose, shown in Table 2). The three H. insolens β-glucosidases had different inhibition constants of glucose: HiBgl3B (55.2 mM) was most resistant to glucose, followed by HiBgl3C (37.1 mM) and HiBgl3A (25.0 mM) using pNPG as the substrate. Their Ki values were much higher than most fungal GH3 β-glucosidases30,31,32,33,34,35, including the most generally used commercial β-glucosidase Novozyme 188(Ki value of 2.7 mM)30. In addition, HiBgl3A and HiBgl3C had higher kcat/Km values (11.1/s/mM and 23.0/s/mM, respectively) towards cellobiose than that of several other β-glucosidases35,38,40, but lower than that of Novozyme 188 (36/s/mM)30. A saccharification experiment was carried out to evaluate the ability of the most efficient HiBgl3C in promoting cellulose degradation. Given the same conditions, supplementation of HiBgl3C yielded more than 80% of the reducing sugars released by commercial Celluclast 1.5 L and Novozyme 188 and achieved almost equivalent glucose conversion rate. It was reported that most microbial β-glucosidases performed worse in synergetic cellulose degradation than Novozyme 188 because of their low inhibition constants7. TaBG3 from Acremonium thermophilum and AtBG3 from Thermoascus aurantiacus had good performances by exhibiting kcat/Km values of 26.2 kJ · mol−1 and 20.5 kJ · mol−1 at 55 °C, respectively, which were also lower than the value of Novozyme 188 (29.5 kJ · mol−1)7. However, NpaBGS from the buffalo rumen fungus Neocallimastix patriciarum W5 had a slightly better efficiency than Novozyme 188 at a lower temperature 40 °C39. It demonstrated that, with outstanding cooperation effect, HiBgl3C from H. insolens Y1 has great application potential to enhance the enzymatic saccharification of biomass.
Conclusions
Three H. insolens β-glucosidases of GH3 were produced in yeasts and systematically characterized. The enzymes are highly tolerant to glucose inhibition (25.0–55.2 mM) and share thermophilic and neutral features, but vary in substrate specificities. HiBgl3A and HiBgl3C are active towards both cellobiose and aryl β-glucoside while HiBgl3B is a typical aryl β-glucosidase. Substitutive mutations proved the vital role of three key residues in recognizing +1 subsite of different substrates. Their catalytic performances showed difference in biological processes. This work revealed the relationship between sequence differences and functional diversity.
Methods
Strains, media, vectors and chemicals
H. insolens Y1 GCMCC 4573 was routinely cultured in the wheat bran medium45. Escherichia coli Trans1-T1 and vector pEASY-T3 (TransGen, Beijing, China) were used for gene cloning. The gene expression vector and heterologous expression host were pPIC9 and P. pastoris GS115 (Invitrogen, Carlsbad, CA), respectively. The DNA purification kit, restriction endonucleases and LA Taq DNA polymerase were purchased from TaKaRa (Otsu, Japan). T4 DNA ligase and the total RNA isolation system kit were purchased from Promega (Madison, WI). The cDNA synthesis kit was purchased from TransGen. Barley β-glucan, Avicel, 4-nitrophenyl β-d-glucopyranoside (pNPG), 4-nitrophenyl β-d-xylopyranoside (pNPX), 4-nitrophenyl α-l-arabinofuranoside (pNPAf), 4-nitrophenyl α-d-galactopyranoside (pNPGal), 4-nitrophenyl α-l-arabinopyranoside (pNPAb), 4-nitrophenyl β-d-cellobioside (pNPC), disaccharides cellobiose, sophorose and gentibiose and soybean flavones daidzin, genistin and glycitin were all purchased from Sigma-Aldrich. Sodium carboxymethylcellulose (CMC-Na), laminarin and lichenin were obtained from Megazyme (Wicklow, Ireland). All other chemicals used were of analytical grade and commercially available.
Gene cloning and sequence analysis
The total RNA of H. insolens Y1 was extracted from the mycelia after3 days’ growth in the inducing wheat bran medium and was reverse transcribed into cDNA by TransScript® One-Step gDNA Removal and cDNA Synthesis SuperMix kit (TransGen). The gene fragments without the signal peptide-coding sequence were amplified using H. insolens Y1 cDNA as the template, at an annealing temperature of 60 °C, with three specific primer sets (shown in Supplementary Table S2). The PCR products were purified and ligated into the pEASY-T3 vector for sequencing. Vector NTI Advance 10.0 software (Invitrogen) was used to analyze the DNA sequence and to predict the molecular weight and pI of proteins and perform multiple sequence alignments. The signal peptide and the potential N-glycosylation sites were predicted by the SignalP 4.1 server (http://www.cbs.dtu.dk/services/SignalP/) and the NetNGlyc 1.0 Server (http://www.cbs.dtu.dk/services/NetNGlyc/), respectively. The neighbor-joining phylogenetic tree based on the coding sequences of was performed by using MEGA (Version 6.0).
Expression and purification of the recombinant enzymes
The cDNA fragments without the signal peptide-coding sequences and the pPIC9 vector were both digested by SpeI and NotI (for Hibgl3A) or EcoRI and NotI (for Hibgl3B and Hibgl3C) and ligated into in-frame fusion of the α-factor signal peptide to construct the recombinant plasmids. The recombinant plasmids were linearized using BglII and transformed into P. pastoris GS115 competent cells by electroporation using Gene Pulser X cell Electroporation System (Bio-Rad). Minimal dextrose medium (MD) plates were prepared for the screening of positive transformants. The positive transformants were transferred to buffered glycerol complex medium (BMGY) and grown at 30 °C for 2 days. The cells were collected by centrifugation and resuspended in buffered methanol complex medium (BMMY) containing 0.5% methanol for induction. The β-glucosidase activities of the culture supernatants were assayed using pNPG as the substrate and the transformants exhibiting the highest β-glucosidase activities were subjected to high level expression in 1-l Erlenmeyer flasks. The culture supernatants of aforementioned recombinant strain were collected by centrifugation (12,000× g, 4 °C, 10 min), followed by concentration through a Vivaflow ultrafiltration membrane (Vivascience) with a molecular weight cut-off of 5 kDa. The crude enzyme was loaded onto a FPLC HiTrap Q Sepharose XL 5 mL column (GE Healthcare) that was equilibrated with 20 mM Tris-HCl (pH 8.0). Proteins were eluted using a linear gradient of NaCl (0–1.0 M) in the buffer mentioned above at a flow rate of 3.0 ml/min. Fractions exhibiting β-glucosidase activities were pooled and subjected to SDS-PAGE analysis. The protein concentration was determined by a Bradford assay with bovine serine albumin as a standard. To remove N-glycosylation during heterologous expression in P. pastoris, proteins were incubated with 500 U of Endo H at 37 °C for 2 h according to the manufacturer’s instructions (New England Biolabs). The deglycosylated enzymes were also analyzed by SDS-PAGE.
Enzyme activity assay
The β-glucosidase activities were assayed using pNPG and cellobiose as the substrate. One unit of β-glucosidase activity was defined as the amount of enzyme that released 1 μmol of products per minute under the assay conditions. For substrate pNPG, the standard reaction system consisted of 250 μl of appropriately diluted enzyme and 250 μl of McIlvaine buffer containing 2 mM pNPG. After incubation at a certain temperature for 10 min, 1.5 mL of 1.0 M Na2CO3 was added into the system to terminate the reaction. The amount of p-nitrophenol released was determined spectrophotometrically by reading the absorbance at 405 nm. Each experiment was performed in triplicate. And for cellobiose, the standard reaction was carried out with 70 μl of appropriately diluted enzyme and 70 μl of McIlvaine buffer containing 2 mM cellobiose for 10 min followed by a boiling water bath to terminate the reaction. GOD-POD coloring solution (2.1 ml) was then added into the system and the absorbance at 520 nm was determined to calculate the amount of released glucose.
Biochemical characterization
The optimal pH for β-glucosidases activities were determined at respective appropriate temperature for 10 min over a pH range of 2.0–11.0. Buffers used for the assays were as following: 100 mM Na2HPO4-citric acid (pH 2.0–8.0), 100 mM Tris-HCl (pH 8.0–9.0) and 100 mM glycine-NaOH (pH 9.0–11.0). To estimate pH stability, the enzymes were pre-incubated in the buffers mentioned above without substrate at 37 °C (physiological temperature) for 1 h and the residual activities were measured under standard conditions (pH 6.0, 60 °C and 10 min). The optimal temperatures were examined at the optimal pH by measuring the enzyme activity over the temperature range of 30 and 90 °C. The thermostability was investigated by measuring the residual enzyme activities after preincubation of the enzymes at 60 °C or 70 °C and optimal pH without substrate for various periods. The samples were collected at 0, 2, 5, 10, 20, 30 and 60 min, respectively. Enzyme activity assays were performed under the standard conditions. The activities of β-glucosidases were measured in the presence of 5 mM of various metal ions and chemical reagents (Ag+, Ca2+, Li+, Co2+, Cr3+, Ni2+, Cu2+, Mg2+, Fe3+, Mn2+, Hg2+, Pb2+, EDTA, SDS or β-mercaptoethanol) to estimate their effects on enzymes. The reaction without any additive was used as a blank control.
Substrate specificity
To investigate the substrate specificity of HiBgl3A, HiBgl3B and HiBgl3C, polysaccharides (barley β-glucan, sodium carboxymethylcellulose, Avicel, laminarin and lichenin), disaccharides (cellobiose, sophorose and gentiobiose), artificial p-nitrophenyl derivatives (pNPG, pNPAf, pNPX, pNPGal, pNPAb, pNPC) and soybean flavones (daidzin, genistin and glycitin) were each used as the substrate to determine the corresponding activities at a final concentration of 1% (w/v) or 1 mM.
Kinetic parameters and glucose inhibition
The Km, Vmax and kcat values of H. insolens β-glucosidases were determined under each optimal conditions for 5 min in 100 mM Na2HPO4-citric acid containing 1–10 mM pNPG or cellobiose as the substrate. The data were plotted according to the Lineweaver-Burk method. To estimate the glucose inhibition, the β-glucosidase activities were determined at two final concentrations of pNPG (0.75 mM and 1 mM) in the presence of different amounts of glucose (5–50 mM) using the standard activity assay. The data were analyzed by a Dixon plot to calculate the inhibition constant (Ki) value of glucose.
Homology-modeling and molecular docking of H. insolens β-glucosidases
The tertiary structure of deduced β-glucosidases were homology-modeled using the Discovery Studio v2.5 software (Accelrys) with the crystal structure of AaBGL1 from A. aculeatus (PDB: 4IIB) as the template for HiBgl3A and HiBgl3B and Cel3A from Hypocrea jecorina for HiBgl3C. The modeled structures were then refined by embedded program energy minimization and Verify-3D. The molecular docking of modeled mutant HiBgl3B-I48W/I278F/T484Y and β-1,2-sophorose was carried out by AutoDock Vina10 to elucidate the substrate binding interaction. The docking grid with an appropriate size of 40 × 40 × 40 of 0.375 Å spacing was set to center the β-carbon atom of Asp252 of HiBgl3B. Three-dimensional molecular visualization and figure preparation were performed with PyMOL version 1.7.2.1 (The PyMOL Molecular Graphics System).
Construction and expression of mutants
All mutants were constructed using overlap extension PCR based on the structural analyses of HiBgl3A and HiBgl3B. Several primer sets (shown in Supplementary Table S2) for site-directed mutagenesis were synthesized. Expression and purification of the mutants were conducted as the same with wild type enzymes.
Enzymatic saccharification
Two cellulose materials, corn stover and Avicel, were pretreated by 1% NaOH solution at 121 °C for 30 min in an autoclave sterilizer, washed with ddH2O, dried and mixed with 20 ml of 100 mM Na2HPO4-citric acid buffer (pH 5.5) in 50 ml shake flasks. Three enzyme combinations, i.e. Celluclast 1.5 L supplemented with the crude enzyme of H. insolens Y1, Novozyme 188 and HiBgl3C, respectively, with the total activities of 5 FPU and 11 BGU per gram dry materials (DM) were added into the cellulose mixture and incubated at 50 °C and 200 rpm in a shaking bath for 96 h. Celluclast 1.5 L alone was used as the control group. Hydrolyzates were collected at several intervals and centrifuged at 12,000 rpm, 4 °C for 10 min. The amounts of reducing sugars and glucose released in the supernatants were determined using the DNS46 and GOD-POD methods, respectively. All these experiments were conducted with three replicates.
Additional Information
Accession codes: The cDNA sequences have been submitted to GenBank and accession numbers are KT203370, KT203372 and KT203372 for Hibgl3A, Hibgl3B and Hibgl3C, respectively.
How to cite this article: Xia, W. et al. Functional diversity of family 3 β-glucosidases from thermophilic cellulolytic fungus Humicola insolens Y1. Sci. Rep. 6, 27062; doi: 10.1038/srep27062 (2016).
References
Bornscheuer, U., Buchholz, K. & Seibel, J. Enzymatic degradation of (ligno)cellulose. Angew. Chem. Int. Ed. 53, 10876–10893 (2014).
Tuck, C. O., Pérez, E., Horváth, I. T., Sheldon, R. A. & Poliakoff, M. Valorization of biomass: deriving more value from waste. Science. 337, 695–9 (2012).
Abdel-Halim, E. S., Alanazi, H. H. & Al-Deyab, S. S. Utilization of olive tree branch cellulose in synthesis of hydroxypropyl carboxymethyl cellulose. Carbohydr. Polym. 127, 124–34 (2015).
Sharma, N., Nainwal, S., Jain, S. & Jain, S. Emerging biorefinery technologies for Indian forest industry to reduce GHG emissions. Ecotoxicol. Environ. Saf. S0147–6513, 00207–9 (2015).
Zhang, Y. H. & Lynd, L. R. Toward an aggregated understanding of enzymatic hydrolysis of cellulose: noncomplexed cellulase systems. Biotechnol. Bioeng. 88, 797–824 (2004).
Singhania, R. R., Patel, A. K., Sukumaran, R. K., Larroche, C. & Pandey, A. Role and significance of β-glucosidases in the hydrolysis of cellulose for bioethanol production. Bioresour. Technol. 127, 500–7 (2013).
Teugjas, H. & Väljamäe, P. Selecting β-glucosidases to support cellulases in cellulose saccharification. Biotechnol. Biofuels. 6, 105 (2013).
Ng, I. S., Tsai, S. W., Ju, Y. M., Yu, S. M. & Ho, T. H. Dynamic synergistic effect on Trichoderma reesei cellulases by novel β-glucosidases from Taiwanese fungi. Bioresour. Technol. 102, 6073–81 (2011).
Henrissat, B. & Davies, G. Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 7, 637–44 (1997).
Cantarel, B. L. et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic. Acids. Res. Database issue, D233–8 (2009).
Langston, J., Sheehy, N. & Xu, F. Substrate specificity of Aspergillus oryzae family 3 β-glucosidase. Biochim. Biophys. Acta. 1764, 972–8 (2006).
Fang, W. et al. Characterization of a novel β-glucosidase from Gongronella sp. W5 and its application in the hydrolysis of soybean isoflavone glycosides. J. Agric. Food. Chem. 62, 11688–95 (2014).
Yeom, S. J., Kim, B. N., Kim, Y. S. & Oh, D. K. Hydrolysis of isoflavone glycosides by a thermostable β-glucosidase from Pyrococcus furiosus. J. Agric. Food. Chem. 60, 1535–41 (2012).
Kudo, K., Watanabe, A., Ujiie, S., Shintani, T. & Gomi, K. Purification and enzymatic characterization of secretory glycoside hydrolase family 3 (GH3) aryl β-glucosidases screened from Aspergillus oryzae genome. J. Biosci. Bioeng. S1389-1723(15)00146-2 (2015).
Suzuki, K. et al. Crystal structures of glycoside hydrolase family 3 β-glucosidase 1 from Aspergillus aculeatus. Biochem. J. 452, 211–221 (2013).
Yoshida, E. et al. Role of a PA14 domain in determining substrate specificity of a glycoside hydrolase family 3 β-glucosidase from Kluyveromyces marxianus. Biochem. J. 431, 39–49 (2010).
Karkehabadi, S. et al. Biochemical characterization and crystal structures of a fungal family 3 β-glucosidase, Cel3A from Hypocrea jecorina. J. Biol. Chem. 289, 31624 (2014).
Seidle, H. F., Allison, S. J., George, E. & Huber, R. E. Trp-49 of the family 3 beta-glucosidase from Aspergillus niger is important for its transglucosidic activity: creation of novel beta-glucosidases with low transglucosidic efficiencies. Arch. Biochem. Biophys. 455, 110–8 (2006).
Bhatia, Y., Mishra, S. & Bisaria, V. S. Microbial β-glucosidases: cloning, properties and applications. Crit. Rev. Biotechnol. 22, 375–407 (2002).
Wilson, D. B. Processive and nonprocessive cellulases for biofuel production—lessons from bacterial genomes and structural analysis. Appl. Microbiol. Biotechnol. 93, 497–502 (2012).
Gusakov, A. V. Alternatives to Trichoderma reesei in biofuel production. Trends. Biotechnol. 29, 419–25 (2011).
Schülein, M. Enzymatic properties of cellulases from Humicola insolens. J. Biotechnol. 57, 71–81 (1997).
Karlsson, J. et al. Enzymatic degradation of carboxymethyl cellulose hydrolyzed by the endoglucanases Cel5A, Cel7B and Cel45A from Humicola insolens and Cel7B, Cel12A and Cel45Acore from Trichoderma reesei. Biopolymers. 63, 32–40 (2002).
Souzaa, F. H. M., Inocentesc, R. F., Warda, R. J., Jorgeb, J. A. & Furriel, R. P. M. Glucose and xylose stimulation of a β-glucosidase from the thermophilic fungus Humicola insolens: A kinetic and biophysical study. J. Mol. Catal. B. Enzym. 94, 119– 128 (2013).
Meleiro, L. P. et al. A novel β-glucosidase from Humicola insolens with high potential for untreated waste paper conversion to sugars. Appl. Biochem. Biotechnol. 173, 391–408 (2014).
Boisset, C., Pétrequin, C., Chanzy, H., Henrissat, B. & Schülein, M. Optimized mixtures of recombinant Humicola insolens cellulases for the biodegradation of crystalline cellulose. Biotechnol. Bioeng. 72, 339–45 (2001).
Varrot, A. et al. Structural basis for ligand binding and processivity in cellobiohydrolase Cel6A from Humicola insolens. Structure 11, 855–64 (2003).
Otzen, D. E., Christiansen, L. & Schülein, M. A comparative study of the unfolding of the endoglucanase Cel45 from Humicola insolens in denaturant and surfactant. Protein. Sci. 8, 1878–87 (1999).
Lambertz, C. et al. Challenges and advances in the heterologous expression of cellulolytic enzymes: a review. Biotechnol. Biofuels. 7, 135 (2014).
Chauve, M. et al. Comparative kinetic analysis of two fungal β-glucosidases. Biotechnol. Biofuels. 3, 3 (2010).
Decker, C. H., Visser, J. & Schreier, P. β-Glucosidases from five black Aspergillus species: study of their physico-chemical and biocatalytic properties. J. Agric. Food. Chem. 48, 4929–36 (2000).
Korotkova, O. G. et al. Isolation and properties of fungal β-glucosidases. Biochem. Mosc. 74, 569–577 (2009).
Langston, J., Sheehy, N. & Xu, F. Substrate specificity of Aspergillus oryzae family 3 β-glucosidase. Biochim. Biophys. Acta. 1764, 972–978 (2006).
Yoon, J. J., Kim, K. Y. & Cha, C. J. Purification and characterization of thermostable β-glucosidase from the brown-rot basidiomycete Fomitopsis palustris grown on microcrystalline cellulose. J. Microbiol. 46, 51–55 (2008).
Karnaouri, A., Topakas, E., Paschos, T., Taouki, I. & Christakopoulos, P. Cloning, expression and characterization of an ethanol tolerant GH3 β-glucosidase from Myceliophthora thermophila. Peer. J. 1, e46 (2013).
Schmid, G. & Wandrey, C. Characterization of a cellodextrin glucohydrolase with soluble oligomeric substrates: experimental results and modeling of concentration-time-course data. Biotechnol. Bioeng. 33, 1445–1460 (1989).
Pei, J., Pang, Q., Zhao, L., Fan, S. & Shi, H. Thermoanaerobacterium thermosaccharolyticum β-glucosidase: a glucose-tolerant enzyme with high specific activity for cellobiose. Biotechnol. Biofuels. 5, 31 (2012).
Zemin, F. et al. Cloning and characterization of β-glucosidase from marine microbial metagenome with excellent glucose tolerance. J. Microbiol. Biotechnol. 20, 1351–1358 (2010).
Chen, H. L. et al. A highly efficient β-glucosidase from the buffalo rumen fungus Neocallimastix patriciarum W5. Biotechnol. Biofuels. 5, 24 (2012).
Del Pozo, M. V. et al. Microbial β-glucosidases from cow rumen metagenome enhance the saccharification of lignocellulose in combination with commercial cellulase cocktail. Biotechnol. Biofuels. 5, 73 (2012).
Christakopoulos, P. et al. Purification and characterisation of an extracellular β-glucosidase with transglycosylation and exo-glucosidase activities from Fusarium oxysporum. Eur. J. Biochem. 224, 379–85 (1994).
Bronnenmeier, K. & Staudenbauer, W. L. Purification and properties of an extracellular β-glucosidase from the cellulolytic thermophile Clostridium stercorarium. Appl. Microbiol. Biotechnol. 28, 380–386 (1988).
Hong, M. R. et al. Characterization of a recombinant β-glucosidase from the thermophilic bacterium Caldicellulosiruptor saccharolyticus. J. Biosci. Bioeng. 108, 36–40 (2009).
Gao, J. & Wakarchuk, W. Characterization of five β-glycoside hydrolases from Cellulomonas fimi ATCC 484. J. Bacteriol. 196, 4103–10 (2014).
Du, Y. et al. Characterisation of three novel thermophilic xylanases from Humicola insolens Y1 with application potentials in the brewing industry. Bioresour. Technol. 130, 161–167 (2013).
Miller, G. L. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal. Chem. 31, 426–428 (1959).
Acknowledgements
This research was supported by the National High Technology Research and Development Program of China (863 Program, 2012AA022105 and 2012AA022208), the China Modern Agriculture Research System (CARS-42) and the Key Scientific and Technological Innovation Group Program “Feed Research and Security” of Zhejiang Province (2011R50025-12).
Author information
Authors and Affiliations
Contributions
W.X. and Y.B. performed the enzyme production, activity assay, acquisition of data and drafted the manuscript. Y.C. and X.X. performed the TLC analysis. P.S. and W.Z. performed the enzymatic saccharification. H.L. and X.Z. performed the data processing and interpretation and participated in revising the manuscript. L.Q. and B.Y. were the corresponding authors; they designed the study and revised the manuscript critically for important intellectual content. All authors read and approved the final version of the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Xia, W., Bai, Y., Cui, Y. et al. Functional diversity of family 3 β-glucosidases from thermophilic cellulolytic fungus Humicola insolens Y1. Sci Rep 6, 27062 (2016). https://doi.org/10.1038/srep27062
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep27062
This article is cited by
-
Unveiling a classical mutant in the context of the GH3 β-glucosidase family in Neurospora crassa
AMB Express (2024)
-
CRISPR/Cas9-mediated genome editing directed by a 5S rRNA–tRNAGly hybrid promoter in the thermophilic filamentous fungus Humicola insolens
Biotechnology for Biofuels (2021)
-
A Novel Glucose-Tolerant GH1 β-Glucosidase and Improvement of Its Glucose Tolerance Using Site-Directed Mutation
Applied Biochemistry and Biotechnology (2020)
-
Metatranscriptomics of the Hu sheep rumen microbiome reveals novel cellulases
Biotechnology for Biofuels (2019)
-
Chaetomella raphigera β-glucosidase D2-BGL has intriguing structural features and a high substrate affinity that renders it an efficient cellulase supplement for lignocellulosic biomass hydrolysis
Biotechnology for Biofuels (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
