
Abstract
To investigate the spatial distribution of microbial communities and their drivers in petroleum reservoir environments, we performed pyrosequencing of microbial partial 16S rRNA, derived from 20 geographically separated water-flooding reservoirs and two reservoirs that had not been flooded, in China. The results indicated that distinct underground microbial communities inhabited the different reservoirs. Compared with the bacteria, archaeal alpha-diversity was not strongly correlated with the environmental variables. The variation of the bacterial and archaeal community compositions was affected synthetically, by the mining patterns, spatial isolation, reservoir temperature, salinity and pH of the formation brine. The environmental factors explained 64.22% and 78.26% of the total variance for the bacterial and archaeal communities, respectively. Despite the diverse community compositions, shared populations (48 bacterial and 18 archaeal genera) were found and were dominant in most of the oilfields. Potential indigenous microorganisms, including Carboxydibrachium, Thermosinus and Neptunomonas, were only detected in a reservoir that had not been flooded with water. This study indicates that: 1) the environmental variation drives distinct microbial communities in different reservoirs; 2) compared with the archaea, the bacterial communities were highly heterogeneous within and among the reservoirs; and 3) despite the community variation, some microorganisms are dominant in multiple petroleum reservoirs.
Similar content being viewed by others

Introduction
Petroleum reservoirs represent unique environments deep under the Earth’s crust, with multiphase fluids containing oil, gas and water. Petroleum reservoirs are composed of multiple oil-bearing strata, of low porosity and low permeability and represent complex oligotrophic ecosystems. Nevertheless, these extreme environments can harbour diverse bacteria and archaea that exhibit strong adaptations to the oligotrophic and oxygen-deficient conditions1,2,3. These microorganisms play key roles in the Earth’s biogeochemical cycles. Studies of oil reservoir ecosystems have improved our understanding of these biogeochemical cycles4,5,6,7,8 and the mechanisms that microorganisms use to tolerate the extreme underground environments9. Some studies have enhanced the potential for biotechnological applications, in particular, the use of biological preservatives10,11,12,13 and microbial enhanced oil recovery14,15. Diverse microbial populations inhabiting these reservoirs have been revealed16,17,18,19,20,21,22,23. However, due to limited sample sources and methodological constraints, few studies have attempted to characterize the large-scale biogeographic distributions of microbial communities in oil reservoirs.
There is growing evidence that free-living microorganisms exhibit non-random distribution patterns across diverse habitats, at various temporal and spatial scales24,25,26,27,28,29,30. Environmental variables, such as day–night differences31, temperature32,33, salinity34, pH24,35 and nutrients15,36 appear to be the major determinants of microbial community composition. The impacts of geographical distance on structuring microbial assemblages have also been observed in several environments, particularly soil and marine environments25,37. However, the complex exploitation history and diverse physicochemical properties of oil reservoirs and the low coverage of traditional detection methods, mean that there is limited knowledge regarding the microbial communities that are present and how they are influenced by spatial and environmental variables (relative to the equivalent knowledge for soil and marine environments)38,39. Previous studies that investigated the microbiology of oil reservoirs generally covered relatively small spatial scales. Therefore, the differences in microbial communities between different reservoir ecosystems and the changes in the communities along environmental gradients, were difficult to elucidate.
Microbial distribution patterns across large spatial scales and their underlying driving mechanisms have become a focus in microbial ecological research. In this study, bacterial and archaeal communities from 20 geographically-separated water-flooding reservoirs and two reservoirs that had not been flooded, in China, were analysed through 454-pyrosequencing of the 16S rRNA gene. Pyrosequencing generates thousands of short sequences representing different biota and significantly improves our ability to compare microbial populations in detail. The aim of this study was to determine the composition and spatial distribution of the bacterial and archaeal communities and determine the major environmental variables (mining patterns, spatial isolation, temperature, salinity and pH) in shaping their composition and structure across a broad range of physical and geochemical reservoirs.
Results
Reservoir characteristics
There were striking differences in both geographic location and environmental characteristics among the 22 oil reservoirs (Fig. 1, Table 1 and Supplementary Table S1). The temperature, salinity and concentrations of ions in the formation brines were significantly different between the reservoirs within most of the oilfields. The distance between each of the reservoirs ranged from tens to thousands of kilometres. Even the reservoirs located the same oilfield generally contained different layers of oil-bearing strata underground. For instance, the DGD, DGX, DGK and DGS reservoir blocks were all located in the DG oilfield, but there were large differences in the layers and the depths of the oil-bearing strata. The mining patterns and history of flooding among the reservoirs were also generally different (Fig. 1 and Table 1). All of the reservoir blocks except YC2 and LHJ had been flooded at some point in the past; the production well sampled in the YC2 reservoir had not previously been flooded with water when the samples were collected, while steam soaking techniques had been conducted in the production wells sampled within the LHJ reservoir block.

Locations of the sampling sites in the 22 geographically-separated reservoirs in China.
The Arabic numerals in the pie charts represent the number of samples; the blue parts of the pie charts represent the reservoirs with temperatures of 22 °C; gray parts represent reservoirs with temperature of 34–45 °C; red parts represent reservoirs with temperatures of 46–73 °C. The map was produced using ‘R (i386 3.1.2)’, with the open source packages ‘maps’, ‘mapdata’ and ‘maptools’.
There was often variation in the physical properties observed within the same reservoir. In the XJL reservoir block, the oil-bearing strata were divided into three: S73−1, S73−2 and S73−3. The SLZ reservoir block included G3, G4 and G6 oil-bearing strata, with depths ranging from 1187 to 1304 m (Table 1). Furthermore, despite being located in the same oil-bearing stratum, the depth of the oil-bearing stratum could vary greatly, for example, 1162 to 2162 m, in DGD block (Table 1). The mining patterns, flooding history and environmental variation suggest that the environmental variables in the reservoirs are complex and volatile, with patchy distributions; this implies that there may be complex microbial community compositions and structures in the reservoirs.
Overall microbial community composition
After filtering the low quality reads and chimeras, an average of 4,261 high-quality bacterial and 2,428 archaeal sequences were obtained in each water sample (Supplementary Tables S2 and S3). These sequences were assigned into 11,195 bacterial OTUs and 3,288 archaeal OTUs (at a genetic distance of 3%), with an average of 312 bacterial OTUs and 159 archaeal OTUs in each sample. The number of OTUs and the Chao 1, ACE, Shannon’s and Simpson’s indices are summarized in Supplementary Tables S2 and S3. The coverage of the sequencing libraries reached 95%, which could reflect the whole of the microbial community in each water sample. However, high numbers of rare OTUs that were represented by one sequence were detected in each water sample. These taxa accounted for approximately 50% of all the bacterial or archaeal OTUs detected, but only accounted for less than 10% of all the sequences in each sample (Supplementary Tables S2 and S3). These data indicate that the reservoirs harbour an abundance of rare microbial organisms from the underground environments.
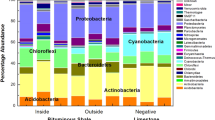
At total of 34 bacterial phyla were identified; Proteobacteria, Firmicutes, Actinobacteria, Bacteroidetes, Thermotogae and Synergistetes were detected, accounting for an average of 93.06% of the whole bacterial community, in every oilfield (Supplementary Fig. S1). At the class level, 53 taxa were identified. Of these, Epsilonproteobacteria, Alphaproteobacteria, Gammaproteobacteria, Betaproteobacteria, Deltaproteobacteria, Clostridia, Bacteroidia, Thermotogae and Synergistia were detected, accounting for an average of 82.47% of the whole bacterial community, in every oilfield (Table 2). Alphaproteobacteria, Gammaproteobacteria and Clostridia were detected in all of the 22 reservoir blocks and accounted for an average of 2.02–68.49% of the bacterial community in each block (Table 2). At the genus level, a total of 380 phylotypes were defined. However, no taxa were detected in every reservoir block, or even across all oilfields. Each reservoir harboured certain unique microbial taxa with higher relative abundances (Fig. 2a). A total of 48 bacterial genera were detected in at least five of the nine oilfields, accounting for an average of 63.01% of the bacterial community in each oilfield (Fig. 3a). The most abundant phylotypes identified were Arcobacter, Pseudomonas, Acinetobacter, Bacillus, Rhodococcus, Dietzia, Paracoccus, Marinobacterium, Rhizobium, Thauera, Rhodobacter, Brevundimonas, Desulfovibrio and Desulforhabdus; the rare genera found included Tepidiphilus, Tistrella and Kosmotoga.

The relative proportion of dominant (a) bacterial and (b) archaeal genera in water samples obtained from the 22 reservoir blocks.
Key: *represent genera for which their abundance was significantly different among oilfields, reservoir blocks, or reservoir temperatures (P < 0.05; see Supplementary Tables S8 and S10).

The (a) bacterial and (b) archaeal genera found in more than five of the nine oilfields. (a) 48 bacterial genera were detected in at least five of the nine oilfields, accounting for an average of 63.01% of the bacterial community in each oilfield; (b) 18 archaeal genera, accounting for 62.17–95.6% of the classified sequences, were observed in at least five of the nine oilfields.
Compared to the bacterial communities, the archaeal communities did not vary very much at the phylum level. An average of 97.86% of the archaeal sequences identified at each oilfield belonged to Euryarchaeota (Supplementary Fig. S1). The archaeal sequences found belonged to the following classes: Methanomicrobia, Methanobacteria, Methanococci, Archaeoglobi, Thermoplasmata, Thermococci, Halobacteria and Thermoprotei. Methanomicrobia, Methanobacteria, Methanococci, Thermoplasmata and Halobacteria were detected in every oilfield, accounting for an average of 88.11% of the archaeal community in each oilfield (Table 2). Among them, Methanomicrobia and Methanobacteria were detected in all of the 22 reservoir blocks, accounting for an average of 70.52% of the archaeal community in each block. In total, 35 archaeal genera were detected in the 22 reservoir blocks (Fig. 2b). Among them, 18 genera, accounting for 62.17–95.6% of the sequences that could be classified, were found in at least five of the nine oilfields (Fig. 3b).
Potential indigenous underground microbial populations
The microbial communities present in reservoirs that had not been flooded with water may reflect the indigenous underground populations. The LHJ reservoir block was a viscous oil reservoir that had been subjected to steam soaking for the exploration of the viscous oil. In this reservoir, Gammaproteobacteria and Alphaproteobacteria were dominant and accounted for an average of 63.1% and 4.7% of the bacterial community, respectively. At the genus level, 83.4% of the sequences present could not be assigned to any taxa, using the RDP Classifier at the 80% threshold; this indicates that the reservoir harbours a large number of unclassified microbial taxa (Supplementary Fig. S2). In the reservoir, 27 genera were detected; among these, 24 genera were detected in at least some of the other reservoirs. However, three genera, including Carboxydibrachium, Thermosinus and Neptunomonas, were detected specifically in LHJ, accounting for 0.02–0.11% of the bacterial community present. In terms of the archaeal community, four methane-producing archaea were detected in the LHJ reservoir. Among them, Methanothermobacter dominated the reservoir and accounted for 82.1% of the whole archaeal community.
The other reservoir that had not been flooded with water previous to sampling was the YC2 reservoir; this is an ultra-low permeability reservoir. Within this reservoir, 96.6% of the bacterial sequences were assigned to the class Epsilonproteobacteria. A total of nine genera, including Arcobacter, Oceanicola, Marinobacterium, Guggenheimella, Geoalkalibacter, Marinobacter, Marinitoga, Sphingopyxis and Rhodococcus, were detected (Supplementary Fig. S2). Among them, Arcobacter accounted for 96.6% of the bacterial community present. Ten archaeal genera were detected in this reservoir. The dominant genera were the methane-producing Methanococcus, Methanobacterium, Methanothermococcus, Methanocalculus and Methanosaeta.
Regional distributions of the microbial populations
The bacterial and archaeal community α-diversity estimates, including the number of OTUs and Shannon’s index, exhibited associations within reservoir blocks. The LHJ, DGD, YC1, YC2 and QH2 reservoir blocks harboured low numbers of bacterial OTUs and had lower Shannon’s indices (Supplementary Fig. S3). The QH2 reservoir had the smallest number of archaeal OTUs and the lowest Shannon’s index (Supplementary Fig. S3). However, the microbial community’s α-diversity (in particular, the archaeal) was not significantly correlated with the chemical parameters measured (P < 0.05; Supplementary Tables S5 and S6).
The microbial community composition and relative abundances of taxa were highly heterogeneous, even for samples retrieved from the same reservoir block (Fig. 2 and Table 2). The phylum Proteobacteria, containing the classes Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria, Deltaproteobacteria and Epsilonproteobacteria, was detected in almost every reservoir. However, the abundances of these taxa differed greatly among the reservoirs (Table 2). For instance, significantly different relative abundances of Epsilonproteobacteria were found among the reservoir blocks (P < 0.05; Supplementary Table S7), whereby taxa in the class were dominant in the XJL, XJQ, DQY, DQS, DQQ, JL, DGK, YC1, YC2 and QH2 reservoir blocks, but only accounted for less than 1% of the bacterial communities in the LHJ, LHS, SLX, SLZ, DGD, DGS, QH1 and HBM reservoir blocks (Table 2). The dominant bacterial and archaeal taxa in each reservoir are summarized in Table 2 and Supplementary Tables S4, S7 and S9.
Although taxa of specific classes dominated multiple reservoir blocks, the dominant genera within the dominant class differed in some cases. In the Bacilli class, Lysinibacillus (37%) was dominant in the LHD block, Bacillus (33.18%) in the DGX block, Paenibacillus (35.04%) in the DGS block and Anaerobacillus (20.07%) was dominant in the HBB block (Fig. 2a and Supplementary Table S4). This phenomenon may be partially explained by the reservoir temperatures among the four blocks: LHD was 38 °C; DGX, 47 °C; DGS, 73 °C; and the HBB block was 58 °C. A similar phenomenon was observed for the class Clostridia. This class was dominant in the QH1 and JL reservoir blocks, accounting for 41.20% and 32.46%, of the bacterial community in each block, respectively (Table 2). However, within Clostridia, Alkalibacter accounted for 16.66% of the bacterial community in the JL block, but was not detected in QH1 block; instead, Desulfotomaculum accounted for 3.66% in the QH1 block and was not detected in the JL block (Fig. 2a). There were bacterial and archaeal taxa that had significantly different relative abundances between the reservoirs (P < 0.05; Supplementary Table S8 and S10); these data indicate that each reservoir harboured specific and dominant microbial taxa.
Through further investigation into the distribution of the reservoirs’ microbial communities, the cluster analysis and weighted UniFrac analysis performed, indicated that the distances between the communities within most of the reservoirs were significant (P < 0.001). Thus, it appears that distinct microbial communities did inhabit each reservoir. Although there are slight variances, predictable beta diversity patterns, for both the bacterial and archaeal communities, were observed. The bacterial and archaeal communities displayed high niche specificity; that is, the microbial communities from the same reservoir block formed clusters that differed from those of other reservoir blocks (Fig. 4 and Supplementary Fig. S4).

Principal coordinates analysis (PCoA) of the (a) bacterial and (b) archaeal communities, based on the UniFrac distance matrix generated from the rarefied taxon abundances and phylogenetic relationships. Points those are closer together on the ordination show communities that are more similar.
Environmental variation and the microbial communities
As mentioned above, the LHJ, DGD, YC1, YC2 and QH2 reservoir blocks harbour low numbers of bacterial OTUs and the QH2 reservoir has the smallest number of archaeal OTUs (Supplementary Fig. S3). Among these, steam-soaking techniques had previously been used in the sampled production wells in LHJ reservoir block; YC1 was an ultra-low permeability reservoir that was only flooded with water for 5 years; YC2 was an ultra-low permeability reservoir that had not been flooded with water previously; DGD was a high temperature and high pressure reservoir; and QH2 was a high temperature, hypersalinity and high pressure reservoir. It appears that, the mining patterns, history of flooding, high temperatures and hypersalinity exerted strong influences on the microbial communities in these reservoirs.
In accordance with these hypotheses, the relative abundance of 48 bacterial and 8 archaeal taxa exhibited strong correlations with reservoir temperature and the formation brines’ pH, salinity, and/or ionic composition (P < 0.05; Supplementary Table S5 and S6). Some of the dominant bacterial and archaeal taxa (that have higher relative abundances) were significantly correlated with certain reservoir physicochemical parameters (Supplementary Fig. S5). Paracoccus, Donghicola, Marinobacterium, Methanococcus, Methanocorpusculum and Methanolinea preferred to inhabit reservoirs with medium and low temperatures, while Methanothermobacter and Thermococcus were dominant in the reservoirs with higher temperatures. Methanothermococcus was found more frequently in reservoirs with high salinity. Pseudomonas was inversely correlated with the formation brine pH, while Desulfuromonas preferred to inhabit the more alkaline reservoirs.
The dominant microbial populations were further clustered along with temperature and pH gradient, to highlight the populations that showed the most variability (Fig. 5 and Supplementary Fig. S6). At the class level, Epsilonproteobacteria were explicitly dominant in reservoirs with a pH of 5.5–6.5; Alphaproteobacteria were dominant in the low and medium temperature reservoirs, with a pH of 7.0–8.0; Actinobacteria were dominant in medium and high temperature reservoirs, with a pH of 7.0–8.0; Gammaproteobacteria and Betaproteobacteria were dominant in the medium and high temperature reservoirs, with with a pH of 5.5–6.5; and Deltaproteobacteria, Bacteroidia, Bacilli, Clostridia, Thermotogae, Mollicutes, Thermoleophilia and Thermodesulfobacteria were clearly dominant in the medium–high temperature reservoirs (Fig. 5a,c). The genera Rhodococcus, Paracoccus, Hyphomonas, Dietzia, Marinobacterium, Microbacterium and Donghicola were more dominant in the medium–low temperature reservoirs, while Bacillus, Anaerobacillus, Thermotoga, Tepidiphilus, Tistrella, Thermodesulfovibrio, Thermodesulfobacterium, Thermus, Thermosyntropha and Kosmotoga had greater abundances in the medium–high temperature reservoirs (Supplementary Fig. S6). It’s worth mentioning that some taxa, including Arcobacter, Pseudomonas, Acinetobacter, Sulfurospirillum, Rhizobium and Sphingomonas were universally detected in reservoirs with a wide range of temperatures. In terms of the archaea genera, Methanolinea, Methanococcus, Methanoculleus, Methanolobus and Methanocorpusculum were dominant in the low and medium temperature reservoirs with alkaline environments; Methanosaeta, Archaeoglobus and Methanocalculus dominated the medium and high temperature reservoirs with alkaline environments; while Methanothermobacter were more frequently detected in in medium and high temperature reservoirs with acidic environments (Fig. 5b,d).

The distributions of dominant bacterial (a,c) and archaeal (b,d) populations against the temperature (a,b) and pH (c,d) gradients.
A multivariate regression tree (MRT) analysis was performed to interpret the relationship between the relative abundances of dominant lineages and the main environmental variables (sampling location, reservoir temperature and the formation brines’ pH, salinity, water type and SO42−, Ca2+ and Mg2+ concentrations) in a visualized tree. The trees collectively explained 60% and 64% of the variance observed in the relative abundances of the bacterial and archaeal taxa, respectively (Fig. 6a,b). In the bacterial MRT, the dominant lineages were first split by sampling location, which explained 16.1% of the variation in community structure. At the second node, the split was determined by temperature, which explained 9.0% of the variation. The communities were then split by salinity and temperature, accounting for 7.9% and 7.6% of the variation in the data, respectively. In terms of the archaeal MRT, the dominant lineages were first split by temperature, which explained 17.1% of the variation in community structure. The following left group was split by sampling location, while the right group was split by salinity, which explained 11.1% and 6.9% of the variation, respectively. The results suggest that spatial isolation, represented by sampling location, temperature and the salinity of the formation brine, explained the variation in the microbial communities well.

The relationship between the microbial community composition and various environmental conditions.
The MRT analysis was performed to interpret the relationship of the relative abundance of dominant (a) bacterial and (b) archaeal lineages with the environmental variables measured. The canonical correspondence analysis (CCA) and redundancy analysis (RDA) used show the relationship between the environmental factors and the (c) bacterial and (d) archaeal communities.
To investigate the relationships between the microbial communities and the environmental variables of the petroleum reservoirs further, canonical correspondence analysis (CCA) and redundancy analysis (RDA) was conducted. The first two axes of the CCA and RDA analysis explained 64.22% and 78.26% of the total variance for the bacterial and archaeal communities, respectively (Fig. 6c,d). The Monte Carlo permutation test showed that temperature, salinity, concentrations of sulphate and pH were significantly correlated with the changes in bacterial composition; temperature, salinity and calcium ion concentrations were significantly correlated with the changes in the archaeal communities (P < 0.05).
Discussion
Microbial alpha-diversity, known as within-habitat diversity, reflects the number of species in a local homogeneous habitat. In petroleum reservoirs, both the bacterial and archaeal community α-diversities were influenced by the extreme environmental conditions; for example, steam-soaking (LHJ), non-water-flooding (YC2), high temperature, high pressure and hypersalinity (QH2) (Supplementary Fig. S3). However, species diversity, in particular that of the archaea, was not strongly correlated with the reservoir temperature and the chemistry of the formation brines (Supplementary Table S5 and S6). Actually, in previous studies, no relationship has been found between microbial α-diversity and either temperature or geographical distance, in other environments, such as in soil38 or acid mine drainage environments24. In terms of the microorganisms in reservoirs, studies have documented diverse microbial communities in Chinese reservoirs across a broad range of temperatures (22–73 °C) and these studies did not observe substantial differences in microbial α-diversity18,19,40. The phenomenon may be related to the fact that a large number of microbial populations, in particular thermophilic and thermotolerant organisms, are prevalent in medium and high temperature reservoirs, while other organisms prefer to inhabit medium and low temperature environments. Furthermore, most of the organisms found in these reservoirs are likely to possess exceptional survival abilities and may be well adapted to, or tolerant of, the diverse reservoir environments.
Based on the phylogenetic analysis, more than 50 bacterial and 8 archaeal classes were identified across the 22 reservoirs. In accordance with previous research, Epsilonproteobacteria, Alphaproteobacteria, Gammaproteobacteria, Betaproteobacteria, Deltaproteobacteria, Bacilli, Clostridia, Actinobacteria, Bacteroidia, Thermotogae, Synergistia, Methanomicrobia, Methanobacteria, Methanococci and Archaeoglobi were most frequently detected16,19,21,40,41,42. Despite the highly diverse microbial communities that inhabit petroleum reservoirs, some specific organisms were found to dominate the microbial communities across the different reservoirs. An increasing amount of studies have focused on the positive correlations between microbial community structure and function. For example, in marine environments39 and sewage treatment plants43; these studies have demonstrated that there are core microbes with specific functions across the different habitats.
In the petroleum reservoirs, 48 bacterial genera were detected and found dominant, in the majority of the oilfields (Fig. 3a). These bacteria were mainly affiliated with the commonly reported hydrocarbon-degraders, surfactant-producers, nitrate-reducers and sulphate-reducers. Among them, Pseudomonas, Rhodococcus, Dietzia, Acinetobacter, Bacillus and Paracoccus are often aerobic organisms and are well known for their ability to degrade hydrocarbon or produce biosurfactants. Most of these populations are common in the microbial-enhanced oil recovery process14,15,44. Pseudomonas, Brevundimonas, Hyphomonas, Arcobacter, Thauera, Rhizobium and Rhodobacter are able to reduce nitrate in either aerobic or anaerobic conditions. Among them, Thauera can anaerobically degrade aromatic compounds and were the main contributor during the mitigation process of biological souring in oil reservoirs3,13. Desulfovibrio and Desulforhabdus are common sulfate-reducers in reservoir environments1,18.
In terms of the archaeal communities, 18 archaeal genera, which accounted for 62.17–95.6% of the classified sequences, were observed in most oilfields (Fig. 3b). Methylotrophic and hydrogenotrophic methanogens include: Methanofollis, Methanocorpusculum, Methanocalculus, Methanothermococcus, Methanobacterium, Methanothermobacter, Methanococcus, Methanoculleus, Methanomethylovorans and Methanolobus18,19,45. These genera were found in abundance in the XJL, XJQ, DQS, DQQ, DQT, JL, LHJ, LHS, SLX, SLZ, DGX, DGK, DGS, YC1, YC2, QH1, QH2 and HBB reservoir blocks. Methanosaeta, which uses only acetate in methane production45, was dominant in the XJL, XJQ, DQY, DQS, DQQ, DQT, JL, LHS, SLX, SLZ, DGX, DGD, YC1, YC2, HBM and HBB reservoir blocks. Therefore, the microbial organisms found across the different petroleum reservoirs may be closely related to the biogeochemical cycles that occur ubiquitously underground; in particular the carbon, nitrogen and sulphur cycles.
Microbial populations in reservoirs that had not previously been subjected to water flooding (LHJ and YC2) were analysed because they may reflect, to some degree, the indigenous populations before mining operations. Most of the taxa observed in the LHJ block were facultative bacteria and methanogens (Fig. 2 and Supplementary Table S4). Furthermore, three genera (Carboxydibrachium, Thermosinus and Neptunomonas) were only detected in the LHJ reservoir. Species of the Carboxydibrachium and Thermosinus genera are anaerobic, thermophilic and facultatively carboxydotrophic bacteria46. Populations of these genera can grow chemolithotrophically on CO, producing equimolar quantities of H2 and CO2. In the YC2 reservoir, nine bacterial genera were detected, with Arcobacter accounting for 96.6% of the bacterial community present. Arcobacter are found in an unusually wide range of habitats, including oil reservoirs and petroleum-contaminated groundwater3,47,48. Therefore, Arcobacter populations may mainly exist in fractures of the oil-bearing strata and could have been introduced by the oil recovery processes. Furthermore, the permeability of the YC2 reservoir was only 0.5 md, which may seriously inhibit microbial migration and dispersion in the oil-bearing strata.
Although they give an indication, the results of this study do not provide sufficient reliable information of the indigenous microbial communities in these habitats. The introduction of exogenous microorganisms in injected water and other sources of contamination that arise from the enhanced oil recovery processes, have made it increasingly difficult to determine whether a microorganism is indigenous. Interestingly, it seems that the reservoirs that had previously been flooded with water had high microbial diversity than the two reservoirs that had not. Furthermore, a large number of aerobes were found to coexist with the facultative and obligate anaerobes in the reservoirs. This phenomenon suggests that the oil exploitation processes may introduce exogenous microorganisms into reservoirs. These exogenous microbial populations may possess exceptional survival abilities and form new communities with the indigenous microorganisms, or they may remain in a dormant state after being introduced into the reservoir strata.
Each reservoir harboured microbial communities with distinct structures; the community composition and the relative abundance of microbial organisms were highly heterogeneous among the different reservoirs. Microbial assemblages in the reservoirs may have been affected by various environmental disturbances; mining patterns, geographical isolation, reservoir temperatures and chemical composition of the formation brine were all found to be important. Many scientists have reported that microbial communities have a close relationship with their geographical position, in both contiguous and island habitats49,50,51. However, similar research of those communities in reservoir environments was scarce. Consistent with the other environments, the bacterial and archaeal communities in the reservoirs displayed high niche specificity. The microbial communities from the same reservoir block and those from reservoir blocks located in the same oil-bearing strata were more similar, structurally. The MRT analyses revealed that the microbial communities in the reservoirs could be distinguished by most of the predefined factors assessed, including location (reservoir block), temperature and the salinity of the formation brine. The CCA and RDA analyses also demonstrated the significant influences that temperature, salinity and pH had on the microbial communities in the reservoirs. Temperature has previously been hypothesized as the main factor that influences microbial communities in water-flooding reservoirs18,19,32,40. pH was previously found to significantly influence soil microbial communities and their metabolisms25,38,52. Another study that found that the major environmental driver of microbial community composition in natural environments was salinity, rather than extremes of temperature, pH, or other physical and chemical factors53.
Diverse microbial community structures in the production wells from the same reservoir were also observed. The differences observed may be related to the isolation of the oil-bearing strata, the depth span of the reservoir strata1,20,23, and/or stochastic processes28,54. The spatial isolation and low permeability of reservoir strata may exert a significant influence on the community composition and structure in oil wells, even those located the same reservoir block. The stochastic processes of microbial community assembling and succession may also lead to the differences in the microbial communities54. As a result, we found that even in the same reservoir block, only a small number of microbial organisms were observed across the water samples from different production wells in the same reservoir. This phenomenon has also been documented by analysing the microbial communities derived from the wellhead and downhole of injection wells and from their effective production wells nearby20.
This study revealed the similarities and differences of the microbial communities in the 22 geographically separated reservoirs, which had diverse physicochemical properties. The results indicate that distinct microbial communities inhabit different reservoirs and that they are mainly driven by mining patterns, geographic isolation, reservoir temperature and the salinity and pH of formation brine. The results expand our knowledge of the broad trends of microbial community distributions in petroleum reservoirs in China. They can be used to guide biotechnological applications in the reservoirs, in particular the biological preservative and microbial enhanced oil recovery. In future work, more comparative analyses, based on time-series sampling at a diverse array of reservoirs will be performed to better determine the community distributions along the reservoir environmental gradients.
Methods
Reservoir information
Samples were collected from 22 geographically separated reservoirs within nine oilfields across China (Fig. 1). The reservoirs, even those located in the same oilfield, represented a broad variety of physical and geochemical conditions, including differences in their exploitation history, temperature and the salinity of the formation brine (Table 1). The DaQing (DQ), JiLin (JL) and LiaoHe (LH) oilfields are located in Northeast China; the ShengLi (SL), DaGang (DG) and HuaBei (HB) oilfields are located in Northern China; the YangChang (YC), QingHai (QH) and XinJiang (XJ) oilfields are located in Northwest China. Among them, the XJ, LH, SL and HB oilfields each included two geographically isolated reservoir blocks, with different reservoir temperatures; the DG oilfield included three geographically isolated reservoir blocks, with different reservoir temperatures; and the DQ oilfield included four geographically isolated reservoir blocks, which could be classed into three groups, based on temperature. The temperatures ranged from 22 °C to 73 °C, while the salinity of the formation water in the reservoirs was 1,195–82,827 mg L−1. The samples were numbered with the name of reservoir block followed by Arabic numerals, such as XJL1, which represented the first water sample obtained from an oil well of Liuzhongqu reservoir block in XinJiang oilfield. The sampling oil wells of the same reservoir block were adjacent, located in a relatively closed site and flooded by the same injected water. The ionic composition of the reservoirs’ formation waters varied greatly; detailed information of the ionic composition and other factors is shown in Supplementary Table S1.
Samples collection and DNA extraction
Samples were collected between July 2011 and September 2012 from the wellheads of the production wells in each reservoir block. Sterilized plastic bottles (15 L) were completely filled, then immediately capped and sealed to avoid contamination and oxygen intrusion. The residual oil in each sample was firstly removed by heating the sample to 60 °C for 15 min and conducting phase separation in sterilized separatory funnels. Microbial cells were then collected from the 5 L water sample by centrifugation (12,000 × g) at 4 °C for 15 min, in a high-speed centrifuge (Beckman, USA). Total genomic DNA was extracted from the cell deposits using methods previously described15. The detailed process is described in the Supplementary text.
PCR amplification and bar-coded pyrosequencing
The widely used universal primers, 27F (5′-AGA GTT TGA TCC TGG CTC AG-3′) and 533R (5′-TTA CCG CGG CTG CTG GCA C-3′), were used to amplify the bacterial 16S rRNA gene55; and the primers 344F (5′-ACG GGG YGC AGC AGG CGC GA-3′) and 915R (5′-GTG CTC CCC CGC CAA TTC CT-3′) were used for the archaeal 16S rRNA gene56. The primer sets have high coverage levels of bacterial and archaeal 16S RNA genes, respectively and are the most effective primers for analyzing the diversity of microbial communities. Amplicon pyrosequencing was performed on a Roche Genome Sequencer GS FLX + platform at Majorbio Bio-Pharm Technology, Shanghai, China. The detailed PCR process and preparation steps for the sequencing of the amplicons is included in the Supplementary text.
The raw data generated were processed and analysed following the MOTHUR pipeline (http://www.mothur.org)57. Sequences with ambiguous bases and low quality scores (<25), those smaller than 200 bp and sequence tags, chimeras and non-ribosomal sequences, were eliminated from the data sets. The remaining sequences were grouped into operational taxonomic units (OTUs) by setting a 0.03 distance limit (equivalent to 97% similarity). Representative sequences were aligned using NAST58. Taxonomic classification of the phylotypes was determined based on the Ribosomal Database Project, at the 80% confidence level59.
Statistical analyses
The relative abundance (%) of individual taxa within each community was estimated by comparing the number of sequences assigned to the specific taxon versus the number of total sequences obtained for that sample. Rarefaction curves were produced based on the identified OTUs, then Shannon’s diversity index and non-parametric measures of species richness (Chao1 and ACE) were calculated for each sample using the MOTHUR pipeline. Finally, clustering analysis and PCoA, based on the UniFrac dissimilarity values, was performed, to interpret the relative similarity of the microbial communities from each sample site.
Linear correlations between the microbial alpha diversity, microbial abundance and environmental factors were examined using Pearson’s correlation analyses. Line charts and Boxplots were used to show the distribution of bacterial and archaeal populations along the environments gradients. Changes in microbial abundance in different reservoirs were compared using One Way Analysis of Variance (ANOVA) with Student-Newmnan-Keuls tests. An MRT analysis was performed using the package ‘mvpart’ within the ‘R’ statistical programming environment, to highlight the main relationships between the biological data and environmental variables60. The ‘1se’ cross-validation process was used to construct the multivariate regression tree. In addition, the correlations between the microbial communities and environmental factors were determined with Canonical correspondence analysis (CCA) and Redundancy analysis (RDA), using the ‘Vegan’ package within R (http://cran.r-project.org/web/packages/vegan). Statistical significance was assessed using the Monte Carlo permutation’s method, based on 999 permutations.
Sequence accession numbers
The raw reads have been deposited in the National Center for Biotechnology Information (BioProject ID: PRJNA 271507, http://www.ncbi.nlm.nih.gov/bioproject/271507).
Additional Information
How to cite this article: Gao, P. et al. Spatial isolation and environmental factors drive distinct bacterial and archaeal communities in different types of petroleum reservoirs in China. Sci. Rep. 6, 20174; doi: 10.1038/srep20174 (2016).
References
Youssef, N., Elshahed, M. S. & McInerney, M. J. Microbial processes in oil fields: culprits, problems and opportunities. Adv. Appl. Microbiol. 66, 141–251 (2009).
Magot, M., O. B. & Patel, B. K. Microbiology of petroleum reservoirs. Anton Leeuw Int J G 77, 103–116 (2000).
Voordouw, G. Production-related petroleum microbiology: progress and prospects. Curr. Opin. Biotechnol. 22, 401–405, 10.1016/j.copbio.2010.12.005 (2011).
Feng, W. W., Liu, J. F., Gu, J. D. & Mu, B. Z. Nitrate-reducing community in production water of three oil reservoirs and their responses to different carbon sources revealed by nitrate-reductase encoding gene (napA). Int Biodeter Biodegr 65, 1081–1086, 10.1016/j.ibiod.2011.05.009 (2011).
Ramos-Padron, E. et al. Carbon and sulfur cycling by microbial communities in a gypsum-treated oil sands tailings pond. Environ Sci Technol 45, 439–446, 10.1021/es1028487 (2011).
Grigoryan, A. & Voordouw, G. Microbiology to help solve our energy needs: methanogenesis from oil and the impact of nitrate on the oil-field sulfur cycle. Ann. N. Y. Acad. Sci. 1125, 345–352, 10.1196/annals.1419.004 (2008).
Siddique, T., Penner, T., Klassen, J., Nesbo, C. & Foght, J. M. Microbial communities involved in methane production from hydrocarbons in oil sands tailings. Environ Sci Technol 46, 9802–9810, 10.1021/es302202c (2012).
Liu, J.-F. et al. Analysis of Microbial Communities in the Oil Reservoir Subjected to CO2-Flooding by Using Functional Genes as Molecular Biomarkers for Microbial CO2 Sequestration. Frontiers in microbiology 6, 10.3389/fmicb.2015.00236 (2015).
Harner, N. K. et al. Microbial processes in the Athabasca Oil Sands and their potential applications in microbial enhanced oil recovery. J Ind Microbiol Biotechnol 38, 1761–1775, 10.1007/s10295-011-1024-6 (2011).
Callbeck, C. M. et al. Microbial community succession in a bioreactor modeling a souring low-temperature oil reservoir subjected to nitrate injection. Appl. Microbiol. Biotechnol. 91, 799–810, 10.1007/s00253-011-3287-2 (2011).
Bødtker, G., Lysnes, K., Torsvik, T., Bjørnestad, E. Ø. & Sunde, E. Microbial analysis of backflowed injection water from a nitrate-treated North Sea oil reservoir. J. Ind. Microbiol. Biotechnol. 36, 439–450, 10.1007/s10295-008-0515-6 (2009).
Kumaraswamy, R., Ebert, S., Gray, M. R., Fedorak, P. M. & Foght, J. M. Molecular- and cultivation-based analyses of microbial communities in oil field water and in microcosms amended with nitrate to control H2S production. Appl. Microbiol. Biotechnol. 89, 2027–2038 (2011).
Gieg, L. M., Jack, T. R. & Foght, J. M. Biological souring and mitigation in oil reservoirs. Appl Microbiol Biotechnol 92, 263–282, 10.1007/s00253-011-3542-6 (2011).
Simpson, D. R., Natraj, N. R., McInerney, M. J. & Duncan, K. E. Biosurfactant-producing bacillus are present in produced brines from Oklahoma oil reservoirs with a wide range of salinities. Appl. Microbiol. Biotechnol. 91, 1083–1093, 10.1007/s00253-011-3326-z (2011).
Li, G. Q. et al. Microbial Abundance and Community Composition Influence Production Performance in a Low-Temperature Petroleum Reservoir. Environ Sci Technol 48, 5336–5344 (2014).
Pham, V. D. et al. Characterizing microbial diversity in production water from an Alaskan mesothermic petroleum reservoir with two independent molecular methods. Environ Microbiol 11, 176–187, 10.1111/j.1462-2920.2008.01751.x (2009).
Li, H., Chen, S., Mu, B. Z. & Gu, J. D. Molecular Detection of Anaerobic Ammonium-Oxidizing (Anammox) Bacteria in High-Temperature Petroleum Reservoirs. Microb Ecol 60, 771–783, 10.1007/s00248-010-9733-3 (2010).
Tang, Y. Q. et al. Microbial communities in long-term, water-flooded petroleum reservoirs with different in situ temperatures in the Huabei Oilfield, China. Plos One 7, e33535 (2012).
Wang, L. Y. et al. Molecular analysis of the microbial community structures in water-flooding petroleum reservoirs with different temperatures. Biogeosciences 9, 5177–5203 (2012).
Gao, P. K. et al. Differences in microbial community composition between injection and production water samples of water flooding petroleum reservoirs. Biogeosciences 12, 3403–3414, 10.5194/bg-12-3403-2015 (2015).
Lenchi, N. et al. Diversity of Microbial Communities in Production and Injection Waters of Algerian Oilfields Revealed by 16S rRNA Gene Amplicon 454 Pyrosequencing. Plos One 8, e66588 (2013).
Lewin, A. et al. The microbial communities in two apparently physically separated deep subsurface oil reservoirs show extensive DNA sequence similarities. Environ Microbiol 16, 545–558, 10.1111/1462-2920.12181 (2014).
Ren, H. Y. et al. Comparison of microbial community compositions of injection and production well samples in a long-term water-flooded petroleum reservoir. Plos One 68, e23258 (2011).
Kuang, J. L. et al. Contemporary environmental variation determines microbial diversity patterns in acid mine drainage. Isme J 7, 1038–1050, 10.1038/ismej.2012.139 (2013).
Ge, Y. et al. Differences in soil bacterial diversity: driven by contemporary disturbances or historical contingencies? Isme J 2, 254–264, 10.1038/ismej.2008.2 (2008).
Briggs, B. R. et al. Seasonal patterns in microbial communities inhabiting the hot springs of Tengchong, Yunnan Province, China. Environ Microbiol 16, 1579–1591, 10.1111/1462-2920.12311 (2014).
Hatosy, S. M. et al. Beta diversity of marine bacteria depends on temporal scale. Ecology 94, 1898–1904 (2013).
Stegen, J. C., Lin, X., Konopka, A. E. & Fredrickson, J. K. Stochastic and deterministic assembly processes in subsurface microbial communities. Isme J 6, 1653–1664, 10.1038/ismej.2012.22 (2012).
Liang, Y. et al. Over 150 years of long-term fertilization alters spatial scaling of microbial biodiversity. Mbio 6, 10.1128/mBio.00240-15 (2015).
Lin, W., Wang, Y., Gorby, Y., Nealson, K. & Pan, Y. Integrating niche-based process and spatial process in biogeography of magnetotactic bacteria. Sci Rep 3, 1643, 10.1038/srep01643 (2013).
Hollibaugh, J. T. et al. Seasonal variation in the metatranscriptomes of a Thaumarchaeota population from SE USA coastal waters. Isme J 8, 685–698, 10.1038/ismej.2013.171 (2014).
Piceno, Y. M. et al. Temperature and injection water source influence microbial community structure in four Alaskan North Slope hydrocarbon reservoirs. Frontiers in microbiology 5, 409, 10.3389/fmicb.2014.00409 (2014).
Sharp, C. E. et al. Humboldt’s spa: microbial diversity is controlled by temperature in geothermal environments. Isme J 8, 1166–1174, 10.1038/ismej.2013.237 (2014).
Auguet, J. C., Barberan, A. & Casamayor, E. O. Global ecological patterns in uncultured Archaea. Isme J 4, 182–190, 10.1038/ismej.2009.109 (2010).
Lauber, C. L., Hamady, M., Knight, R. & Fierer, N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microbiol 75, 5111–5120, 10.1128/aem.00335-09 (2009).
N’Guessan, A. L. et al. Molecular analysis of phosphate limitation in Geobacteraceae during the bioremediation of a uranium-contaminated aquifer. Isme J 4, 253–266, 10.1038/ismej.2009.115 (2010).
Martiny, J. B., Eisen, J. A., Penn, K., Allison, S. D. & Horner-Devine, M. C. Drivers of bacterial beta-diversity depend on spatial scale. Proc Natl Acad Sci USA 108, 7850–7854, 10.1073/pnas.1016308108 (2011).
Fierer, N. & Jackson, R. B. The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci USA 103, 626–631, 10.1073/pnas.0507535103 (2006).
Gibbons, S. M. et al. Evidence for a persistent microbial seed bank throughout the global ocean. Proc Natl Acad Sci USA 110, 4651–4655, 10.1073/pnas.1217767110 (2013).
Zhao, L. X. et al. Characterization of microbial diversity and community in water flooding oil reservoirs in China. World. J. Microb. Biot. 28, 3039–3052, 10.1007/s11274-012-1114-2 (2012).
Al-Bahry, S. N. et al. Microbial consortia in oman oil fields: a possible use in enhanced oil recovery. J. Microbiol. Biotechnol. 23, 106–117 (2013).
Hubert, C. R. et al. Massive dominance of Epsilonproteobacteria in formation waters from a Canadian oil sands reservoir containing severely biodegraded oil. Environ Microbiol 14, 387–404, 10.1111/j.1462-2920.2011.02521.x (2012).
Zhang, T., Shao, M.-F. & Ye, L. 454 pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. 6, 1137–1147 (2012).
Zhang, F. et al. Impact of an indigenous microbial enhanced oil recovery field trial on microbial community structure in a high pour-point oil reservoir. Appl. Microbiol. Biotechnol. 95, 811–821 (2012).
Liu, Y. C. & Whitman, W. B. Metabolic, Phylogenetic and Ecological Diversity of the Methanogenic Archaea. Ann. N.Y. Acad. Sci. 1125, 171–189 (2008).
Sokolova, T. G. et al. Thermosinus carboxydivorans gen. nov., sp. nov., a new anaerobic, thermophilic, carbon-monoxide-oxidizing, hydrogenogenic bacterium from a hot pool of Yellowstone National Park. Int J Syst Evol Microbiol 54, 2353–2359, 10.1099/ijs.0.63186-0 (2004).
Watanabe, K., Watanabe, K., Kodama, Y., Syutsubo, K. & Harayama, S. Molecular characterization of bacterial populations in petroleum-contaminated groundwater discharged from underground crude oil storage cavities. Appl Environ Microbiol 66, 4803–4809 (2000).
Donachie, S. P., Bowman, J. P., On, S. L. W. & Alam, M. Arcobacter halophilus sp. nov., the first obligate halophile in the genus Arcobacter. Int J Syst Evol Micr 55, 1271–1277, 10.1099/ijs.0.63581-0 (2005).
Green, J. L. et al. Spatial scaling of microbial eukaryote diversity. Nature 432, 747–750, 10.1038/nature03034 (2004).
Bell, T. et al. Larger islands house more bacterial taxa. Science 308, 1884, 10.1126/science.1111318 (2005).
Horner-Devine, M. C., Lage, M., Hughes, J. B. & Bohannan, B. J. M. A taxa-area relationship for bacteria. Nature 432, 750–753 (2004).
Liu, B., Frostegard, A. & Bakken, L. R. Impaired reduction of N2O to N2 in acid soils is due to a posttranscriptional interference with the expression of nosZ. Mbio 5, e01383–01314, 10.1128/mBio.01383-14 (2014).
Lozupone, C. A. & Knight, R. Global patterns in bacterial diversity. Proc Natl Acad Sci USA 104, 11436–11440, 10.1073/pnas.0611525104 (2007).
Zhou, J. et al. Stochasticity, succession and environmental perturbations in a fluidic ecosystem. Proc Natl Acad Sci USA 111, E836–845, 10.1073/pnas.1324044111 (2014).
Watanabe, K., Kodama, Y. & Harayama, S. Design and evaluation of PCR primers to amplify bacterial 16S ribosomal DNA fragments used for community fingerprinting. J Microbiol Meth 44, 253–262 (2001).
Casamayor, E. O. et al. Changes in archaeal, bacterial and eukaryal assemblages along a salinity gradient by comparison of genetic fingerprinting methods in a multipond solar saltern. Environ Microbiol 4, 338–348, 10.1046/j.1462-2920.2002.00297.x (2002).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541 (2009).
DeSantis, T. Z. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72, 5069–5072, 10.1128/aem.03006-05 (2006).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267, 10.1128/Aem.00062-07 (2007).
De’ath, G. Multivariate regression trees: a new technique for modeling species–environment relationships. Ecology 83, 1105–1117 (2002).
Acknowledgements
We thank Mr. Jianjun Le and Ms. Fang Liu for sampling the production water in Daqing Oilfield, Mr. Haitao Ran for sampling the production water in Jilin Oilfield, Mr. Ben Zhao for sampling the production water in Liaohe Oilfield, Mr. Hongtao Qu for sampling the production water in Huabei Oilfield, Mr. Gangzheng Sun for sampling the production water in Shengli Oilfield, Mr. Kaiqiang Liang for sampling the production water in Yanchang Oilfield, Mr. Lixin Huang for sampling the production water in Qinghai Oilfield, Mr. Xuecheng Dai for sampling the production water in Xinjiang Oilfield, Mr. Qingxian Feng for sampling the production water in Dagang Oilfield. This study was supported by the National High Technology Research and Development Program of China (Grant No. 2013AA064402), the National Natural Science Foundation of China (Grant No. 41373074 and 31500414) and the Chinese Postdoctoral Science Foundation (Grant No. 2014M561175).
Author information
Authors and Affiliations
Contributions
P.K.G., G.Q.L. and T.M. designed the work and wrote the manuscript. P.K.G., H.M.T., Y.S.W., Y.S.L. and Y.L. performed the experiments and analyzed the data. J.X.X., B.Z., J.F.Z., G.Q.L. and T.M. carried out some experiments. All authors discussed and reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Gao, P., Tian, H., Wang, Y. et al. Spatial isolation and environmental factors drive distinct bacterial and archaeal communities in different types of petroleum reservoirs in China. Sci Rep 6, 20174 (2016). https://doi.org/10.1038/srep20174
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep20174
This article is cited by
-
Bacterial and Archaeal Community Distribution in Oilfield Water Re-injection Facilities and the Influences from Microorganisms in Injected Water
Microbial Ecology (2022)
-
Nitrogen has a greater influence than phosphorus on the diazotrophic community in two successive crop seasons in Northeast China
Scientific Reports (2021)
-
Bacterial and archaeal diversity in oil fields and reservoirs and their potential role in hydrocarbon recovery and bioprospecting
Environmental Science and Pollution Research (2021)
-
The linkage between methane production activity and prokaryotic community structure in the soil within a shale gas field in China
Environmental Science and Pollution Research (2020)
-
Metabolic capability and in situ activity of microorganisms in an oil reservoir
Microbiome (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
