
Abstract
Under 971 nm excitation, bright green and red emissions from Yb3+/Er3+ co-doped Ba5Gd8Zn4O21 phosphor can be observed, especially the intense red emission in highly doped samples. The experimental results indicate that Ba5Gd8Zn4O21:Yb3+, Er3+ emits stronger upconversion luminescence than NaYF4:Yb3+, Er3+ under a low excitation power and a maximum upconversion power efficiency of 2.7% for Ba5Gd8Zn4O21:Yb3+, Er3+ was achieved. More significantly, to explain the red emission enhanced with the dopant concentration, this paper presents a possible cross-relaxation process and demonstrates it based on the rate equation description and temporal evolution. In view of the strong upconversion luminescence, colour tunable ability and stable chemical nature, Yb3+/Er3+ co-doped Ba5Gd8Zn4O21 phosphor could be an excellent candidate for efficient upconversion luminescence generation.
Similar content being viewed by others
Introduction
Upconversion (UC) is a nonlinear optical process characterized by the successive absorption of multiple pump photons via metastable states, followed by the emission of the luminescence at a shorter wavelength than the excitation wavelength1. In view of the unique optical properties, UC luminescence (UL) materials doped with rare earth (RE) ions have been utilized in potential applications, such as biological diagnosis, data storage, solar cells, solid display technique, solid-state lasers and sensor technology2,3,4,5,6,7,8,9,10,11,12. However, one outstanding roadblock still exists: most of the UL materials fail to produce strong optical emission under low excitation power, which greatly limits the practical application in different fields. For example, owing to the weak emission signal, which limits the penetration depth to a few centimeters in bio-labeling and bio-imaging, UC luminescence bio-imaging could be used to obtain anatomical and physiological details only in small animals. To increase the penetration depth of UL bio-imaging in vivo, a high excitation power density is required. Paradoxically, further increasing the laser power may cause marked overheating effects, which can lead to possible scalding of animal tissue upon continuous irradiation8,9. Moreover, infrared solar photons with wavelength greater than 1 μm are unabsorbed by solar cells working in the visible and near-infrared regions, thus UL materials provide a solving method by turning infrared solar photons into visible photons, leading to increased photoelectric conversion efficiency of the solar cells in theory11,12. However, sun-light is usually not sufficiently strong to activate the UC process. To solve this type of practical problem, research on highly efficient UL materials induced by low excitation power is an urgent task.
The Yb3+-Er3+ codoped UC system has been most commonly used to obtain efficient UL materials, mainly owing to the large absorption cross section of approximately 980 nm in Yb3+, the relatively long lifetime energy levels in Er3+ and the good energy level match between these two types of RE ions13,14,15,16,17,18,19. Herein, we conducted a series of research on Yb3+, Er3+ codoped materials and found that one type of UL material, Ba5Gd8Zn4O21:Yb3+, Er3+, could realize bright UC emissions even stronger than β-NaYF4:Yb3+, Er3+ when the excitation power is not high and its UC optical properties are detailed in this paper.
More interestingly, by increasing the doping concentrations of Ba5Gd8Zn4O21:Yb3+, Er3+ samples, the red emission intensity originating from 4F9/2 in Er3+ increased dramatically and the red to green emission power ratio went up from lower than 1 to approximately 10. We have noticed that this dynamic change regularity exists not only in Ba5Gd8Zn4O21:Yb3+, Er3+, but also in many other Yb3+, Er3+ co-doped UL materials such as β-NaYF4, NaGdF4, Y2O3, BaIn6Y2O13, BaGd2ZnO5 and Y2Sn2O713,14,15,16,17,18. Considering this fact, it is most likely that in the Yb3+-Er3+ co-doped UC system the red emission have more efficient UC routes, which can contribute when the doping concentration is sufficiently high; however, this conjecture has not yet been confirmed. In this paper, we successfully proved the existence of CR by establishing the rate equations of the energy levels. In addition, the temporal evolutions of UC emissions from a series of Ba5Gd8Zn4O21:Yb3+, Er3+ samples were recorded and the experimental results can also explained by the CR process.
Results and Discussion
Crystal Structure
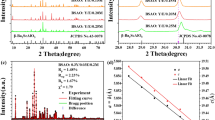
Figure 1 shows the XRD pattern of the as-synthesized Ba5Gd8Zn4O21:Yb3+, Er3+. It can be observed that the diffraction peaks of the sample coincide well with the standard data on Ba5Gd8Zn4O21 (JCPDS NO. 51–1686) and no extra peaks from any impurities are seen, which indicates that the dopants Yb3+ and Er3+ completely enter the lattice of the host. The crystalline structure is tetragonal with space group I4/m.

The X-ray powder diffraction.
The XRD pattern of the as-synthesized Ba5Gd8Zn4O21:Yb3+, Er3+.
Spectroscopic Characterizations and UC Power Efficiency
The UC optical properties depend on dopant concentrations. Under 971 nm CW laser excitation at r oom temperature, the selected Ba5Gd8Zn4O21:x% Yb3+, 1% Er3+ (x = 1, 3, 6, 9, 12, 15, 20) samples yield green to red emission. In Fig. 2, the spectra exhibit three broad emission bands centered at 548 nm, 557 nm and 672 nm, which are ascribed to the 2H11/2→4I15/2, 4S3/2→4I15/2 and 4F9/2→4I15/2 energy transitions in Er3+, respectively. The inset of Fig. 2 shows the red to green UL intensity ratio for the seven selected samples. By increasing the Yb3+ concentration and fixing the Er3+ concentration to 1%, the red to green emission intensity ratio increases strongly and reaches 0.47, 0.51, 0.72, 1.40, 3.22, 6.54, 7.07 and 9.61 for the samples doped with 1%, 3%, 6%, 9%, 12%, 15% and 20% Yb3+ respectively and there exists a similar rule in samples with a fixed Yb3+ concentration: the red emission intensity grows much faster and higher than that of the green emission by increasing Er3+ concentration, as shown in Figure S1. The dependence of UC optical properties on the doping concentration indicates that the red emission level has a more efficient means to be filled than the green emission levels especially in doped samples with Yb3+ or Er3+ concentration.

PL upconversion spectra of the sample.
UC spectra of seven selected Ba5Gd8Zn4O21:x% Yb3+, 1% Er3+ (x = 1, 3, 6, 9, 12, 15, 20) samples under the 971 nm CW laser excitation (18.6 W/cm2), the inset presents the red to green emission intensity ratio of the samples.
The UC power efficiencies of Ba5Gd8Zn4O21:Yb3+, Er3+ samples, including one commercial NaYF4:Yb3+, Er3+ phosphor as a contrast, were measured at room temperature under 971 nm excitation. As shown in Fig. 3, the UC power efficiency exhibited a similar changing trend: increasing under low excitation power up to ~0.7 W and then decreasing at higher excitation power. A maximum UC power efficiency of 2.7% was obtained with Ba5Gd8Zn4O21:12% Yb3+, 4% Er3+ and Ba5Gd8Zn4O21:15% Yb3+, 5% Er3+ under 0.67 W excitation. It is worth noting that the Ba5Gd8Zn4O21:Yb3+, Er3+ sample has a higher UC power efficiency than the commercial β-NaYF4:Yb3+, Er3+ phosphor when excitation power is low, despite the fact that the β-NaYF4 host lattice doped with RE ions is regarded as a high efficiency phosphor and is currently the most widely used. For the emission power of each Ba5Gd8Zn4O21:Yb3+, Er3+ sample please refer to Table S1.

UC power efficiency test.
UC power efficiency of several Ba5Gd8Zn4O21:Yb3+, Er3+ phosphors under different excitation power, the UC power efficiency of commercial NaYF4:Yb3+, Er3+ phosphor is also provided as a contrast.
Many reports have regarded high UC efficiency as a measure of UC materials with strong luminescence16,20,21. Generally, UC efficiency takes three forms: actual UC power efficiency (UC emission power/excitation power), UC power efficiency (UC emission power/absorbed excitation power) and UC quantum efficiency (UC emission quantum numbers/absorbed excitation quantum numbers). In our previous research, we found that the absorptivity to laser radiation varies greatly among different types of phosphors. For example, the phosphor CaIn2O4:Yb3+, Ho3+ has a higher UC power efficiency (approximately 5%) than Ba5Gd8Zn4O21:Yb3+, Er3+ 20, whereas the emission power of CaIn2O4: Yb3+, Ho3+ is much lower than that of Ba5Gd8Zn4O21:Yb3+, Er3+ under the equal power excitation, as shown in Fig. 4 (a,b). The CaIn2O4: Yb3+, Ho3+ is more efficient because of its much lower absorptivity to 971 nm laser radiation than Ba5Gd8Zn4O21:Yb3+, Er3+; the absorptivity of Ba5Gd8Zn4O21:Yb3+, Er3+ is approximately 30% and that for CaIn2O4:Yb3+, Ho3+ is only approximately 5%. Obviously, CaIn2O4:Yb3+, Ho3+ does not have high emission power and judging only from the UC efficiency will mislead us in finding the appropriate UL materials. Actually, how much excitation power could be converted into UC emission power is our focus, which has nothing to do with the absorptivity of the material. The core intent here is to find phosphors with high emission power in low excitation power regions. Thus, we deem it necessary to also note the actual UC power efficiency for UC material studies, which considers only the relationship between the emission power and the pumping power. The results in Fig. 4 (c) indicate that the highest actual UC power efficiency for Ba5Gd8Zn4O21:Yb3+, Er3+ is 0.87% when the excitation power is approximately 0.67 W and it can produce stronger UL than the other two materials in Fig. 4. More details of the actual UC power efficiency for Ba5Gd8Zn4O21:Yb3+, Er3+ can be seen in Figure S2.

Comparison among three types of UC materials.
The comparison of (a) emission power, (b) UC power efficiency and (c) actual UC power efficiency among three types of UL materials: Ba5Gd8Zn4O21:15% Yb3+, 5% Er3+ (red line), CaIn2O4:10% Yb3+, 0.5% Ho3+ (green line) and β-NaYF4:18% Yb3+, 2% Er3+ (blue line). The excitation source is a 971 nm power-controllable diode laser.
The x and y color coordinates associated with the emission of the Ba5Gd8Zn4O21:Yb3+, Er3+ phosphor family are positioned on a CIE x, y chromaticity diagram in Fig. 5; the excitation power is 0.65 W. It can be seen that when the Yb3+ or Er3+ concentration reaches a higher value, the color of the UC emission turns from yellowish green to reddish orange, which indicates that the Ba5Gd8Zn4O21:Yb3+, Er3+ phosphor has excellent color-tunable properties.

The CIE chromaticity diagram.
CIE chromaticity diagram and x, y color coordinates (cross mark) of several Ba5Gd8Zn4O21:Yb3+, Er3+ phosphors, the percentages of each group represent the concentration of Yb3+, Er3+, respectively.
UC Mechanisms
The pump power dependence of the green and red emissions was investigated at room temperature under 971 nm excitation. Notably, under high excitation power, the slope l decreased to less than 1 as a result of the competition between linear decay and UC processes for the depletion of the intermediate excited states according to M. Pollnau’s report and our previous work21,22. Please refer to the Supporting Documents for more details.
Ground state absorption (GSA), subsequent UC by excited state absorption (ESA) and energy transfer (ET) UC were the main UC mechanisms in the population for excited states, additional processes such as CR between two doped ions or an avalanche process may occur as well23. The GSA/ESA process involves a single ion, so it is the main possible UC process that occurs in materials with low dopant concentrations, whereas ET involves two neighboring ions and will be dominant in the materials with high doping concentrations by shortening the average distance between dopant ions and enhancing the interionic interaction.
Figure 6 presents the energy level diagrams of Yb3+ and Er3+ ions24,25 and the proposed UC routes in Ba5Gd8Zn4O21:Yb3+, Er3+ are also listed. Because Yb3+ ions have a larger absorption cross-section at the NIR region and a high doping concentration, the ET process from Yb3+ to Er3+ makes prominent contributions to the population of the emission energy levels. The UC route for the green emission is simple and we do not discuss it here. For the red emission originating in the energy level 4F9/2, most of the relevant literature asserted that are two different routes. One route is non-radiative relaxation from the upper level 4S3/2 to 4F9/2 and the second route is to populate the long-lived 4I13/2 level via non-radiative relaxation of 4I11/2 → 4I13/226,27, followed by an ET process: 2F5/2 (Yb3+) + 4I13/2 (Er3+) → 2F7/2 (Yb3+) + 4F9/2 (Er3+) or an ESA process: photon + 4I13/2 (Er3+) → 4I15/2(Er3+) + 4F9/2 (Er3+). In fact, the energy gap between 4I13/2 and 4F9/2 is approximately 8700 cm−1 and the energy of one 971 nm photon is approximately 10300 cm−1, which is much higher than the energy gap; therefore, the ET and ESA processes from the 4I13/2 level will be restricted owing to the absence of the energy match condition. In addition, the positions of the energy level for Yb3+ and Er3+ ions in different hosts remain almost the same because of the shielding effect of their outer closed 5 s2 and 5 p6 shields.

The energy level diagram.
Energy level diagram of Yb3+ and Er3+ and the proposed UC mechanism in Ba5Gd8Zn4O21:Yb3+, Er3+.
As noted above, CR occurs between two ions and requires a good coincidence between the energy gaps involved in this process. From the energy level diagram of Er3+, we found that the energy gap between 4F7/2 and 4F9/2 (approximately 5200 cm−1) matches well with the energy gap between 4F9/2 and 4I11/2 (approximately 5100 cm−1)24. Moreover, both 4F7/2 and 4I11/2 could be directly filled by the ET process, which guarantees major populations in the two energy levels. In fact, Liao reported that this cross-relaxation process may result in an enhanced population of 4F9/217. Unfortunately, they have not provided a more detailed explanation.
As shown in Fig. 6, the CR process between Er3+ ions is expressed as follows: 4F7/2 (Er3+) + 4I11/2 (Er3+) → 4F9/2 (Er3+) + 4F9/2 (Er3+); the doubling of the ions in 4F9/2 offers a cogent reason for the strong red emission. As seen in the previous section, when Yb3+ concentration is lower, the absorbed excitation power is too low to maintain plentiful populations in 4I11/2 and 4F7/2 and the ions in 4F7/2 will relax to the green emission levels preferentially. The CR process only negligibly populates the red emission level. Thus, the green emission is stronger than the red emissions owing to the low Yb3+ concentration. In contrast, because the Yb3+ concentration increases to a higher value, the ET process from Yb3+ to Er3+ becomes more active to populate the excited levels, which guarantees a greater contribution made by the three red level population routes, especially doubling the number of ions in 4F7/2 caused by CR processes. In view of this fact, the red emission increased dramatically with the Yb3+ concentration. For the samples with fixed Yb3+ concentration, high Er3+ concentration will shorten the average distance between Er3+ ions and enhance the CR process; thus, the increasing intensity of red emission rises with increasing Er3+ concentration can be explained.
The Rate Equation Model
To prove the existence of the CR process in Er3+ ions, we have simplified the Yb3+-Er3+ UC system in Ba5Gd8Zn4O21:Yb3+, Er3+, as shown in Fig. 7 and listed the following assumptions to clarify the rate equation concisely:
-
1
UC steps between sequential excited states take place through ET, GSA or ESA, the main population route in excited states of Er3+ is ET from Yb3+ ions, which has been demonstrated22,28.
-
2
Radiative transition is the main depopulation route in green and red emission levels E4 and E5.

The energy level diagram for rate equation analysis.
The simplified Yb3+-Er3+ UC system in Ba5Gd8Zn4O21:Yb3+, Er3+.
Initially the rate equations describing the excitation mechanisms in this system can be written as21,22,28:




where Ni is the population in energy level i, σi is the absorption cross section of state i at the pump wavelength, ϕ is the luminous flux of the pump light, τi is the lifetime of state i (the excited states i decay with rate constants τi−1) and α is the relaxation rate from E3 to E4. The energy transfers from state i to state j are described by the factor WkNiNj, in which the constant Wk represents the energy transfer rate from state i to state j.
According to the assumptions, the rate equations above could then be rewritten as




where Ai represents the radiative transition probabilities of state i.
For the rate equation of red emission level E5, there are three limiting cases corresponding to three means of populating E5:
(1) If red emission level E5 is populated only by the proposed CR process between states E2 and E3 in Er3+ ions, the rate equation of red emission level E5 is

which can be rewritten as:

Under steady-state excitation, Equation 5, Equation 6, Equation 8 and Equation 10 yield:




In this situation, the red to green emission intensity ratio R can be expressed as:

where Ci represent a constant. If the Yb3+ concentration is fixed, then both NY1 and NY2 are constants based on Equation 11 and it follows from Equation 15 and Equation 12 that

If the Er3+ concentration is fixed such that NE1 is constant, it follows from Equation 15, Equation 12 and Equation 11 that

As a result, the ground state populations NE1 and NY1are proportional to the dopant concentration and based on Equation 16 and Equation 17, we could reach the conclusion that the red to green emission intensity ratio R is proportional to the dopant concentration of Yb3+ and Er3+. This result agrees with experimental data.
(2) If the red emission level E5 is populated only by the ET process from Y2 to E6, the rate equations of E5 and E6 are


both of which could be rewritten as


where β represents the relaxation rate from E2 to E6.
Under steady-state excitation, Equation 7, Equation 20 and Equation 21 yield



R can then be expressed as

In this situation, R is a constant and the red to green emission intensity ratio will not vary with Yb3+ or Er3+ concentration, which contradicts the experimental results.
(3) If red emission level E5 is populated only by the relaxation from E3, the rate equation of E5 is

where γ represents the relaxation rate from E3 to E5, under steady-state excitation, this yields

and R can be expressed as:

Obviously, R will not vary with Yb3+ or Er3+ concentration, which contradicts the experimental results.
According to the rate equation analysis, the CR process as the third means to generate the red emission is reasonable and necessary.
Lifetime Measurements
The temporal evolutions of 2H11/2→4I15/2 (green) and 4F9/2→4I15/2 (red) transitions of Er3+ ion will also be helpful in understanding the UC processes, as shown in Fig. 8. In fact, all decay curves can be fitted by solving the rate equations. In view of the complexity, we can obtain the lifetime of red and green emission by fitting the decay parts of the lifetime curve with a single exponential function. According to Equation 10, after the green level stops being populated, the change in the population of the green level is written as

The lifetime measurements for Ba5Gd8Zn4O21:Yb3+, Er3+.
The temporal evolutions of the green emission at 548 nm and red emission at 672 nm under 971 nm pulsed laser excitation and the fitted curve of Ba5Gd8Zn4O21:15% Yb3+, 1% Er3+. The inset presents the calculated lifetime for both the green emission and the red emission of the selected Ba5Gd8Zn4O21:x% Yb3+, 1% Er3+ (x = 1, 3, 6, 9, 12, 15, 20) samples under the 971 nm pulsed laser excitation.

This could be rewritten as

In the same way, after the red level stops being populated, the population of the red level can be written as

Thus, the emission decay curves of the green and red emission can be well fitted with a single exponential function. The inset presents the calculated lifetime for both the green and red emissions of different samples. Before specifically analyze on the temporal evolutions, we summarize the key findings in the lifetime measurements:
-
1
For both green and red emissions, there exist a delayed rise and a decay in the emission intensity after the end of the excitation pulse.
-
2
The red emission, in contrast, has a longer rise time than the green emission.
-
3
The lifetime of both green and red emissions decreases with increasing Yb3+ doping concentration.
-
4
The green and red emissions have similar lifetime in the sample doped with a low concentration of Yb3+, but once the Yb3+ concentration reaches to a higher value, the lifetime of green emission decreases more quickly than the lifetime of red emission.
Observation (1) clearly indicates the existence of an energy transfer process. The ET process from Yb3+ to Er3+ and the proposed CR from Er3+ to Er3+ can still happen when the excitation is complete, which leads to a delayed rise of the emission intensity. Observation (2) suggests that the red emission level 4F9/2 requires longer time than the green emission level to be populated. According to the UC mechanisms described above, CR occurs after the population of Er3+ energy levels, which means that the red emission caused by CR requires additional time after ET from Yb3+ to Er3+. The green emission does not require such a long time owing to the lack of additional ET or CR processes required. Observation (3) can be explained by the back energy transfer (BET) from Er3+ to Yb3+ 19. Once the excitation is complete, the high energy state 4F7/2 and 2H11/2 in Er3+ can transfer energy to Yb3+ ions in the ground state and the higher the Yb3+ concentration is, the stronger BET is likely to take place; the lifetime of both green and red emissions will then decrease with increasing Yb3+ concentration. The related data can be seen in Figure S5. Observation (4) once again proved the existence of the proposed CR process. According to Equation 30, the lifetime τG of green emission can be written as

The lifetime τR of red emission can be written as:

The lifetime τG of green emission will decrease with increasing Yb3+ concentration. However, the lifetime τR of red emission does not change with increasing Yb3+ concentration. Thus, the lifetime of green emission decreases more quickly than the lifetime of red emission when the Yb3+ concentration increases.
The key point of the CR process is the multiplier effect to the population of 4F9/2 in Er3+, which is similar to the photon avalanche (PA) as shown in Figure S4(a). However, PA suffered from some drawbacks29,30,31,32. One is that the pump wavelength does not match the energy gap between the ground state and the intermediate excited level, possibly leading to the intermediate excited level being populated only by weak GSA initially. Another disadvantage is that such a process always requires high pump powers to reach the threshold condition, below which the UC luminescence intensity is weak. Moreover, PA has a longer rise time because many looping cycles are required to achieve avalanche. Compared with the PA process, the proposed CR process combines the advantages of efficient ET in a Yb3+, Er3+ co-doped system with the multiplier effect to populate the red emission level. Especially when the doping concentration is sufficiently high, frequent ET from Yb3+ to Er3+ guarantees a substantial population in the excited states of Er3+, promoting CR between Er3+ ions. Moreover, the multiplier effect caused by PA takes place in the intermediate excited level, whereas in the CR process the multiplier effect benefits the UC emission level directly. More significantly, the proposed CR process does not require a pumping power threshold condition to have an immediate effect on the UC emission.
Conclusion
UC properties of the oxide material Ba5Gd8Zn4O21:Yb3+, Er3+ were investigated as a function of Yb3+ and Er3+ dopant concentration under 971 nm excitation. The phosphors exhibit strong UC emissions, including a green emission band at 548 nm and a predominant red emission band at 672 nm. Based on the power dependence studies, we provided a model for the UC mechanisms involved in these materials. The UC efficiency testing indicated that Ba5Gd8Zn4O21:Yb3+, Er3+ had a maximum UC power efficiency of 2.7% under low power excitation and is thus more efficient than the commercial NaYF4:Yb3+, Er3+ phosphor. Together with the colour-turnable property and the stable nature of oxide, Ba5Gd8Zn4O21:Yb3+, Er3+ has the potential to be an ideal UC phosphor.
In particular, we demonstrated that the CR process occurred between two Er3+ ions through rate equations; based on this fact, the strong red emission appearing in different Yb3+, Er3+ codoped UL materials can be fully explained for the first time and the temporal evolution of the green and red emission improved this idea.
Methods
Compounds Synthesis
The compound Ba5Gd8−x−yYbxEryZn4O21 was prepared by the typical high-temperature solid state method, in which Yb3+ and Er3+ were added as a sensitizer and activator, respectively. The stoichiometric amount of starting materials BaCO3 (Alfa Aesar, 99.99%), ZnO (Fisher Scientific, 99.5%), Gd2O3 (Alfa Aesar, 99.99%), Yb2O3 (Alfa Aesar, 99.99%) and Er2O3 (Alfa Aesar, 99.99%) were thoroughly mixed, ground together in an agate mortar and filled into an aluminum oxide crucible. The mixture was then sintered at 1400 °C for 3 h, followed by cooling to room temperature spontaneously. After a second grinding stage we obtained the final product of Ba5Gd8Zn4O21:Yb3+, Er3+ power.
XRD Characteristic
X-ray powder diffraction patterns were measured by a Bruker D8 advance X-ray diffractometer (Bruker Optics, Germany) with Cu Kα radiation in the range 10° ≤ 2θ ≤ 70°. The UC luminescence spectra were recorded on an Andor SR-500 i spectrometer (Andor Technology Co., UK) equipped with a Hamamatsu R928 photomultiplier. A power-controllable 971 nm laser diode (BWT Beijing Ltd., China) was used as the excitation source, which can produce both continuous-wave and pulsed laser radiation. For the UC luminescence lifetime measurements, the UC emissions under the 971 nm pulse laser (pulse width = 50 μs) were passed through the Andor SR-500 i spectrometer and detected by the Hamamatsu R928 photomultiplier connected to a 1 GHz Tektronix digital oscilloscope.
Optical Measurements
The excitation source used for the UC efficiency measurement was a 971 nm controlled temperature CW semiconductor diode laser (BWT Beijing Ltd., China) with Pmax = 3 W. The copper sample holder in the middle of the integrating sphere was excited by the diode laser. After multiple reflections in the integrating sphere, the emitted UC light passed through an optical fiber and was analyzed with a spectrometer (380–800 nm) and a relative luminance meter. The laser output power PL under different currents was measured by a LP-3A laser power meter (Physcience Opto-Electronics Co., China). After initial calibration of the setup, the efficiency was determined in two steps. For the first measurement, the copper sample holder in the integrating sphere was left empty and the laser spectrum was obtained by a spectrometer. From this step, we obtained the integrated intensity IL over the range 950–1000 nm. For the second measurement, the copper sample holder in the integrating sphere was filled with the sample. From this step, we obtained the UC emission power Pem in the range 380–800 nm and the integrated intensity Iunabs in the range 950–1000 nm. Finally the UC power efficiency ηUC was calculated as the ratio of the luminescence power Pem emitted by sample over the power Pabs absorbed in the infrared range 950–1000 nm:

and the actual UC power efficiency ηAUC is defined as:

which represents the ratio of the UC emission power to the laser output power. We can calculate the excitation density based on the following formula: the excitation density = excitation power/laser spot area. We measured the excitation power by the laser power meter and the laser spot area could be calculated by the formula S = πR2sinθ, where R and θ represent the radius of the laser spot and the angle between the sample surface and the fiber end, respectively. R equals the distance between the sample surface and the fiber end multiplied by 0.22 (provided by the manufacturer) and θ is 45°.
Additional Information
How to cite this article: Mi, C. et al. Efficient upconversion luminescence from Ba5Gd8Zn4O21:Yb3+, Er3+ based on a demonstrated cross-relaxation process. Sci. Rep. 6, 22545; doi: 10.1038/srep22545 (2016).
References
Auzel, F. Upconversion and anti-Stokes processes with f and d ions in solids. Chem. Rev. 104, 139–173 (2004).
Wang, G., Peng, Q. & Li, Y. Lanthanide-doped nanocrystals: Synthesis, optical-magnetic properties and applications. Acc. Chem. Res. 44, 322–332 (2011).
Liu, Q., Feng, W. & Li, F. Water-soluble lanthanide upconversion nanophosphors: Synthesis and bioimaging applications in vivo, Coordin. Chem. Rev. 100, 273–274 (2014).
Wang, Y., Wu, Z. & Liu, Z. Upconversion fluorescence resonance energy transfer biosensor with aromatic polymer nanospheres as the lable-free energy acceptor. Anal. Chem. 85, 258–264 (2013).
Zhou, J., Liu, Z. & Li, F. Upconversion nanophosphors for small-animal imaging. Chem. Soc. Rev. 41, 1323–1349 (2012).
Yang, Y. et al. Optical thermometry based on the upconversion fluorescence from Yb3+/Er3+ codoped La2O2S phosphor. Ceramics International. 40, 9875–9880 (2014).
Lim, M. E., Lee, Y. L., Zhang, Y. & Chu, J. J. H. Photodynamic inactivation of viruses using upconversion nanoparticles. Biomaterials. 33, 1912–1920 (2012).
Wang, R., Li, X., Zhou, L. & Zhang, F. Epitaxial seeded growth of rare earth nanocrystals with efficient 800 nm near infrared to 1525 nm short wavelength infrared downconversion photoluminescence. Angew. Chem. Int. Ed. 53, 12282–12286 (2014).
Wang, F. et al. Upconversion nanoparticles in biological labeling, imaging and therapy. Analyst. 135, 1839–1854 (2010).
Xia, L. et al. An upconversion nanoparticle–Zinc phthalocyanine based nanophotosensitizer for photodynamic therapy. Biomaterials. 35, 4146–4156 (2014).
Huang, X., Han, S., Huang, W. & Liu, X. Enhancing solar cell efficiency: the search for luminescent materials as spectral converters. Chem. Soc. Rev. 42, 173–201 (2013).
Wang, X. et al. Photon energy upconversion through thermal radiation with the power efficiency reaching 16%. Nat. Commun. 5, 5669 (2014).
Page, R. H. et al. Upconversion-pumped luminescence efficiency of rare-earth-doped hosts sensitized with trivalent ytterbium. J. Opt. Soc. Am. B. 15, 996–1008 (1998).
Zhang, J. et al. White up-conversion luminescence power and efficiency in Yb3+-, Er3+- and Tm3+-doped BaIn6Y2O13 . Dalton Trans. 44, 1093–1101 (2015).
Wang, J. et al. Enhancing multiphoton upconversion through energy clustering at sublattice level. Nat. Mater. 13, 157–162 (2014).
Etchart, I. et al. Oxide phosphors for efficient light upconversion: Yb3+ and Er3+ co-doped Ln2BaZnO5 (Ln = Y, Gd). J. Mater. Chem. 20, 3989–3994 (2010).
Liao, J. S., Nie, L. L., Liu, S. H. & Wen, H. R. Yb3+ concentration dependence of up conversion luminescence in Y2Sn2O7:Yb3+/Er3+ nanophosphors. J Mater Sci. 49, 6081–6086(2014).
Li, A. H., Sun, Z. J. & Lü, Q. Laser heating effect on the power dependence of upconversion luminescence in Er3+-doped nanopowders. J Nanopart Res. 15, 1377 (2013).
Yang, Y. et al. Ultraviolet C upconversion fluorescence of trivalent erbium in BaGd2ZnO5 phosphor excited by a visible commercial light-emitting diode. Opt. Lett. 39, 2000–2003 (2014).
Li, T. et al. Efficient green up-conversion emission in Yb3+/Ho3+ co-doped CaIn2O4 . Acta Mater. 61, 7481–7487 (2013).
Pollnau, M., Gamelin, D. R., Lüthi, S. R. & Güdel, H. U. Power dependence of upconversion luminescence in lanthanide and transition-metal-ion systems. Phys. Rev. B. 61, 3337–3346 (2000).
Yang, Y. et al. Preparation and upconversion luminescence dynamic process of Yb3+/Er3+ co-doped BaGd2ZnO5 . Spectroscopy and Spectral Analysis. 32, 2637–2641 (2012).
Lakshminarayana, G. et al. Cross relaxation in rare-earth-doped oxyfluoride glasses. J. Lumin. 139, 132–142 (2013).
Carnall, W. T., Crosswhite, H. & Crosswhite, H. M. In Energy level structure and transition probabilities in the spectra of the trivalent lanthanides in LaF3 (report, Argonne National Laboratory, 1978).
Dieke, G. H. In Spectra and energy levels of rare earth ions in crystals (eds Crosswhite, H. M. et al.), Ch. 13, 298–302 (Interscience Publishers, 1968).
Yang, Y. et al. Dependence of optical properties on the composition in Er3+-doped xNaPO3–(80−x) TeO2–10ZnO–10Na2O glasses. Opt. Mater. 32, 133–138 (2009).
Yang, Y., Yang, Z. & Chen, B. Investigate on the feasibility of introducing dopants with high phonon enegy into tellurite host to enhance 1.5 μm emission efficiency. Photonics and Optoelectronics, SOPO,1–4 (2009).
Yang Y. et al. Dynamic process of green upconversion emission from Ho3+ under square wave excitation. Chinese Journal of Luminescence. 34, 866–871 (2013).
Chivian, J. S., Case, W. E. & Eden, D. D. The photon avalanche: a new phenomenon in Pr-based infrared quantum counters. Appl. Phys. Lett. 35, 124–126 (1979).
Guy, S., Joubert, M. F. & Jacquier, B. Photon avalanche and the mean-field approximation. Phys. Rev. B. 55, 8240–8248 (1997).
Joubert, M. F. Photon avalanche upconversion in rare earth laser materials. Opt. Mater. 11, 181–203 (1999).
Sivakumar, S., van Veggel, F. C. J. M. & May, P. S. Near-Infrared (NIR) to red and green up-conversion emission from silica sol-gel thin films made with La0.45Yb0.50Er0.05F3 nanoparticles, hetero-looping-enhanced energy transfer (hetero-LEET): a new up-conversion process. J. Am. Chem. Soc. 129, 620–625 (2007).
Acknowledgements
This work was supported by the National Science Foundation of China (no. 11474083), the Natural Science Foundation of Hebei province (no. E2010000283) and Key Technology R&D Program of Hebei province Baoding City (no. 15ZG044, 15ZG045).
Author information
Authors and Affiliations
Contributions
Y.Y. and C.M. developed the idea and Y.Y. supervised the project. C.M. and J.W. conducted most of the experiments, C.M. wrote the paper and B.H. and J.W. helped with material preparation. All authors discussed the results.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Mi, C., Wu, J., Yang, Y. et al. Efficient upconversion luminescence from Ba5Gd8Zn4O21:Yb3+, Er3+ based on a demonstrated cross-relaxation process. Sci Rep 6, 22545 (2016). https://doi.org/10.1038/srep22545
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep22545
This article is cited by
-
A study of virescent emission from Er3+-activated ternary gadolinium-based nanophosphor system applicable for current pc-WLEDs and solid-state lightings
Indian Journal of Physics (2024)
-
Green Upconversion of a SrLaAlO4:Yb,Er Phosphor and Its Application for LED Illumination
Journal of Electronic Materials (2023)
-
Effect of Yb/Er/Li on structure and up-conversion luminescence properties of NaSc(WO4)2
Journal of Materials Science: Materials in Electronics (2022)
-
Transmittance, Photoluminescence and Electrical Properties in Er-Doped 0.98K0.5Na0.5NbO3-0.02Sr(Yb0.5Ta0.5)O3 Ferroelectric Ceramics
Journal of Electronic Materials (2022)
-
Investigation of new color-tunable up-conversion phosphors and their long-persistent luminescence properties for potential biomedical applications
Applied Physics A (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
