
Abstract
Hybridization and polyploidization are considered important driving forces that form new epigenetic regulations. To study the changing patterns of expression accompanying hybridization and polyploidization, we used RNA-seq and qRT-PCR to investigate global expression and homoeologue expression in diploid and tetraploid hybrids of Carassius auratus red var. (♀) (R) and Cyprinus carpio (♂) (C). By comparing the relative expression levels between the hybrids and their parents, we defined the expression level dominance (ELD) and homoeologue expression bias (HEB) in liver tissue. The results showed that polyploidization contributed to the conversion of homoeologue ELD. In addition, hybridization had more effect on the change in HEB than polyploidization, while polyploidization had more effect on the change of global gene expression than hybridization. Meanwhile, similar expression patterns were found in growth-related genes. The results suggested that hybridization and polyploidization result in differential degrees of maternal HEB in three tissues (liver, muscle and ovary) tested. The results of this study will increase our understanding of the underlying regulation mechanism of rapid growth in diploid hybrids and allotetraploids. The differential degrees of global expression and homoeologue expression contribute to growth heterosis in newly formed hybrids, ensuring the on-going success of allotetraploid speciation.
Similar content being viewed by others

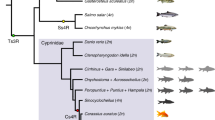

Introduction
Hybridization and polyploidization are fundamental processes in evolution that result in the emergence of novel genotypes from the merger of two or more different genomes1,2,3,4. Many studies have focused on global expression between the parents and hybrid offspring to determine the mechanism of expression regulation in allopolyploids5. This phenomenon has been described as the evolution of gene expression, which is considered useful for adaptation and speciation6. Meanwhile, two sets of homoeologous genes and duplicated pairs may lead to changes in the expressions of some genes related to phenotypic differences in allopolyploids7,8. Thus, a study of homoeologue expression would provide a useful platform to investigate genomic divergence in hybrids and polyploids.
Carassius auratus red var. (R) and Cyprinus carpio (C), which belong to different genera, are the most predominant and widespread form of cyprinid fish, and contain 100 chromosomes. After selective breeding, diploid hybrid offspring (2n = 100) were produced with 50 chromosomes from R and 50 from C9. Fertile allotetraploid hybrids (4n = 200) were obtained on a large scale by crossing F2 diploid hybrids10, and now have reproduced successfully up to generation 25. Fluorescence in situ hybridization (FISH) results showed that allotetraploid fish contained two sets of R and C genomes, respectively (unpublished data). The two hybrid populations that originated from R and C provide us with a platform to study the regulation of homoeologue expression by hybridization and polyploidization.
Hybrid fish are widely distributed as a result of artificial or natural interspecies hybridization. Upon crossing the interspecies barrier, the newly formed progeny display heterosis, such as fast growth. Recent studies have focused on expression level dominance (ELD) and homoeologue expression bias (HEB) to analyse gene regulation patterns and their underlying mechanisms11,12,13. Other studies have shown that allelic interactions and gene redundancy result in heterosis in allopolyploids relative to non-coding RNA, DNA, methylation and transcriptome changes14,15. Although previous studies in teleost hybrids were largely based on global expression8,16, determining homoeologue expression is a promising way to study the regulation of the underlying expression mechanisms. In particular, analysis of the regulation of sets of growth-related genes is crucial to decipher the genomic basis of growth heterosis8.
An increasing number of studies of homoeologue expression have used RNA-seq to investigate gene expression patterns between hybrids and their parents. RNA-seq is regarded as an efficient method to examine overlapping hybridization among homoeologues12,13,17. Meanwhile, in non-model organisms, the identification of homoeologue-specific single nucleotide polymorphisms (SNPs) in the two different genomes is also useful18. Homoeologue expression is then estimated by relative expression using real-time quantitative PCR (qRT-PCR)18. In this study, we combined RNA-seq and qRT-PCR to investigate the ELD and HEB relative to hybridization (genome merger) and polyploidization (genome doubling).
To investigate changes in homoeologue expression levels related to heterosis, particularly the underlying growth regulation mechanism, we used diploid and tetraploid hybrids of C. auratus red var. (♀) and C. carpio (♂) in our study. By comparing with the relative expression levels between the hybrids and their parents, we defined the ELD and HEB in liver tissue by RNA-seq. Meanwhile, the expression silencing of R/C homoeologues originated from R/C genomes was identified for certain genes, revealing epigenetic changes and underlying regulation mechanisms after genome merger and genome doubling. Seven key growth-regulated genes were studied in various tissues using qRT-PCR. The results showed that R-bias was predominant in the F1 diploid hybrid of C. auratus red var. (♀)× C. carpio (♂) (F1) and the eighteen generations of tetraploid hybrids of C. auratus red var. (♀)× C. carpio (♂) (F18). Our goal was to assess the magnitude and directionality of ELD and HED relative to heterosis in different ploidy level hybrids. Therefore, these data provided a novel perspective to study expression patterns of homoeologous genes under genome merger and genome doubling, and gave us an insight into the regulation mechanism that contributed to heterosis.
Results
Statistical mapping of RNA-seq data
To investigate how hybridization and polyploidization affect growth regulatory mechanism, we used the allotetraploid line of C. auratus red var. ×C. carpio to study the pattern of global expression and homoeologue expression in two different ploidy level hybrids (Fig. 1). The F1 diploid hybrid and F18 allotetraploid individuals were sexually mature cyprinid fish that possess hybrid traits10. All short-read data have been deposited at the Short Read Archive (SRA) under accession numbers SRX668436, SRX175397, SRX668453, SRX177691, SRX671568, SRX671569 and SRX668467 (same material: Liu et al. (2016)) and SRX1610992. We then annotated the exons of R and C using BLASTX alignment (e-value ≤ 1e−6) with protein databases (Supplementary Table S1). 20,169 genes were identified in the R genome assembly and 20,365 genes in the C genome. Meanwhile, 739 million (M) clean reads (76.8%) from 12 libraries were surveyed to map to the two references sequences (Supplementary Tables S1 and S2). The liver transcriptome results showed that approximately 17,275 genes were expressed in four kinds of fish (Table 1). Notably, slightly more genes were expressed in the hybrids than in both of their diploid parents. This phenomenon also reflected the coexistence of R- and C-genomes in hybrid individuals.

(A) 100 Chromosomes were observed in C. auratus red var. (B) 100 Chromosomes were observed in C. carpio. (C,D) After hybridization, F1-F2 diploid hybrids (C) and F3-F25 allotetraploid (D) were obtained. The observation of chromosomes showed that duplication of genome was occurred in F3-F25 relative to F1-F2.
Differential gene expression, novel expression and silencing
To study gene expression patterns in F1 diploid hybrids and F18 allotetraploids, we performed pairwise comparisons between the diploid parents to assess pre-existing differential gene expression (Fig. 2). Approximately 5,104 genes (33.32%) were differentially expressed between the diploid parents (P < 0.05 in comparisons; Fisher’s exact test). In all comparisons, the percentage of genes showing differential expression between F1 or F18 and their two parents was asymmetric (P < 0.05; Fisher’s exact tests). Meanwhile, the differentially expressed genes exhibited a bias toward the different parents. For example, global expression of F1 was closer to the maternal R than to paternal C. Approximately 18.31% of genes were differentially expressed between F1 and R, whereas the number of differentially expressed genes was 26.45% relative to C (P < 0.05 in comparisons; Fisher’s exact test). Conversely, the global expression patterns in F18 were closer to the paternal C than to the maternal R.

(A) Bold text exhibits the total number and fraction of genes differentially expressed in each contrast. Also shown for each contrast is the partitioning of the total number of differentially expressed genes into the direction of upregulation. For example, 5,104 genes are indicated as being differentially expressed between C. auratus red var. and C. carpio. Of these, 3,200 are upregulated in C. auratus red var., and 1,904 genes are upregulated in C. carpio. The asymmetry between differential expression between the progeny and its diploid parents corresponds to genome-wide ELD toward one parental genome. The left figure show an interspecific diploid hybrid F1 generated from the diploid parents C. auratus red var. (R) and C. carpio (C). The middle of figure show that F18 allotetraploid was generated from duplication of genome of diploid hybrids. The right figure exhibits that F18 genome was consist of C. auratus red var. homoeologue and C. carpio homoeologue. (B) Bold text exhibits the 118 growth genes number and fraction of genes differentially expressed in each contrast. Also shown for each contrast is the partitioning of the growth genes number of differentially expressed genes into the direction of upregulation.
In the expression comparison, only 13 genes (0.08%) exhibited novel expression in F1. However, novel expression increased with polyploidization: 44 (0.25%) genes exhibited novel expression in F18 (Table 2). We then evaluated homoeologue silencing in total expressed genes. There were 38 (0.22%) cases of R homoeologue silencing in F1 and 26 (0.15%) cases in F18. Nineteen (0.11%) C homoeologues were silenced in F1 and 46 (0.27%) in F18 (Table 2). These results suggested that polyploidization accelerates the occurrence of homoeologue silencing.
Expression level dominance in the liver transcriptome
To study ELD in F1 diploid hybrids and F18 allotetraploids, we performed pairwise comparisons between the hybrid offspring with the diploid parents to assess differentially expressed genes. Compared with the maternal R, 2,805 (18.31%) of F1 genes were identified as significantly differentially expressed, and 3,618 (23.61%) genes were identified in F18 (P < 0.05 in comparisons; Fisher’s exact test) (Fig. 2). For genes pairs between the hybrid and paternal C, 4,051 (26.45%) differentially expressed genes were detected in F1, and 2,184 (14.19%) genes in F18 (P < 0.05 in comparisons; Fisher’s exact test) (Fig. 2). To better study the ELD, we binned gene pairs from the hybrids into 12 categories including mid-parents (XI and XII), up/down expression (I, II, III, IV, V, and VI), and ELD (VII, VIII, IX and X) (see Methods). Categories VII and X represented gene pairs showing upregulated ELD in the hybrids. For example, our results showed that maternal effect played prominent role in F1 (R vs. C = 1,277 vs. 517), and paternal effect predominated in F18 (R vs. C = 779 vs. 1,061) (Fig. 3). Conversely, categories VIII and IX represented the gene pairs showing downregulated ELD in the hybrids (Fig. 3).

Roman numerals indicate the same categorization as used in Rapp et al. (2009), with figures schematizing their respective gene expression pattern for the R-genome in diploid maternal parent (♀), R and C-genome in F1 or F18 (P) and C-genome in diploid paternal parent (♂).
Homoeologue expression bias in different ploidy levels
According to the report of Rappet et al. (2009), the expression categorisation would not only help in the study of ELD, but also provides an insight into the HEB in the hybrids. The unbalanced gene number (VII and X vs. IX and X) reflected a preference toward the paternal or maternal expression in the hybrids. For example, among the 15,316 expression pairs of F18, we determined that approximately 13.69% of all genes (categories VII and VIII) showed C-ELD, and 7.40% (categories IX and X) showed R-ELD, which indicated the phenomenon of C-HEB in F18. Likewise, we examined F1 for evidence of R-HEB, in which 2,120 genes (13.84% of all genes) (categories IX and X) fell into the R-ELD category (Fig. 3). Additionally, we examined the upregulated genes (IV, V, VI, X, and XII) and downregulated genes (I, II, III, IX, and XI) in the hybrids compared with the paternal C and compared the upregulated genes (IV, V, VI, VII, and XI) and downregulated genes (I, II, III, VIII, and XII) in the hybrids compared with the maternal R (Fig. 3). In these comparisons, the number of significantly differentially expressed gene (up vs. down = 352 vs. 391 in F18, up vs. down = 200 vs. 207 in F1) was similar (P < 0.05 in comparisons; Fisher’s exact test).
To address whether the observed category of HEB really reflects the HEB in F1 diploid hybrids and F18 allotetraploids, we compared 3,540 genes with homoeologue-specific SNPs on a case-by-case basis between the parental diploids and their diploid hybrid and polyploids. As shown in Table 3, the patterns observed in the diploid parents were often conserved in F1 and F18. For example, the first three rows in Table 3 show that the parental expression patterns were maintained for greater than half of all genes in this analysis: 74.8% (in F1) to 77.6% (in F18) (P < 0.05 in comparisons; Fisher’s exact test). Rows 4 and 5 represent the second most common class of genes, representing 13.9–15.4% of the 3,540 genes. In these cases, pre-existing expression bias in the parental homoeologue reverted to non-differential expression of the homoeologous copies in the diploid hybrids and allotetraploids (P < 0.05 in comparisons; Fisher’s exact test). A small numbers of genes were detected as having novel patterns that accompanied the genome merger or doubling. These cases suggested novel regulatory and/or evolutionary interactions in the hybrid offspring. We also collected genes with significant HEB in F1 and F18 (rows 11 and 12) (Table 3 and Fig. 4). In addition, to further detect the R-/C-biased in hybrids, we assessed the potential bias based on the ratio of R/C homoeologue expression levels (Table 3 rows 13 and 14). These genes helped us to understand the origin of some of the genetic traits in the hybrid offspring.

(A) The maternal HEB in total genes is estimated by the gene number of R homoeologue to C homoeologue in F1 and F18. (B) The maternal HEB in growth-regualted genes is estimated by the gene number of R homoeologue to C homoeologue in both of hybrid offsprings.
For the 15,316 gene expressed in F1, F18 and their original parents, we analysed the differential expression between the hybrids with in silico mid-parent expression values (MPV) that replaced the expression level of both of the parents. The three categories comparison showed that only 2.8% of the genes (430 out of 15,316 genes) changed their expression patterns in response to genome merger (Table 4). As a result of genome doubling, 1,893 (12.4%) genes changed their expression patterns. The results showed that genome doubling had more effect on global expression changes than the genome merger. Among the 3,541 homoeologue-specific SNPs-containing genes, 75.09% (2,659 genes) show no change in expression level compared with the R/C patents. However, among those that did change, the genome merger resulted in more genes with changed expression levels (13.9%) compared with genome doubling (7.4%) (P < 0.05 in comparisons; Fisher’s exact test, Table 4).
As to investigate of functional enrichment related to differential expression under the effect of hybridization and polyploidization, GO analysis was used to collect the possible functions of significantly differentially expressed among the two hybrid offspring and MPV. Among of pair comparisons, change expressed genes were enriched in main GO categories including cell part, binding, catalytic, biological regulation, cellular process, developmental process and metabolic process (Supplementary Fig. S1). The down-regulated genes in both of two hybrids were enriched in antioxidant, rhythmic process and viral reproduction (Supplementary Fig. S1).
The expression pattern of growth-regulated genes using RNA-seq
To investigate how hybridization and polyploidization affect the growth regulatory mechanism in different ploidy level individuals, we used RNA-seq and qRT-PCR to detect HEB in the allotetraploid line of C. auratus red var. ×C. carpio. The five growth-related genes were obtained from the analysis of novel expression and expression silencing pattern (Supplementary Table S3). Then, as to analyse the 180 growth-regulated genes, we used the 12 categories of expression patterns to obtain the information on the differential regulation between the hybrids and both parents (up: down = 6: 1 in F1, up: down = 2: 8 in F18) (P = 0.015 in comparisons; Fisher’s exact test) (Fig. 2). These results reflected a growth-regulated mRNA preference toward upregulation in F1 and downregulation in F18 compared with the parents. Additionally, we examined percent of growth-related genes in categories VII and VIII and percent in categories IX and X. As a result, R-HEB was observed in F1, and C-HEB in F18 (Fig. 3).
To further investigate the regulation of HEB related to growth function, all 34 growth-regulated genes were collected from the 3,540 genes under HEB analysis (Table 3). Some categories had no statistical significance because of the number of genes selected was a small percentage of the total. However, similar ratios were shown in the other categories. Ultimately, only four R/C-biased growth-regulated genes were identified in F1 and F18 (Fig. 4). Additionally, a similar situation was observed in the analysis of their expression patterns, in which the MPV was used as a reference point in comparisons with hybrids (Table 4). Among the 180 growth-regulated genes, 71.7% exhibited no expression change in both F1 and F18 (Table 4). Thus, global expression and homoeologue expression analysis of growth-regulated genes provided an insight into how changes in expression levels were induced by genome doubling or genome merger and the underlying regulation mechanism.
Determination of homoeologue expression bias in seven genes using qRT-RCR
To validate whether the patterns of HEB observed above reflected the growth regulation in F1 and F18, we detected the HEB of seven key growth-related genes (igf1, igf2, ghr, tab1, bmp4 and mstn) in three tissues (liver, muscle and ovary) using homoeologue-specific qRT-PCR. Interestingly, two scenarios were observed: (1) the silencing of the C homoeologous transcripts of the mstn gene was detected in the liver of F1 and F18 and the muscle of F18 (Fig. 5). (2) Different degrees of HEB were observed in the three tissues (Fig. 6). However, R-HEB was observed in most tissues in F1 and F18. Compared with the RNA-seq results, homoeologue expression was only verified for the igf2 genes using qRT-PCR. The results did show similar HEBs between the two methods (Fig. 6 and Supplementary Table S4). In addition, as to the detected by using the two methods, the homoeologous expression of bmp4 gene in RNA-seq were positively correlated with one’s in qRT-PCR (P = 0.087, R = 0.998, Supplementary Fig. S2), while igf2 showed little a positive correlation between the two methods (P = 0.134, R = 0.996, Supplementary Fig. S2).

The transcripts with a high expression level are indicated in yellow, and those with a low expression level are indicated in blue. Additionally, silenced transcripts are shown in green.

The CT ratio of R homoeologue to C homoeologue in various tissues of the two hybrids. (A) The CT ratio of R homoeologue to C homoeologue in igf1 gene. (B) The CT ratio of R homoeologue to C homoeologue in ghr gene. (C) The CT ratio of R homoeologue to C homoeologue in igf2 gene. (D) The CT ratio of R homoeologue to C homoeologue in tab1 gene. (E) The CT ratio of R homoeologue to C homoeologue in bmp4 gene. (F) The CT ratio of R homoeologue to C homoeologue in vasa gene. (G) The CT ratio of R homoeologue to C homoeologue in mstn gene. **indicates CT ratio >1, which suggests R homoeologue expression bias. *indicates C homoeologue expression bias. Comparative analysis reveals significant differences in gene expression (P < 0.05) (n = 3 for each group).
The R to C homoeologue expression level ratio suggested that HEB existed in the different hybrids. We used the ratio to classify the seven homoeologues in the three tissues (Fig. 6). For example, C-HEB of the igf1 gene was detected in the ovary and R-HEB was detected in liver and muscle (Fig. 6A). R-HEB of the ghr gene was observed in F18, but F1 showed C-HEB (Fig. 6B). Interestingly, silencing of C homoeologue expression of the mstn transcripts was observed in the liver of F1, and liver and muscle of F18, which represented overall R-HEB in the progeny. Overall, the phenomenon of R-HEB was obvious in F1 and F18 (Supplementary Fig. S3). The expression levels of the R and C homoeologues allowed us to determine how the genetic effect from either of the parents affected F1 and F18 (Supplementary Fig. S4).
Discussion
In this study, distinct genomes of C. carpio and C. auratus red var. were merged through hybridization in F1 diploid hybrid, while F18 allotetraploids represented the genome doubling of the diploid hybrids10,19,20. Here, we used two approaches (RNA-seq and qRT-PCR) to study the ELD and HEB for total genes and growth-related genes. Our results demonstrated that a decrease in unbalanced ELD and more HEB accompanied hybridization and polyploidization, respectively. The evolution of global expression and R/C homoeologue expression was accompanied by increased HEB and novel expression, as well as increasing levels of silencing of homoeologue expression. A similar analysis was performed on growth-related genes to investigate the relationship between the regulation of growth and homoeologue expression, which provided an insight into growth heterosis under the effect of genome merger and doubling, respectively.
As to the two genomes of the different genera were merged into one cell nucleus, the expression level status from either parent was destroyed. The new expression levels were described as the ELD, where the global expression level resembles that of one of the two parents. Our results demonstrated that the average change in expression level was 22.38% in F1 (vs. R = 18.31% and vs. C = 26.45%) (Fig. 2). After the two types of genome merged, most gene expression levels maintained a steady state. However, the maternal R dominated compared with the paternal C. This phenomenon is frequently observed in hybrid fish, including hybrid Megalobrama amblycephala × Culter alburnus11, hybrid Oncorhynchus mykiss21 and hybrid Salmo salar22. The new expression levels of F1 were close to MPV (Fig. 2). The similar expression levels provided an insight into the character of the hybrid related to heterozygosity, in which two different alleles from different species cooperate in the control of regulatory function.
The study of homoeologue expression level is also an important way to detect the effect of genome merger11,12. The co-regulated expression of R and C homoeologues would result in different functions in the hybrids. A previous report on mRNA and microRNA showed that mid-parent expression rarely occurs in genes related to growth and adaptability11,12. Thus, the diversified homoeologue expression benefits the combination of advantageous traits in hybrid individuals. Our result for F1 showed no bias of homoeologue expression in 13.9% genes (Table 3), while the majority of genes obtained either of the parental traits after the genome merger. In addition, 15.7% of homoeologue-specific SNPs genes were categorized as overall R/C-biased in F1 (Table 3), represent the heterozygosity in most of traits in the hybrid.
The F18 allotetraploid is considered as suitable material to study the ELD and HEB under polyploidization, while the genome doubling occurred in F1 diploid hybrids. Changes in the expression levels of 3502 (25.5%) genes were identified in the comparison between F18 and F1, which suggested that genome doubling alters the transcriptome more than genome merger. However, comparing the hybrid expression with both of the parents, we detected 18.9% genes as having significant differences in expression in F18 compared with 22.3% in F1 (Fig. 2). This suggested that the pattern of expression levels after the genome doubling had been rebuilt. However, the changes in F18 did not simply originate from accumulation of genome merger and genome doubling. To address the dimension of expression evolution, we compared MPV expression levels to those actually observed in F1 (9.6%) and F18 (15.1%). Our analysis showed that the change in global expression in F18 represented the combined effects of genome doubling and genome merger. Meanwhile, our result showed that the R-ELD in F1 transform to C-ELD in F18 (Fig. 3), in contrast to the results for HEB (Table 4). A similar study showed the same trends in polyploid cotton12. These results suggested the reasonable conclusion that genome merger plays the dominant role in the changes in HEB compared with global expression analysis, which was mostly affected by genome doubling. In terms of the scope of transcriptome alterations, we suspect that most changes in gene expression reflect the downstream consequences of the regulatory networks that subtly responded to the stress of the merger of doubling process.
Allopolyploid fish are distributed worldwide and result from artificial or natural selection. Upon crossing the interspecies barrier, the newly formed progeny always display heterosis, such as rapid growth. For the allotetraploid line of C. auratus red var. ×C. carpio, rapid growth was observed in hybrid offspring compared with both parents (Supplementary Fig. S5). However, there has been no study on the underlying mechanism related to growth heterosis. Recent studies have focused on ELD and HEB to analyse the regulation pattern and their underlying mechanisms11,12,13. These findings show that allelic interactions and gene redundancy result in heterosis in allopolyploids relative to non-coding RNA, DNA, methylation and transcriptome changes14,15. In contrast to global expression analysis in teleost hybrids8,16, the study of homoeologue expression is a promising method to determine the regulation of growth heterosis8.
In the RNA-seq analysis on 118 growth-related genes in the hybrids compared with the MPVs (in silico), the study of global expression suggest that 10.0% of growth-related genes in F1 were upregulated, which was higher than that in the F18 (3.0% in total genes) (Fig. 2). Moreover, the expressions of growth-related gene were downregulated in 10% in F1, which was lower than that in F18 (18.3% of total genes) (Fig. 2). In addition, the differential expression analysis between F1 and F18 not only suggested that the effects of genome doubling and genome merger cooperate to form a new pattern of growth regulation in the hybrid populations, but also showed that genome doubling resulted in a reduction in growth-regulated gene expression. Previous studies on homoeologous genes support this non-additive expression after genome doubling in allopolyploid wheat23 and fish, including carp11, salmon18 and cichlid24. The differentially expressed genes between F1 and F18 were placed in 12 categories of expression patterns: upregulated (IV, V and VI) and downregulated (I, II, III) growth genes contributed to the lower expression level of homoeologous transcripts in allotetraploids (Fig. 3). This result might provide an insight into the rapid growth in F1 compared with F18 (Supplementary Fig. S5).
Maternal-specific expression is observed not only in hybrid plants, but also in lower vertebrates25,26. In the analysis of the categories of growth-related homoeologous genes, the analysis of HEB provided an insight into effect of originating from either of maternal R or paternal C, respectively. The analysis of overall bias identified four genes (pdgfaa, igfbp2a, igfbp1a-a and igfbp1a-b) from the 34 homoeologue-specific growth-related genes. The result of R bias analysis in F1 (R vs. C = 4.0 vs. 0) and F18 (R vs. C = 3.0 vs. 1.0) suggested that homoeologue expression of maternal R plays a major role in the liver transcriptome (Fig. 4). Compared with maternal R, the rapid growth characteristics were detected in paternal C. Meanwhile, the joint expression of R/C homoeologues of igf1 and ghr increases the expression diversity and play an important role in promoting the growth ratio in the hybrids8. However, our results for igf1, igf2 and ghr suggested that C-HEB might contribute to rapid growth. Meanwhile, other key growth-related genes (tab1, bmp4, mstn and vasa) were used to detect R-/C- HEB (Fig. 6), in which regulation of growth was accompanied by different levels of R/C-homoeologue bias. In the R/C bias analysis, although few significant differential homoeologue expression genes were detected in our study, the consequence of potential R-biased was still identified in the analysis of 34 homoeologue-specific growth-related genes (Table 3). The biases of homoeologue-specific genes observed here suggested a role for epigenetic modulation in growth. This phenomenon suggested that the changes in homoeologue expression might contribute to enhance growth and accelerated body development.
Interestingly, silencing of C homoeologue was observed for the growth-related gene mstn (Fig. 5). One explanation for this observation could be genomic imprinting, implying that gene expression control would be mediated by one parental genome, whereas the genetic material inherited from the other parents is silenced in the hybrid27. Some genes always exhibit single-genome-mediated expression in hybrids28. In addition, the silencing of homoeologue has been considered as the transition period of the loss of homoeologue just after the genome merge and duplication. The comparative genome analysis on clupeocephalan teleosts reveals that whole-genome duplication accelerated the formation of new species accompanied with the loss of 1,100 homoeologues29. A recent study demonstrated that mutations in the mstn gene resulted in increased muscle mass and strength in vertebrates, making these individuals considerably stronger than their peers30. The observation that larger individuals are always seen in hybrid fish populations supports these findings31,32. However, further study is necessary to verify the homoeologue silencing and its relationship with epigenetic traits associated with genome merger and genome doubling.
Materials and Methods
Animals
All experiments, performed from 2012–2014, were approved by the Animal Care Committee of Hunan Normal University. The Administration of Affairs Concerning Animal Experimentation guidelines stated approval from the Science and Technology Bureau of China. The methods were carried out in accordance with the approved guidelines. Experimental individuals were fed in a pool with suitable illumination, water temperature, dissolved oxygen content, and adequate forage in the Engineering Center of Polyploidy Fish Breeding of the National Education Ministry located at Hunan Normal University, China. Approval from the Department of Wildlife Administration is not required for the experiments conducted in this paper. Fish were deeply anesthetized with 100 mg/L MS-222 (Sigma-Aldrich) before dissection.
Four female individuals including a diploid C. auratus red var. (R), a diploid C. carpio (C), a interspecific diploid F1 hybrid of C. auratus red var. (♀) × C. carpio (♂)10, and a allotetraploid of C. auratus red var. (♀) × C. carpio (♂)10 (2-year-old individuals) were collected for our study. Body traits (body length, height and weight) were recorded once every month (Supplementary Fig. S5). To measure the DNA content of the erythrocytes from the above samples, 1–2 ml of blood was drawn from the caudal vein using syringes containing 200–400 units of sodium heparin. The blood samples were subjected to nuclei extraction and 40, 6-diamidino-2-phenylindole DNA-staining with cysteine DNA 1 step (Partec). The DNA contents of the erythrocytes were then detected by flow cytometry in each sample. In addition, to detect the ploidy levels of each sample, the red blood cells were cultured in nutrient solution at 25.5 °C and 5% CO2 for 68–72 h, and then colchicine was added 3.5 h before harvest. Cells were harvested by centrifugation, followed by hypotonic treatment with 0.075M KCl at 26 °C for 25–30 min, fixed in methanol–acetic acid (3:1, v/v) with three changes. Cells were dropped onto cold slides, air-dried and stained for 30 min in 4% Giemsa solution. Good-quality pictures of the metaphase spreads from 12 individuals were observed under a microscope (Fig. 1)33.
Illumina sequencing
After anesthetizing the fish with 2-phenoxyethanol, liver, muscle and ovary tissues were excised and immediately placed into RNALater (AM7021, Ambion Life Technologies, Carlsbad, CA, USA) following the manufacturer’s instructions, for storage. Total RNA was extracted from the three tissues after the RNALater was removed. RNA was isolated according to the standard Trizol protocol (Invitrogen) and quantified with an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA).
After the isolation of 2 μg mRNA using the beads with oligo (dT) Poly (A), fragmentation buffer was added for interrupting mRNA to short fragments. After taking these short fragments as templates, cDNA was then synthesized using buffer, dNTPs, RNaseH, and DNA polymerase I. Short fragments were purified with the QiaQuick PCR extraction kit (Qiagen) and resolved with elution buffer. These fragments were performed with agarose gel electrophoresis after adding sequencing adapters. PCR amplification templates of the suitable fragment were selected as PCR amplification templates. The stage of quality control was performed with the Agilent 2100 Bioanaylzer and ABI StepOnePlusTM Real-Time PCR System. Finally, cDNA libraries were sequenced using Illumina HiSeq 2000.
Mapping and differential expression
The read adaptors and low quality reads were removed from the raw reads and the clean reads from each library were examined using software FastQC (version 0.11.3). Principal component analysis (PCA) of the twelve liver transcriptomes was applied to examine the contribution of each transcript to the separation of the classes34,35. Then, fastq formatted reads from the two diploid parents and two hybrid offspring were mapped to the reference genome using TopHat236,37. We utilized the gynogenetic C. auratus red var. genome assembly ( http://rd.biocloud.org.cn/) (39,069 transcripts) and the C. carpio genome assembly ( http://www.carpbase.org/) (52,610 transcripts) as the reference genomes because these transcripts databases were built from genome sequencing (Supplementary table S1). To identify putative orthologues between R and C, the two sets of sequences were aligned using the reciprocal BLAST (BLASTN) hit method, with an e-value cut off of 1e−20 38. Two sequences were defined as orthologues if each of them was the best hit of the other and if the sequences were aligned over 300 bp. After identifying SNPs between the R and C orthologues, we mapped our reads from R and C to compare the mapping results. Reads with SNPs that differed between the R- and C-genome in the progenitors were parsed into R and C homoeologue-specific bins using custom perl scripts.
To calculate expression levels, the replicates were normalized using Cufflink (version 2.1.0)36 and then, using the overall expression levels of both homoeologues of a gene, differential expression was assessed between the different ploidy levels relative to their diploid parents, using Fisher’s exact tests39. The mapping results were analysed with the DEGseq package in the R software version 2.13 (R Foundation for Statistical Computing, Vienna, Austria)39. To remove the negative effect of expression noise, we restricted the analysis to genes have read counts (≥1) in all biological replicates. The abundance or the coverage of each transcript was determined by read counts and normalized using the number of reads per kilobase exon per million mapped reads (RPKM)40. The RPKM value of the reads was calculated to obtain the gene expression level. The false discovery rate (FDR) was used to determine the threshold P value in multiple tests and analyses. Meanwhile, the unigenes with FDR ≤ 0.05 and fold change >2 were considered as differentially expression genes. In addition, Gene Ontology was performed to illustrate the functional annotation of the differential expression genes among samples. GO enrichment analysis was carried out with WEGO41.
Analyses of expression level dominance and homoeologue expression bias
We identified candidate novel expressions (new expression of a gene in liver) and silencing in the hybrids according to the standards of Yoo et al.12. The number of novel expression and silenced genes was screened in the categories of global expression and growth-related genes (Table 2 and Table S3). We then focused on genes that were expressed in both the diploid parents and in the hybrid offspring to analyse the ELD.
In the hybrid offspring, genes that were identified as differentially expressed in the hybrid relative to the diploid parents were binned into 12 possible differential expression categories (Fig. 3), ELD, mid-parents, and up/down expression (outside the range of either parent), according to Rappet et al. (2009). Briefly, genes were parsed into these 12 categories (using Roman numerals; see Fig. 3), depending on the relative expression levels between the hybrid and the diploid parents. In this manner, genes may display mid-parent (XI and XII), paternal C-ELD (VII and VIII), maternal R-ELD (IX and X), expression lower than both parents (I, II, and III), or expression higher than both parents (IV, V, and VI).
To describe the extent and direction of HEB in response to hybridization and evolution at different ploidy levels, we analysed the differential expression across the F1 diploid hybrid, F18 allotetraploid, and the in silico MPVs. Values from the three biological replicates of each parent were averaged to calculate the MPV and then analysed in the same manner as described above.
Expression of growth-related genes in RNA-seq and qRT-PCR
Among the 3,540 genes used in the study of HEB in hybrids, thirty-four growth-regulated genes were selected and analysed to help us understanding the effect from either parent based on the RNA-seq data (Supplementary Table S4).
To further validate the HEB related to growth regulation in F1 and F18, we selected seven key growth-regulated genes and subjected than to homoeologue-specific qRT-PCR18. Total RNA was extracted from the three tissues and first-strand cDNA was synthesized using AMV reverse transcriptase (Fermentas, Canada) with an oligo (dT)12–18 primer at 42 °C for 60 min and 70 °C for 5 min. The conserved region of the teleost orthologues’ vasa genes was used as a template to design universal primers (Supplementary Table S5). The PCR products were cloned using appropriate primers and sequences in six parental samples and six hybrid samples. The sequences of other genes (igf1, igf2, ghr, tab1, bmp4, and mstn) were obtained from the assembly of liver transcriptome data.
Comparison of the sequences was done using Bioedit ver. 7.0.9, and an analysis of cDNA polymorphisms in the transcripts revealed R and C homoeologue expressed in hybrid. SNPs between the R and C homoeologues were obtained from one gonad-specific gene (vasa), a housekeeping gene (β-actin), and ubiquitously expressed gene (igf1, igf2, ghr, tab1, bmp4, and mstn). The SNP regions were used to design R/C homoeologue-primers for qRT-PCR (Supplementary Fig. S6 and Supplementary Table S6). The R and C homoeologue-specific primers were obtained to permit the detection of only R or C homoeologues by qRT-PCR using the ABI Prism 7500 Sequence Detection System (Applied Biosystems, USA) (Supplementary Table S7). Amplification conditions were as follows: 50 °C for 5 min, 95 °C for 10 min, and 40 cycles at 95 °C for 15 s and 60 °C for 45 s. Each test was performed three times to improve the accuracy of the results. Finally, relative quantification was performed and melting curve analysis was used to verify the generation of a single product at the end of the assay. Triplicates of each sample were used both for standard curve generation and during experimental assays. After obtaining the R and C homoeologue expression levels of the seven genes, the relative expression of each homoeologous gene was calibrated with β-actin, and the relative mRNA expression data were determined using the 2−ΔΔCt method42. The expression level of the reference gene β-actin in the hybrids was estimated using the ratio of the transcript abundance to the gene copy using PCR and qRT-PCR of co-extracted DNA and RNA from the ovaries of diploid and allotetraploid individuals. β-actin expression is the same between fish of different ploidy and genome constitution, and in somatic organs and gonads16,43,44,45. In addition, we performed the multiple linear regression analysis on mstn and igf2 gene between the method of RNA-seq and qRT-PCR (Supplementary Fig. S2).
Additional Information
How to cite this article: Ren, L. et al. Homoeologue expression insights into the basis of growth heterosis at the intersection of ploidy and hybridity in Cyprinidae. Sci. Rep. 6, 27040; doi: 10.1038/srep27040 (2016).
References
Dawley, R. M. Hybridization and polyploidy in a community of three sunfish species (Pisces: Centrarchidae). Copeia 1987, 326–335 (1987).
Schultz, R. J. Hybridization, unisexuality, and polyploidy in the teleost Poeciliopsis (Poeciliidae) and other vertebrates. Am Nat 103, 605–619 (1969).
Salmon, A., Ainouche, M. L. & Wendel, J. F. Genetic and epigenetic consequences of recent hybridization and polyploidy in Spartina (Poaceae). Mol Ecol 14, 1163–1175 (2005).
Dufresne, F. & Hebert, P. D. Hybridization and origins of polyploidy. P Roy Soc B-Biol Sci 258, 141–146 (1994).
Comai, L. The advantages and disadvantages of being polyploid. Nat Rev Genet 6, 836–846 (2005).
Wolf, J. B. et al. Nucleotide divergence vs. gene expression differentiation: comparative transcriptome sequencing in natural isolates from the carrion crow and its hybrid zone with the hooded crow. Mol Ecol 19 Suppl 1, 162–175 (2010).
Zhou, Y. et al. Elevated expression of Piwi and piRNAs in ovaries of triploid crucian carp. Mol Cell Endocrinol 383, 1–9, 9 (2014).
Zhong, H. et al. Elevated expressions of GH/IGF axis genes in triploid crucian carp. Gen Comp Endocr 178, 291–300 (2012).
Liu, S. Distant hybridization leads to different ploidy fishes. Sci China Life Sci 53, 416–425 (2010).
Liu, S. et al. The formation of tetraploid stocks of red crucian carp × common carp hybrids as an effect of interspecific hybridization. Aquaculture 192, 171–186 (2001).
Zhou, Y. et al. Global transcriptional and miRNA insights into bases of heterosis in hybridization of Cyprinidae. Scientific reports 5, 13847 (2015).
Yoo, M. J., Szadkowski, E. & Wendel, J. F. Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity 110, 171–180 (2013).
Rapp, R. A., Udall, J. A. & Wendel, J. F. Genomic expression dominance in allopolyploids. BMC Biol 7, 18 (2009).
Michalak, P. Epigenetic, transposon and small RNA determinants of hybrid dysfunctions. Heredity 102, 45–50 (2009).
Ng, D. W., Lu, J. & Chen, Z. J. Big roles for small RNAs in polyploidy, hybrid vigor, and hybrid incompatibility. Curr Opin Plant Biol 15, 154–161 (2012).
Liu, Z. et al. Characterization and dietary regulation of glutamate dehydrogenase in different ploidy fishes. Amino acids 43, 2339–2348 (2012).
Udall, J. A., Swanson, J. M., Nettleton, D., Percifield, R. J. & Wendel, J. F. A novel approach for characterizing expression levels of genes duplicated by polyploidy. Genetics 173, 1823–1827 (2006).
Pala, I., Coelho, M. M. & Schartl, M. Dosage Compensation by Gene-Copy Silencing in a Triploid Hybrid Fish. Curr Biol 18, 1344–1348 (2008).
Liu, S. et al. Establishment of the diploid gynogenetic hybrid clonal line of red crucian carp × common carp. Sci China Life Sci 50, 186–193 (2007).
Yan, J. et al. RAPD and microsatellite analysis of diploid gynogens from allotetraploid hybrids of red crucian carp (Carassius Carassius auratus) ×common carp (Cyprinus carpio). Aquaculture 243, 49–60 (2005).
White, S. L., Sakhrani, D., Danzmann, R. G. & Devlin, R. H. Influence of developmental stage and genotype on liver mRNA levels among wild, domesticated, and hybrid rainbow trout (Oncorhynchus mykiss). BMC Genomics 14, 673 (2013).
Debes, P. V., Normandeau, E., Fraser, D. J., Bernatchez, L. & Hutchings, J. A. Differences in transcription levels among wild, domesticated, and hybrid Atlantic salmon (Salmo salar) from two environments. Mol Ecol 21, 2574–2587 (2012).
Pumphrey, M., Bai, J., Laudencia-Chingcuanco, D., Anderson, O. & Gill, B. S. Nonadditive expression of homoeologous genes is established upon polyploidization in hexaploid wheat. Genetics 181, 1147–1157 (2009).
Albertson, R. C. & Kocher, T. D. Genetic architecture sets limits on transgressive segregation in hybrid cichlid fishes. Evolution 59, 686–690 (2005).
Michalak, P. Evidence for maternal imprinting of 45S ribosomal RNA genes in Xenopus hybrids. Dev Genes Evol 224, 125–128 (2014).
McKeown, P. C. et al. Identification of imprinted genes subject to parent-of-origin specific expression in Arabidopsis thaliana seeds. BMC Plant Biol 11, 113 (2011).
Martin, C. C. & McGowan, R. Parent‐of‐origin specific effects on the methylation of a transgene in the zebrafish, Danio rerio . Dev Genet 17, 233–239 (1995).
McGowan, R. & Martin, C. DNA methylation and genome imprinting in the zebrafish, Danio rerio: some evolutionary ramifications. Biochem Cell Biol 75, 499–506 (1997).
Inoue, J., Sato, Y., Sinclair, R., Tsukamoto, K. & Nishida, M. Rapid genome reshaping by multiple-gene loss after whole-genome duplication in teleost fish suggested by mathematical modeling. PANS 112, 14918–14923 (2015).
Schuelke, M. et al. Myostatin mutation associated with gross muscle hypertrophy in a child. New Engl J Med 350, 2682–2688 (2004).
Yu, F. et al. Rapid growth and sterility of growth hormone gene transgenic triploid carp. Chinese Sci Bull 56, 1679–1684 (2011).
Shen, J. M. et al. A new type of triploid crucian crap-red crucian carp (female) x allotetraploid (male). Prog Nat Sci 16, 1348–1352 (2006).
Xiao, J. et al. The fertility of the hybrid lineage derived from female Megalobrama amblycephala × male Culter alburnus . Anim Reprod Sci 151, 61–70 (2014).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol 11, R106 (2010).
Reeb, P. D. & Steibel, J. P. Evaluating statistical analysis models for RNA sequencing experiments. Frontiers in genetics 4, 178 (2013).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36 (2013).
Blanc, G. & Wolfe, K. H. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. The Plant cell 16, 1667–1678 (2004).
Wang, L., Feng, Z., Wang, X., Wang, X. & Zhang, X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138 (2010).
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5, 621–628 (2008).
Ye, J. et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34, W293–297 (2006).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (San Diego, Calif.) 25, 402–408 (2001).
Long, Y. et al. Differential expression of Gnrh2, Gthβ, and Gthr genes in sterile triploids and fertile tetraploids. Cell Tissue Res 338, 151–159 (2009).
Tao, M. et al. The cloning of Dmc1 cDNAs and a comparative study of its expression in different ploidy cyprinid fishes. Sci China Life Sci 51, 38–46 (2008).
Liu, D. et al. Identification and Expression Analysis of Genes Involved in Early Ovary Development in Diploid Gynogenetic Hybrids of Red Crucian Carp × Common Carp. Mar Biotechnol 12, 186–194 (2010).
Acknowledgements
This research was supported by National Natural Science Foundation of China Grants 30930071, 91331105, 31360514, 31430088, and 31210103918, the Cooperative Innovation Center of Engineering and New Products for Developmental Biology of Hunan Province (20134486), the Construction Project of Key Discipline of Hunan Province and China, the National High Technology Research and Development Program of China (Grant No. 2011AA100403).
Author information
Authors and Affiliations
Contributions
L.R. and G.L. carried out bioinformatics analyses and wrote the manuscript. L.R. and W.L. contributed to the qRT-PCR experiment. J.C., Q.Q., W.L., X.T. and J.X. provided assistance extracting the raw material and collected the photographs. J.X. provided the photo of fish. J.L., C.T., M.T., Y.Z., W.D., Y.H. and C.Z. modified the manuscript. S.L. contributed to the conception and design of the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ren, L., Li, W., Tao, M. et al. Homoeologue expression insights into the basis of growth heterosis at the intersection of ploidy and hybridity in Cyprinidae. Sci Rep 6, 27040 (2016). https://doi.org/10.1038/srep27040
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep27040
This article is cited by
-
Homoeolog gene expression analysis reveals novel expression biases in upland hybrid cotton under intraspecific hybridization
Functional & Integrative Genomics (2022)
-
Diverse transcriptional patterns of homoeologous recombinant transcripts in triploid fish (Cyprinidae)
Science China Life Sciences (2021)
-
Optimal reference genes for gene expression analysis in polyploid of Cyprinus carpio and Carassius auratus
BMC Genetics (2020)
-
Establishment and application of distant hybridization technology in fish
Science China Life Sciences (2019)
-
Targeted disruption of tyrosinase causes melanin reduction in Carassius auratus cuvieri and its hybrid progeny
Science China Life Sciences (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
