
Abstract
Retinitis pigmentosa (RP) is a genetically highly heterogeneous retinal disease and one of the leading causes of blindness in the world. Next-generation sequencing technology has enormous potential for determining the genetic etiology of RP. We sought to identify the underlying genetic defect in a 35-year-old male from an autosomal-dominant RP family with 14 affected individuals. By capturing next-generation sequencing (CNGS) of 144 genes associated with retinal diseases, we identified eight novel DNA variants; however, none of them cosegregated for all the members of the family. Further analysis of the CNGS data led to identification of a recurrent missense mutation (c.403C > T, p.R135W) in the rhodopsin (RHO) gene, which cosegregated with all affected individuals in the family and was not observed in any of the unaffected family members. The p.R135W mutation has a reference single nucleotide polymorphism (SNP) ID (rs104893775) and it appears to be responsible for the disease in this large family. This study highlights the importance of examining NGS data with reference SNP IDs. Thus, our study is important for data analysis of NGS-based clinical genetic diagnoses.
Similar content being viewed by others


Introduction
Retinitis pigmentosa (RP) is the most common form of inherited retinopathy, with a prevalence of approximately 1 in 35001. It is a clinically and genetically heterogeneous group of eye diseases and one of the leading causes of blindness in the world. Clinically, the age at onset of symptoms is highly variable and ranges from childhood to mid-adulthood. Genetically, RP displays all three modes of Mendelian inheritance—autosomal dominant (adRP), autosomal recessive (arRP) and X-linked (XLRP)—, as well as digenic and mitochondrial inheritance modes2. Thus far, at least 280 genes have been identified as the cause of one form or another of inherited retinal disease2,3,4,5. This complex of genes and clinical features complicates the corresponding clinical diagnoses. For example, although rhodopsin mutations usually cause dominant RP, other rare rhodopsin mutations cause arRP6. Mutations in ABCA4 are found in most patients with autosomal-recessive Stargardt disease and studies have shown that mutations in the ABCA4 gene are responsible for a wide variety of other retinal dystrophy phenotypes, such as cone-rod dystrophy (CRD) and RP7,8.
Approaches based on stem cell and gene therapy hold a great deal of promise for treatment of retinal disease; however, currently, no effective therapies have been identified3,9,10,11. Thus, identifying new genes and mutations for clinical genetic diagnosis and prenatal diagnosis of RP is crucial. Because at least 280 genes have been identified as the cause of various forms of inherited retinal disease, identifying underlying mutations with traditional methods has been challenging3,12. Next-generation sequencing (NGS) is a highly versatile and effective approach to detecting novel disease-causing genes and mutations in families with different diseases, including retinopathies13,14. Our previous study provided support for using NGS as an effective approach to distinguishing cone-rod dystrophy and Stargardt disease, two inherited retinopathies with overlapping clinical symptoms7.
In the present study, patients and unaffected individuals from a five-generation family with fourteen individuals diagnosed with RP were recruited. We sought to identify the underlying genetic defect in this family by capturing next-generation sequencing (CNGS) and Sanger sequencing. These approaches led to identification of eight novel DNA variants, but the variants did not cosegregate with all affected individuals in the family, indicating none of them was the disease-causing mutation. Thus, re-analysis of the data was necessary because the disease-causing gene may already be identified or be outside of the panel of the captured genes. To rule out the first possibility, we retrieved the data from the CNGS. We observed a recurrent mutation (p.R135W) in the rhodopsin (RHO) gene with reference SNP ID (rs104893775, http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?rs=104893775).
Results
Clinical data
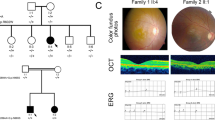
The family in this RP study included 14 affected individuals in a five-generation family (Fig. 1A) originating from Henan province, China. The proband was a 35-year-old male (indicated with black arrow in Fig. 1A). His decimal best-corrected visual acuity (BCVA) was 0.3 (Table 1). Fundus examination revealed slight intraretinal bone spicule pigmentation (green arrow in Fig. 1B). 3-D optical coherence tomography (OCT) scans revealed the degree of thinning of the outer retinal layer (red arrow in Fig. 1C), indicating the dysfunction of the photoreceptors. Electro-retinogram (ERG) results showed both cone and rod functions were seriously affected (Fig. 1D). Night-blindness was also reported. The clinical data of the proband and family members are shown in Fig. 1, Figure S1 and Table 1. According to the available clinical data, three affected individuals had RP-specific symptoms, including night blindness, thinning of the outer retinal layer and serious effects of both cone and rod functions. As for the early stages of the disease, recordable cone function may still be observed (Figure S1). All three affected individuals had similar levels of best corrected visual acuities while the corresponding astigmatism was dramatically different. Thus, based on the clinical manifestations, the diagnosis of RP is appropriate.

Pedigree and clinical manifestations.
(A) Pedigree of autosomal-dominant RP family. Black circles (females) and black squares (males) represent affected individuals. Unaffected individuals are not shaded. Black lines denote deceased. An individual is identified by generation number and the numbers below the symbols. The arrow marks the proband. (B) Color fundus photographs demonstrate slight intraretinal bone spicule pigmentation has been observed. (C) Outer retina layers of the patient on OCT are disrupted and disorganized, indicating the dysfunction of the photoreceptors. (D) Non-recordable ERG has been detected, indicating the functions of both cone and rod are severely reduced.
DNA variants in CNGS
We used a CNGS platform for screening 144 genes associated with retinal diseases. After initial analysis (http://122.228.158.106/exomeassistant) of candidate mutations, we identified eight distinct DNA variants in the samples (Table S1). We then performed cosegregation analysis to determine whether these DNA variants could be responsible for the disease. Sanger sequencing of the amplified fragments harboring the candidate mutations was performed to test for cosegregation. None of the eight mutations cosegregated with all affected individuals in the family.
RHO mutation screening
Because no cosegregating mutation was identified in the initial sequencing data analysis, we re-analyzed the CNGS data from the CNGS experiment. Because it has been reported that mutations in the RHO gene account for 7.7% to 40% of all adRP cases in different populations6,15,16, we investigated whether the mutation in the family of study was located in the area of missing sequence coverage in the RHO gene. This strategy was similar to one used in our recent study in which we successfully filled in missing sequence data for the OPA1 gene from CNGS data while searching for disease-causing genes linked with autosomal dominant optic atrophy17. In this present study, the CNGS data showed good coverage of the RHO gene (Table S2). A single RHO DNA variant was detected in the sequence (c. 403C >T, p.R135W), with a reference SNP ID (rs104893775).
Surprisingly, it has been reported that the p.R135W mutation in RHO is a disease-causing RP mutation in different populations18,19,20,21,22. Thus, we used Sanger sequencing to confirm that the proband had the mutation (Fig. 2A). We found that codon 135, where the mutation (p.R135W) occurred, was located within a phylogenetically conserved region (Fig. 2B). Then, we performed a cosegregation study using the restriction fragment length polymorphism (RFLP) method. Specifically, fragment harboring the mutation was amplified with genomic DNA from the family members. The purified PCR products were digested with Bsrb 1, which causes differential patterns of bands on agarose gels between the unaffected and affected individuals. Compared to the unaffected individuals with fragments of 241 and 169 bp, affected individuals had 241,169 and 410 bp fragments (Fig. 2C). RFLP analysis confirmed this mutation cosegregated with all affected individuals in the family and was not present in any of the unaffected family members (Fig. 2D) or the 1402 normal controls (from in-house exome databases with 1402 samples from a Chinese population).

Mutation analysis.
(A) DNA-sequence chromatograms of the affected family members with RP disease. The heterozygous peak of the mutation is indicated by red arrows. (B) Multiple-sequence alignment in RHO from different species indicating the mutation (p.W135R) in RP patient is located within a highly conserved region. (C) Schematic diagram of RFLP for the affected and unaffected individuals. (D) RFLP results showed p.W135R mutation in RHO cosegregated with all affected individuals in the family and was not observed in any of the unaffected family members. The purified PCR products were digested with Bsrb 1, showing the unaffected individual with 240 and 169 bp fragments as opposed to the affected individual with 241, 169 and 410 bp.
In addition, we screened the remainder of the coding sequence of RHO (Table S3) and no additional sequence changes were detected. Furthermore, we used online bioinformatics software tools designed to distinguish between functionally neutral and deleterious amino acid changes in mutagenesis studies and human polymorphisms. We predicted the amino acid substitution in RHO could have a phenotypic effect and found that the substitution at position 135 from R to W to be deleterious (Table S4). Thus, the p.R135W mutation in RHO is a disease-causing mutation in this family.
Discussion
NGS offers a cost-effective approach to detecting disease-causing genes for clinically and genetically heterogeneous disorders or drug resistant genes23,24. However, analytical data analysis for identifying mutation from NGS data remains a challenge because NGS could provide massive amounts of data25. In addition, the success of using NGS data may be inconsistent because of different disease inheritance modes. Specifically, because the pathogenic mutations for autosomal-recessive disorders may be recorded in the dbSNP database, DNA variants with SNP IDs could be assigned as candidate mutations26,27. As for autosomal-dominant disorders, traditionally, to identify a mutation, novel DNA variants from NGS data are prioritized for selection as disease-causing genes because only a single mutation would be responsible for the disease. In this study, we showed ignoring DNA variants with reference SNP IDs may lead to missing the disease-causing mutation. The incorrect conclusion—that no mutation is present—may lead to the further time and expense of whole-exome or whole-genome sequencing. This study highlights the importance of suitable treatment of NGS data with reference SNP IDs for clinical genetic diagnosis.
For clinical laboratories, determining which sequence variants are pathogenic is difficult, particularly if no simple functional assays are readily available to determine the phenotypic effects of specific variations28. The RHO DNA mutation (c. 403C >T, p.R135W) has been reported in at least five studies with different populations18,19,20,21,22. In this present case, it cosegregates with all affected individuals in the large family examined and was not observed in any of the unaffected family members or normal controls. The functional studies also showed that Arg135 residue is an important interaction site with transducin29. The bovine p.R135W rhodopsin mutant was unable to activate the G protein in vitro, contrary to the wild-type rhodopsin30. In addition, the glycosylation state of the p.R135W rhodopsin mutant is the most defective19. As for the two different mutations at Arg135 residues—R135L and R135W—, both result in diffuse severe RP disease, but R135W causes more severe and a more rapidly progressive RP than does R135L. Therefore, p.R135W could be considered a causative mutation beyond reasonable doubt, despite having a reference SNP ID (rs104893775). For newly discovered or rare gene variants with a reference SNP ID, we still need to examine the candidate gene carefully. As more data are deposited and more papers published, the assignment of the mutation will become easier.
In our previous study, we showed missing sequence coverage of some exons in the PROM1 gene from CNGS-based molecular diagnosis of putative Stargardt disease, one of the most common genetic forms of juvenile or early adult onset macular degeneration7. Further, with deep sequencing, missing coverage of some regions is common31. Therefore, before searching for the disease-causing mutations, the coverage of the targeted sequence should be checked. Missing coverage may vary case by case, depending on the gene. In this study, we showed the RHO gene, when examined with CNGS, was well covered in our data set (Table S2).
More than 280 genes are linked with RP, including 25 genes are known to cause adRP3,5. Over 1000 mutations have been reported in these 25 genes5. Theoretically, each one accounts for a small percentage of all disease-causing genes. While, RHO gene has a pretty high frequency of 7.7–25% in adRP6,15,16; therefore, it could be treated as a “major” gene for adRP. In terms of diagnosis of the autosomal-dominant inherited disease, this study highlights the “major” gene should be prioritized for Sanger sequencing before NGS if its size is small because it has a relatively good chance of being easily identified. The human RHO gene has only five exons and four pairs of primers could be used for amplifying the whole coding sequence and flanking intron sequences (Table S3). Four sequencing reactions could fully cover all the coding and flanking intron sequences, thus being a cost-effective approach for adRP diagnosis.
To our knowledge, this is the first case to report that a disease-causing mutation for autosomally dominant inherited disease could also have a reference SNP ID. In addition, we suggest the screening strategy for autosomally dominant inherited disease should be to screen the major genes for mutations before performing NGS. Thus, the findings of this study are applicable to screening mutations for RP, paving the way for gene therapy and prenatal RHO-causing RP diagnosis.
Methods
Patient Recruitment
This study conformed to the tenets of the Declaration of Helsinki. It was approved by the Ethics Committee of our hospital. Written informed consent was obtained from the recruited individuals. The study included 16 participants, including seven affected individuals, seven unaffected individuals and two spouses (Fig. 1). All experiments were performed in accordance with the approved guidelines. Optical coherence tomography (OCT), ERG and fundus examinations were performed as routine retinal ophthalmic examinations. A five ml venous blood sample was drawn into an ethylenediaminetetraacetic acid (EDTA) sample tube. Genomic DNA was extracted from peripheral blood leukocytes using the standard phenol/chloroform extraction protocols.
Capturing Next-Generation Sequencing and Bioinformatics Analysis
Capturing next-generation sequencing was performed as previously described7,17. The enriched libraries were sequenced on an Illumina Solexa HiSeq 2000 sequencer for paired-end reads of 100 bp. Briefly, we used the Solexa QA, the cutadapt (http://code.google.com/p/cutadapt/), SOAP aligner32, BWA (bio-bwa.sourceforge.net/)33 and GATK programs (https://www.broadinstitute.org/gatk/)34, to retrieve, align and identify SNPs and insertions or deletions (InDels). The GRCh37/hg19 version of the human genome was used for sequence alignment. SNPs and InDels were annotated using the exome-assistant program (http://122.228.158.106/exomeassistant). Nonsynonymous variants were evaluated as previously described7,17. Specifically, nonsynonymous variants were evaluated through three algorithms—SIFT (http://sift.jcvi.org/), PolyPhen (http://genetics.bwh.harvard.edu/pph2/) and PANTHER (http://www.pantherdb.org/tools/csnpScoreForm.jsp)—to determine pathogenicity. Finally, from the Chinese population, 1402 individuals without obvious eye diseases were used as normal controls.
Cosegregation study
Primers (RHO E2F/2R, Table S3) were used for amplifying the fragment harboring the mutation. PCR was performed with the genomic DNA from the family and its product was purified using standard protocols. Sanger sequencing and RFLP were performed as standard protocols. Specifically, concerning RFLP, the purified PCR products were digested with Bsrb 1, which indicated the unaffected individual with 241 and 169 bp fragments, as compared to the affected individual with 241,169 and 410 bp.
Nomenclature of mutations
For mutations, nucleotide numbering reflects cDNA numbering, with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence, according to journal guidelines (www.hgvs.org/mutnomen). The initiation codon is codon 1.
Multiple-sequence alignment and mutation analysis
Using the NCBI and UCSC websites, we obtained multiple-sequence alignment of RHO protein in various species with DNAMAN biosoftware (Lynnon Biosoft, Quebec, Canada), including Homo sapiens, Bos taurus, Rattus norvegicus, Mus musculus and Danio rerio.
Additional Information
How to cite this article: Yu, X. et al. Identification of a rhodopsin gene mutation in a large family with autosomal dominant retinitis pigmentosa. Sci. Rep. 6, 19759; doi: 10.1038/srep19759 (2016).
References
Sullivan, L. S. et al. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: a screen of known genes in 200 families. Invest Ophthalmol Vis Sci 47, 3052–64 (2006).
Chakarova, C. F. et al. Mutations in HPRP3, a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa. Hum Mol Genet 11, 87–92 (2002).
Benjaminy, S., Kowal, S. P., MacDonald, I. M. & Bubela, T. Communicating the promise for ocular gene therapies: challenges and recommendations. Am J Ophthalmol 160, 408–415 e2 (2015).
Daiger, S. P., Sullivan, L. S. & Bowne, S. J. Genes and mutations causing retinitis pigmentosa. Clin Genet 84, 132–41 (2013).
Daiger, S. P., Bowne, S. J. & Sullivan, L. S. Genes and Mutations Causing Autosomal Dominant Retinitis Pigmentosa. Cold Spring Harb Perspect Med (2014).
Daiger, S. P., Bowne, S. J. & Sullivan, L. S. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol 125, 151–8 (2007).
Zhang, X. et al. Molecular diagnosis of putative Stargardt disease by capture next generation sequencing. PLoS One 9, e95528 (2014).
Maugeri, A. et al. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am J Hum Genet 67, 960–6 (2000).
Schwartz, S. D. et al. Embryonic stem cell trials for macular degeneration: a preliminary report. Lancet 379, 713–20 (2012).
Maguire, A. M. et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med 358, 2240–8 (2008).
Maguire, A. M. et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet 374, 1597–605 (2009).
Daiger, S. P. et al. Targeted high-throughput DNA sequencing for gene discovery in retinitis pigmentosa. Adv Exp Med Biol 664, 325–31 (2010).
Renkema, K. Y., Stokman, M. F., Giles, R. H. & Knoers, N. V. Next-generation sequencing for research and diagnostics in kidney disease. Nat Rev Nephrol 10, 433–44 (2014).
Ratnapriya, R. & Swaroop, A. Genetic architecture of retinal and macular degenerative diseases: the promise and challenges of next-generation sequencing. Genome Med 5, 84 (2013).
Yang, G. et al. Spectrum of rhodopsin gene mutations in Chinese patients with retinitis pigmentosa. Mol Vis 20, 1132–6 (2014).
Katagiri, S. et al. RHO Mutations (p.W126L and p.A346P) in Two Japanese Families with Autosomal Dominant Retinitis Pigmentosa. J Ophthalmol 2014, 210947 (2014).
Zhang, L. et al. A recurrent deletion mutation in OPA1 causes autosomal dominant optic atrophy in a Chinese family. Sci Rep 4, 6936 (2014).
Kim, C. et al. Microarray-based mutation detection and phenotypic characterization in Korean patients with retinitis pigmentosa. Mol Vis 18, 2398–410 (2012).
Iannaccone, A. et al. Retinitis pigmentosa associated with rhodopsin mutations: Correlation between phenotypic variability and molecular effects. Vision Res 46, 4556–67 (2006).
Jacobson, S. G., Kemp, C. M., Sung, C. H. & Nathans, J. Retinal function and rhodopsin levels in autosomal dominant retinitis pigmentosa with rhodopsin mutations. Am J Ophthalmol 112, 256–71 (1991).
Pannarale, M. R. et al. Autosomal-dominant retinitis pigmentosa associated with an Arg-135-Trp point mutation of the rhodopsin gene. Clinical features and longitudinal observations. Ophthalmology 103, 1443–52 (1996).
Wu, J. et al. Whole exome sequencing reveals genetic predisposition in a large family with retinitis pigmentosa. Biomed Res Int 2014, 302487 (2014).
Roosing, S. et al. Disruption of the basal body protein POC1B results in autosomal-recessive cone-rod dystrophy. Am J Hum Genet 95, 131–42 (2014).
Thress, K. S. et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med 21, 560–2 (2015).
Rusk, N. Focus on next-generation sequencing data analysis. Forward. Nat Methods 6, S1 (2009).
Gilissen, C., Hoischen, A., Brunner, H. G. & Veltman, J. A. Disease gene identification strategies for exome sequencing. Eur J Hum Genet 20, 490–7 (2012).
Nishiguchi, K. M. & Rivolta, C. Genes associated with retinitis pigmentosa and allied diseases are frequently mutated in the general population. PLoS One 7, e41902 (2012).
Rivera, A. et al. A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am J Hum Genet 67, 800–13 (2000).
Li, J., Edwards, P. C., Burghammer, M., Villa, C. & Schertler, G. F. Structure of bovine rhodopsin in a trigonal crystal form. J Mol Biol 343, 1409–38 (2004).
Shi, W. et al. Rhodopsin arginine-135 mutants are phosphorylated by rhodopsin kinase and bind arrestin in the absence of 11-cis-retinal. Biochemistry 37, 4869–74 (1998).
Ross, M. G. et al. Characterizing and measuring bias in sequence data. Genome Biol 14, R51 (2013).
Li, R., Li, Y., Kristiansen, K. & Wang, J. SOAP: short oligonucleotide alignment program. Bioinformatics 24, 713–4 (2008).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–60 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–303 (2010).
Acknowledgements
We thank the patients for their participation in this project. We are indebted to Dr. Jinyu Wu (Institute of Genomic Medicine, Wenzhou Medical University) for bioinformatics analysis. This work was supported by grants from the Chinese National Program on Key Basic Research Project (973 Program, 2013CB967502, F.G.), the Natural Science Foundation of China (81201181/H1818, F.G.), Zhejiang provincial & Ministry of Health research fund for medical sciences (WKJ2013-2-023, F.G.), Wenzhou city grant (Y20140633 F.G.), Wenzhou Medical University grant (QTJ12011, F.G.) and Eye Hospital at WenZhou Medical University (YNCX201309,JL.X., YNZD201502, F.G.).
Author information
Authors and Affiliations
Contributions
F.G. conceived the idea, X.P.Y., W.S., L.L.C., Y.F.W., D.C., X.T.H., J.L.X., L.M.X. and Y.M.W. performed the experiments, J.Q. and F.G. performed data analyses and F.G. wrote the manuscript. All authors have read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yu, X., Shi, W., Cheng, L. et al. Identification of a rhodopsin gene mutation in a large family with autosomal dominant retinitis pigmentosa. Sci Rep 6, 19759 (2016). https://doi.org/10.1038/srep19759
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep19759
This article is cited by
-
Potential therapeutic strategies for photoreceptor degeneration: the path to restore vision
Journal of Translational Medicine (2022)
-
Replication of a rare risk haplotype on 1p36.33 for autism spectrum disorder
Human Genetics (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
