
Abstract
Nucleic acid amplification testing (NAAT) enables rapid and sensitive diagnosis of tuberculosis (TB), which facilitates treatment and mitigates transmission. Nucleic acid extraction from sputum constitutes the greatest technical challenge in TB NAAT for near-patient settings. This report presents preliminary data for a semi-automated sample processing method, wherein sputum is disinfected and liquefied, followed by PureLyse® mechanical lysis and solid-phase nucleic acid extraction in a miniaturized, battery-operated bead blender. Sputum liquefaction and disinfection enabled a >104 fold reduction in viable load of cultured Mycobacterium tuberculosis (M.tb) spiked into human sputum, which mitigates biohazard concerns. Sample preparation via the PureLyse® method and a clinically validated manual method enabled positive PCR-based detection for sputum spiked with 104 and 105 colony forming units (cfu)/mL M.tb. At 103 cfu/mL sputum, four of six and two of six samples amplified using the comparator and PureLyse® method, respectively. For clinical specimens from TB cases and controls, the two methods provided 100% concordant results for samples with  1 mL input volume (N = 41). The semi-automated PureLyse® method therefore performed similarly to a validated manual comparator method, but is faster, minimally instrumented and can be integrated into TB molecular diagnostic platforms designed for near-patient low-resource settings.
1 mL input volume (N = 41). The semi-automated PureLyse® method therefore performed similarly to a validated manual comparator method, but is faster, minimally instrumented and can be integrated into TB molecular diagnostic platforms designed for near-patient low-resource settings.
Similar content being viewed by others

Introduction
Tuberculosis (TB) is a major health threat worldwide with an estimated 9 million incident active TB cases and 1.5 million TB-associated deaths in 20131, mainly in resource-limited settings within high-burden countries in Africa, Asia and Eastern Europe. Despite international efforts2, active TB continues to be significantly under-diagnosed, or is diagnosed long after becoming symptomatic and infectious, which compromises the ability to effectively treat patients and curb transmission3,4,5.
In most endemic regions, diagnosis of active pulmonary TB relies on smear microscopy, which is relatively simple and low cost, but suffers from insufficient sensitivity and specificity6,7. Culture-based methods, the current gold standard for TB diagnosis, are highly sensitive but expensive, require biosafety level 3 (BSL3) laboratory infrastructure and the time to result is usually several weeks. As a result, neither microscopy nor culture is sufficient for effective case finding, patient management and containment of TB transmission.
Nucleic acid amplification testing (NAAT) is becoming more integral to TB diagnostics in developed and developing countries8. NAAT enables sensitive and specific TB diagnosis4,9,10, can identify drug resistance mutations and in principle can provide results during the same patient visit, which reduces loss to follow-up. However, worldwide implementation of TB NAAT is hampered by its relatively high cost and complexity8. TB NAATs in use or in the development pipeline vary in complexity11 and entail either laboratory-developed tests12,13,14, or commercial systems9,15,16,17,18,19 based on the polymerase chain reaction (PCR)9,12,19,20,21, or isothermal amplification methods, such as loop-mediated isothermal amplification (LAMP)15,22, transcription-mediated amplification (TMA)18, cross-priming amplification (CPA)16, or helicase-dependent amplification (HDA)17,23.
The GeneXpert MTB/RIF is the most widely used TB NAAT system, following WHO endorsement for TB diagnosis and rifampin resistance testing and global roll-out starting in 201124. This highly automated platform performs nucleic acid sample preparation and hemi-nested PCR amplification with real-time detection25,26, with all reagents on board. The GeneXpert is recommended for use in district and sub-district level laboratories of countries where TB and MDR-TB are prevalent. However, the instrument and consumables cost remains substantial and the infrastructure required cannot be accommodated in low-resource microscopy centers and primary care settings that serve the majority of the affected patient population. Several other NAAT systems are in late-stage development or on the market for TB diagnosis8,15,16. However, these systems lack integration of sample preparation with amplification and detection in a fully automated format, posing implementation challenges in low-resource, remote primary care settings. Methods with cumbersome and lengthy multi-step processes are error-prone, require skilled users and in most cases additional consumables and laboratory instrumentation (e.g. centrifuges and heat baths) which may not be readily available27. Furthermore, low-resource settings often do not have uninterrupted power, which is required to operate such instruments7. The integration of sample preparation therefore is a significant challenge and bottleneck in enabling TB NAAT in near-patient low-resource settings. Minimally instrumented, fully enclosed and battery-operated sample preparation technologies with a small footprint are ideal for such settings.
Many current TB NAAT methods process sputum specimens analogous to culture-based protocols26. First, raw sputum is liquefied, usually using N-acetyl-L-cysteine (NALC) and/or NaOH, to enable manipulation of the viscous matrix. Culture-based TB diagnosis requires sample decontamination, which preserves at least some live mycobacteria but inactivates other microorganisms which would otherwise overgrow the slow-growing mycobacteria. However, to protect the operator in low-resource settings with limited biosafety precautions, it is preferable for TB NAAT to disinfect the sputum, i.e. to render all pathogens including mycobacteria non-viable. Several reagents have been reported to effectively render Mycobacterium tuberculosis (M.tb) non-viable including sodium hypochlorite, povidone iodine and hydrogen peroxide28,29, or in the case of the GeneXpert MTB/RIF assay, isopropanol with NaOH26. For some TB NAAT methods, the liquefied sputum is centrifuged to pellet bacteria12,13,22 and the pellet is then washed and re-suspended, to remove impurities and in some cases concentrate the bacteria in a smaller volume30. This approach resembles culture- and microscopy-based TB diagnostic methods, but centrifugation is difficult to automate. In the GeneXpert MTB/RIF assay, bacteria in the processed sputum are separated and concentrated through automated filtration and washing9,25.
Mycobacteria have thick waxy cell envelopes that are resistant to many conventional chemical or enzymatic lysis methods. Lysis of mycobacteria in most cases entails either heating the sample to ≥80 °C for 20 min to 1 h12,31, or mechanical disruption through sonication25,32,33 or high-energy bead beating32,34. If mycobacteria have been isolated from sputum prior to lysis, the lysed material is typically used directly for nucleic acid amplification. Alternatively, if mycobacteria are lysed in the sputum sample, the DNA can be purified by removing inhibitors e.g. through adsorption to a zeolite matrix15, through standard solid-phase extraction methods13,14 or sequence-specific capture23,35.
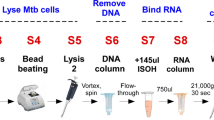
Claremont BioSolutions (ClaremontBio) developed a miniaturized, battery-operated bead beating system for mechanical pathogen lysis, called the OmniLyse® device. In previous work, this device was shown to effectively disrupt tough-walled microorganisms, such as M.tb complex bacteria and Bacillus subtilis spores and liberate their nucleic acids in a format suitable for PCR amplification34. ClaremontBio’s miniaturized bead beating system (Fig. 1) can also perform solid-phase DNA extraction, using the PureLyse® technology36, which does not require chaotropic salts or organic solvents that can inhibit downstream polymerase amplification37,38.

Disposable miniaturized battery-operated PureLyse® bead blender for mechanical cell lysis and solid-phase nucleic acid extraction.
Sample in binding buffer, wash and elution buffer are introduced at the sample inlet (a) and flow through the PureLyse® chamber (b) containing beads, micro-motor and impeller, exiting via the outlet (c). Battery pack (Bat-Pac) (d) connected to blender motor via connectors (e, f). Printed with permission from Claremont BioSolutions.
This report describes a novel nucleic acid sample preparation method from sputum which can be coupled with PCR to detect M.tb genomic DNA. The method incorporates sample disinfection and liquefaction, followed by mechanical lysis and solid-phase extraction of liberated nucleic acids using the PureLyse® technology. This semi-automated approach is compared to a clinically validated manual sample preparation method of sputum liquefaction, isolation of bacteria via centrifugation and heat lysis to liberate nucleic acids, developed by the Wadsworth Center at the New York State Department of Health12. The method described herein can be completed in <20 min, much faster than the comparator method, uses disposable battery-operated components, protects users by disinfecting samples at the outset and is suitable for automation. In ongoing efforts, we are integrating this method with DNA amplification and detection in a disposable cartridge and portable battery-operated instrument39,40, which has the potential to facilitate near-patient diagnosis of TB in resource-limited settings.
Results
We developed a sputum disinfection and liquefaction method, based on trisodium phosphate (TSP) as liquefaction reagent33 and povidone iodine (PVI) as disinfectant28,29. Various formulations of these components were explored, along with necessary incubation times to achieve sample liquefaction and mycobactericidal properties. We conducted a kill study to quantify the effectiveness of the protocol at inactivating M.tb complex cells in sputum, as described in the Methods section. Sputum samples spiked with M.tb H37Rv were treated using the disinfection/liquefaction protocol and replicates of undiluted and 10-fold diluted samples were plated. Treated spiked sputum samples contained a few M.tb colonies (<10) on some plates streaked with undiluted sample, but no colonies were observed for the diluted samples (Table 1). Based on control experiments in buffer without disinfection, each spiked sample contained >106 cfu/mL M.tb H37Rv in the final suspension, of which >105 cfu M.tb H37Rv were plated for the undiluted samples. Therefore, we obtained a >4-log reduction in viability relative to these controls. Furthermore, non-mycobacterial colonies observed in unprocessed sputum controls were not observed in the disinfected samples, suggesting a broad microbicidal effect.
In conjunction with this sputum disinfection and liquefaction method, a rapid sputum sample preparation method was employed, using ClaremontBio’s PureLyse® system to lyse M.tb and extract nucleic acids in a miniaturized and minimally instrumented format, with a 3-step protocol that takes less than 10 minutes to complete. The PureLyse® cartridge (Fig. 1) contains a micro-motor equipped with a precision-cut impeller capable of operating at up to 30,000 rpm with power supplied by a 6 V battery pack. The cartridge is packed with beads to generate shear forces sufficient for mechanical lysis of tough-walled organisms and to bind and release DNA under specific buffer conditions which enables solid-phase nucleic acid extraction34,36.
The PureLyse® protocol (liquefaction, disinfection and nucleic acid extraction) was compared to an established and clinically validated protocol for nucleic acid extraction from sputum for molecular TB diagnosis, developed by Halse et al.12. For initial analytical validation, cultured M.tb H37Ra was spiked into M.tb-negative human sputum at three concentrations and processed using the PureLyse® and comparator sample preparation methods. Nucleic acid from samples purified by each method were amplified and detected via the qPCR protocol of Halse et al. (Fig. 2)12.

Schematic diagram of the experimental design comparing the comparator12 and PureLyse® sample preparation methods.
TB-negative pooled sputum (~1 mL) samples were spiked with 105, 104, 103, or 0 cfu M.tb H37Ra and processed by both methods. Extracted nucleic acids were then amplified and detected using qPCR, as described by Halse et al.12.
The PureLyse® and comparator sample preparation methods performed comparably for sputum samples spiked with 104 and 105 cfu/mL H37Ra (Fig. 3). At these two concentrations, 100% of the samples (N = 6) amplified by both methods, with comparable Cq values [no statistically significant difference, p = 0.283 (104 cfu/mL) and p = 0.054 (105 cfu/mL), paired sample T-test, 2-tailed]. At 103 cfu/mL, four of six samples amplified using the comparator method whereas two of six samples amplified using the PureLyse® method. A sample’s extract was considered positive if at least one of two qPCR technical replicates yielded a positive result. We observed no substantial inhibition for either extraction method, as the average internal amplification control Cq’s were comparable for samples processed via the PureLyse® method (30.37 ± 0.31), comparator method (29.99 ± 0.24) and for the DNA standards and no template controls (29.83 ± 0.15), with no statistically significant difference (p = 0.0564 (PureLyse®), p = 0.328 (comparator), One-way ANOVA]. None of the negative controls showed false amplification.

qPCR detection of M.tb H37Ra spiked into sputum, extracted via the comparator (black diamond) and PureLyse® methods (white square): mean and standard deviation from six biological replicates, with two technical replicates each.
At 105 and 104 cfu/mL, all replicate samples amplified. At 103 cfu/mL, four of six samples and two of six samples amplified for the comparator and PureLyse® methods, respectively.
To test the performance of the PureLyse® protocol on clinical sputum specimens from TB positive and negative patients, a similar experimental approach was used to analyze 46 sputum specimens collected from patients of the Seattle-King County Tuberculosis Control Clinic. Smear and culture results were not available for each of these individual specimens. Smear and culture analysis was only performed on three initial samples used for laboratory diagnosis, which were collected before the study samples were obtained. Some of the study samples from patients with active TB were therefore expected to be negative for M.tb DNA by the Purelyse® and comparator methods due to normal fluctuations in bacillary load. Of the 30 specimens obtained from TB positive subjects, 24 samples were positive and five samples were negative following sample preparation by the comparator method. One sample was excluded from further analysis due to a technical error during processing. All 16 specimens from TB-negative subjects gave negative qPCR results using both methods.
Of the 24 samples determined to be positive based on the comparator method, 22 yielded positive qPCR results following PureLyse® sample preparation, while two samples yielded negative qPCR results. Both of the PureLyse® method “false negatives” had sample input volumes <1 mL (a deviation from the standard protocol) and also gave weak signals after extraction via the comparator method. Overall, this pilot clinical study resulted in a true positive fraction (sensitivity relative to the comparator method) of 91.7% (95% Confidence Interval (CI): 71.5–98.5%) when considering all samples (Table 2, “All Samples”), or 100% (95% CI: 80–100%) when considering only samples with input volume >1mL (Table 2, “Samples of ≥1 mL volume”). The true negative fraction (specificity) relative to the comparator method was 100% in both cases (95% CI: 80.8–100%). Comparable average internal amplification control Cq values were observed for the PureLyse® method (28.33 ± 0.42), comparator method (28.04 ± 0.78) and for the DNA standards and no template controls (28.25 ± 0.34), which indicates negligible inhibition for either sample extraction method.
Discussion
This report presents preliminary analytical and clinical studies, comparing semi-automated PureLyse® sputum sample preparation to an established and clinically validated manual sample preparation method described by Halse et al.12. In the analytical evaluation, the comparator method performed slightly better for sputum spiked with M.tb H37Ra at 103 cfu/mL, the lowest concentration tested, while identical performance was observed at 104 and 105 cfu/mL. Both methods gave concordant results when performed on patient-derived clinical samples, except when the sputum sample input volume was less than 1 mL, for which some samples did not amplify following PureLyse® sample preparation and amplified late following the comparator sample preparation method. Since substantial inhibition was not observed using either method, discrepancies are likely linked to the DNA extraction yield. Ongoing optimization to improve the sensitivity of the PureLyse®-based sputum sample preparation method achieved promising results through refinements in disinfection and liquefaction composition, lysis and binding configuration and buffer composition.
The PureLyse® sputum sample preparation method described herein improves the occupational safety of DNA extraction from sputum for molecular diagnosis of TB. In addition to a >104-fold reduction in M.tb viability, other microorganisms in sputum were rendered uncultivable on Middlebrook media after <10 minutes of treatment. In contrast, many sputum sample preparation protocols, including the comparator method, are not microbicidal until further downstream in the process. The GeneXpert MTB/RIF assay includes a sample disinfection step, enabling >106 fold reduction in viable M.tb bacterial load26. Through further improvements to our current disinfection method, we anticipate that a similar efficacy can be obtained.
While there are limitations attributed to small data sets for both the analytical and clinical studies and further validation of the PureLyse® extraction system is required following subsequent optimization efforts, the preliminary data demonstrate the potential utility in TB NAAT diagnostics. The PureLyse® sputum sample preparation method is battery powered, minimally instrumented and does not require additional equipment such as centrifuges and heat baths. In ongoing efforts, the PureLyse® protocol and mechanical design are being incorporated into a cartridge to enable fully integrated sample preparation, isothermal amplification and lateral flow detection, controlled by a portable, inexpensive instrument39,40. Such devices could enable rapid TB diagnosis at the point-of-care in low-resource, high-burden areas globally.
Methods
Sample Sources and General Study Design
Analytical studies were performed using as matrix pooled human sputum purchased from BioreclamationIVT (Westbury, NY), confirmed by us to be negative for M.tb complex genomic DNA via the qPCR method of Halse et al.12. M.tb H37Ra (ATCC 25177, cultured in Difco Middlebrook 7H9 broth with 0.2% glycerol, 40 mM sodium pyruvate, 10% [v/v] ADC enrichment and 0.05% Tween 80) was spiked into TB-negative sputum to a final concentration of 103, 104, or 105 cfu/mL. For each concentration, plus a TB-negative sputum control spiked with media only, six 1 mL aliquots were processed via the PureLyse® and comparator sample preparation method,12 followed by amplification and detection using qPCR12, as described below.
Clinical sputum specimens were collected at the Public Health–Seattle & King County TB Clinic (Seattle, WA) from de-identified male and female subjects 18 years or older able to provide spontaneous sputum specimens. Protocols and consent forms were approved by Institutional Review Boards at the University of Washington and Claremont Graduate University. Informed consent was obtained from all subjects and sample collection was carried out in accordance with the approved protocol. Up to ten specimens were collected per subject, with at least eight hours between each sample. For subjects with active TB, samples were collected within seven days of treatment initiation. Of the 46 specimens included in this study, 30 were obtained from 17 subjects subsequently diagnosed by microbiological culture and clinical criteria as active pulmonary TB cases; and 16 specimens came from ten patients who microbiologically and clinically did not have active TB. Each clinical sputum sample was divided into two aliquots of equal volume, typically ~1 mL, which were then processed respectively via the PureLyse® and comparator sample preparation method12, followed by qPCR amplification and detection12. For five of the 46 samples, aliquots <1 mL (750–900 μL) were used due to insufficient total sample volume. One low-volume specimen was subsequently excluded from data analysis due to a technical problem during process execution.
PureLyse® Sputum Nucleic Acid Sample Preparation Method
Samples were processed using the PureLyse® kit (Claremont BioSolutions LLC), modified for sputum nucleic acid sample preparation to enable sputum liquefaction and disinfection, followed by miniaturized mechanical cell disruption and solid-phase nucleic acid extraction. Sputum liquefaction and disinfection: A disinfection/liquefaction reagent containing PVI (Rite Aid Corporation) and TSP was prepared within ≤2 min before use and vortexed within ≤10 sec before adding 0.334 mL to each ~1 mL sputum sample. Sample tubes were then vortexed briefly and incubated at room temperature for 10 minutes. Nucleic acid extraction: An equal volume of 2× binding buffer was added to the disinfected and liquefied sample, followed by vortexing. The sample was then pumped through the PureLyse® blender using a programmable syringe pump (KD Scientific, KDS-250) at a flow rate of 1 mL/min, with the PureLyse® motor activated, enabling cell lysis and DNA binding to the beads. Next, 4 mL wash buffer (0.2× binding buffer) was pumped through the PureLyse® blender at a flow rate of 0.75 mL/min with the motor activated, followed by 1 mL of air purging, with the motor de-activated. To elute the nucleic acids from the beads, the PureLyse® chamber was filled with ~150 μL elution buffer, the motor was activated for 30 seconds and then the syringe pump was used to pump another ~150 μL elution buffer through the chamber at a flow rate of 0.33 mL/min. The final eluate was stored on ice for immediate downstream amplification by qPCR.
Verification of Sample Disinfection
To evaluate the effectiveness of sputum sample disinfection, M.tb H37Rv (ATCC 25618, cultured in Difco Middlebrook 7H9 broth with 10% [v/v] ADC enrichment and 0.05% Tween 80) was spiked into pooled TB-negative human sputum to a final concentration of ~107 cfu/mL (estimated by optical density) and 1 mL spiked sputum aliquots were processed using the sputum liquefaction and disinfection protocol. Disinfection was stopped after the 10 min incubation by diluting the sample to 50 mL with sterile phosphate buffered saline (PBS), followed by inversion and vortexing. Cells and other particulates were pelleted at 4000 × g for 20 minutes, supernatants were removed and the pellets re-suspended in 1 mL PBS. Ten-fold dilution series of each cell suspension were prepared in PBS. Six replicate 100 μL aliquots (3 replicates each of 2 separate experiments) of undiluted and diluted samples were plated on Difco Middlebrook 7H10 agar with 10% (v/v) OADC enrichment, incubated at 37 °C for 6–8 weeks, then colonies were counted.
In parallel with the spiked sputum experiments, the number of M.tb H37Rv cells remaining after identical processing but in the absence of disinfection was determined microbiologically. These controls were designed to isolate the effect of disinfection from confounding causes of cell loss (e.g. adherence to sample tubes and pipette tips, aspiration, etc.) and thereby quantify the effect of disinfection alone. Sputum was not used as matrix since overgrowth with non-mycobacterial organisms present in sputum would confound the results. Here, M.tb H37Rv was spiked into 1 mL PBS aliquots and processed as described above. Six replicate 100 μL aliquots (3 replicates each of 2 separate experiments) of undiluted and 10× diluted samples were plated and incubated and colonies were counted. Six unprocessed sputum controls (unspiked and untreated) were also plated directly. After incubation for 6–8 weeks, these unprocessed sputum samples formed diverse colonies too numerous to count.
Comparator Method for Sputum Nucleic Acid Sample Preparation
As a comparative nucleic acid extraction method, sputum samples were processed as described by Halse et al.12. Briefly, an equal volume of 3.5% NaOH was added to each ~1 mL sputum sample. Samples were vortexed and incubated for 15 min at room temperature to liquefy and decontaminate the sputum. Decontamination was stopped by diluting the samples with PBS (pH 6.8) to a final volume of 50 mL, followed by inversion mixing. The contents were pelleted by centrifugation (15 min, 3000 × g) and supernatants were carefully removed by aspiration. Pellets were resuspended in 1 mL PBS, heated at 80 °C for 1 hr to lyse the mycobacteria and stored on ice until qPCR was performed.
PCR Amplification and Detection
We used a qPCR method for detecting M. tuberculosis complex (MTBC) DNA, with internal amplification control, as described by Halse et al.12, with 0.8 mg/mL BSA added to the master-mix. This qPCR method targets the MTBC-specific insertion element IS6110 and a sequence within an engineered internal control plasmid (pIC) flanked with identical primer binding sites. The IS6110 and pIC amplicons can be differentiated with FAM and VIC labeled sequence-specific hydrolysis probes. Each sample was analyzed in two separate reactions: one with only the IS6110-FAM probe, the other with the IS6110-FAM probe, the pIC-VIC probe and 1 fg/μL pIC. The degree of PCR inhibition was assessed by comparing Cq values for the internal amplification control (pIC-VIC probe) relative to those in uninhibited reactions receiving water or purified genomic DNA as template. PCR amplification and detection was executed at two sites, using either a CFX96 (Bio-Rad) or StepOnePlus (Applied Biosystems).
Additional Information
How to cite this article: Ferguson, T. M. et al. Pilot study of a rapid and minimally instrumented sputum sample preparation method for molecular diagnosis of tuberculosis. Sci. Rep. 6, 19541; doi: 10.1038/srep19541 (2016).
References
World Health Organization. Global tuberculosis report 2014. (World Health Organization, 2014). at http://apps.who.int/iris/bitstream/10665/137094/1/9789241564809_eng.pdf?ua=1. Date of Access: 03/09/2015.
World Health Organization. The Global Plan to Stop TB 2011-2015. (WHO, 2011). at http://www.stoptb.org/assets/documents/global/plan/TB_GlobalPlanToStopTB2011-2015.pdf. Date of Access: 01/24/2014.
McNerney, R. & Daley, P. Towards a point-of-care test for active tuberculosis: obstacles and opportunities. Nat Rev Micro 9, 204–213 (2011).
Perkins, M. D. & Cunningham, J. Facing the Crisis: Improving the Diagnosis of Tuberculosis in the HIV Era. The Journal of Infectious Diseases 196, S15–S27 (2007).
Uys, P. W., Warren, R., Helden, P. D., van, Murray, M. & Victor, T. C. Potential of Rapid Diagnosis for Controlling Drug-Susceptible and Drug-Resistant Tuberculosis in Communities Where Mycobacterium tuberculosis Infections Are Highly Prevalent. J. Clin. Microbiol. 47, 1484–1490 (2009).
Urdea, M. et al. Requirements for high impact diagnostics in the developing world. Nature 444, 73–79 (2006).
Yager, P., Domingo, G. J. & Gerdes, J. Point-of-Care Diagnostics for Global Health. Annual Review of Biomedical Engineering 10, 107–144 (2008).
Niemz, A. & Boyle, D. S. Nucleic acid testing for tuberculosis at the point-of-care in high-burden countries. Expert Review of Molecular Diagnostics 12, 687–701 (2012).
Boehme, C. C. et al. Rapid Molecular Detection of Tuberculosis and Rifampin Resistance. New England Journal of Medicine 363, 1005–1015 (2010).
Niemz, A., Ferguson, T. M. & Boyle, D. S. Point-of-care nucleic acid testing for infectious diseases. Trends Biotechnol. 29, 240–250 (2011).
Pai, M. & Schito, M. Tuberculosis Diagnostics in 2015: Landscape, Priorities, Needs and Prospects. J Infect Dis. 211, S21–S28 (2015).
Halse, T. A. et al. Combined Real-Time PCR and rpoB Gene Pyrosequencing for Rapid Identification of Mycobacterium tuberculosis and Determination of Rifampin Resistance Directly in Clinical Specimens. J. Clin. Microbiol. 48, 1182–1188 (2010).
Aldous, W. K., Pounder, J. I., Cloud, J. L. & Woods, G. L. Comparison of six methods of extracting Mycobacterium tuberculosis DNA from processed sputum for testing by quantitative real-time PCR. J. Clin. Microbiol. 43, 2471–2473 (2005).
Gomez, D. I. et al. Rapid DNA extraction for specific detection and quantitation of Mycobacterium tuberculosis DNA in sputum specimens using Taqman assays. Tuberculosis 91 Suppl 1, S43–48 (2011).
Mitarai, S. et al. Evaluation of a simple loop-mediated isothermal amplification test kit for the diagnosis of tuberculosis. The International Journal of Tuberculosis and Lung Disease 15, 1211–1217 (2011).
Fang, R. et al. Cross-Priming Amplification for Rapid Detection of Mycobacterium tuberculosis in Sputum Specimens. J. Clin. Microbiol. 47, 845–847 (2009).
Ao, W. et al. Rapid Detection of rpoB Gene Mutations Conferring Rifampin Resistance in Mycobacterium tuberculosis. J. Clin. Microbiol. 50, 2433–2440 (2012).
Pfyffer, G. E., Kissling, P., Wirth, R. & Weber, R. Direct detection of Mycobacterium tuberculosis complex in respiratory specimens by a target-amplified test system. J. Clin. Microbiol. 32, 918–923 (1994).
Hida, Y. et al. Rapid Detection of the Mycobacterium tuberculosis Complex by Use of Quenching Probe PCR (geneCube). J. Clin. Microbiol. 50, 3604–3608 (2012).
Choi, Y. J. et al. Evaluation of peptide nucleic acid probe-based real-time PCR for detection of Mycobacterium tuberculosis complex and nontuberculous mycobacteria in respiratory specimens. Ann Lab Med 32, 257–263 (2012).
Bogard, M. et al. Multicenter Study of a Commercial, Automated Polymerase Chain Reaction System for the Rapid Detection of Mycobacterium tuberculosis in Respiratory Specimens in Routine Clinical Practice. Eur J Clin Microbiol Infect Dis 20, 724–731 (2001).
Boehme, C. C. et al. Operational Feasibility of Using Loop-Mediated Isothermal Amplification for Diagnosis of Pulmonary Tuberculosis in Microscopy Centers of Developing Countries. J. Clin. Microbiol. 45, 1936–1940 (2007).
Torres-Chavolla, E. & Alocilja, E. C. Nanoparticle based DNA biosensor for tuberculosis detection using thermophilic helicase-dependent isothermal amplification. Biosens Bioelectron 26, 4614–4618 (2011).
Lawn, S. D. et al. Advances in tuberculosis diagnostics: the Xpert MTB/RIF assay and future prospects for a point-of-care test. The Lancet Infectious Diseases 13, 349–361 (2013).
Blakemore, R. et al. Evaluation of the Analytical Performance of the Xpert MTB/RIF Assay. J. Clin. Microbiol. 48, 2495–2501 (2010).
Helb, D. et al. Rapid Detection of Mycobacterium tuberculosis and Rifampin Resistance by Use of On-Demand, Near-Patient Technology. J. Clin. Microbiol. 48, 229–237 (2010).
Denkinger, C. M., Kik, S. V. & Pai, M. Robust, reliable and resilient: designing molecular tuberculosis tests for microscopy centers in developing countries. Expert Rev. Mol. Diagn. 13, 763–767 (2013).
Best, M., Sattar, S. A., Springthorpe, V. S. & Kennedy, M. E. Comparative mycobactericidal efficacy of chemical disinfectants in suspension and carrier tests. Appl. Environ. Microbiol. 54, 2856–2858 (1988).
Rikimaru, T. et al. Efficacy of common antiseptics against multidrug-resistant Mycobacterium tuberculosis. Int. J. Tuberc. Lung Dis. 6, 763–770 (2002).
Warren, J. R., Bhattacharya, M., De Almeida, K. N., Trakas, K. & Peterson, L. R. A minimum 5.0 ml of sputum improves the sensitivity of acid-fast smear for Mycobacterium tuberculosis. Am. J. Respir. Crit. Care Med. 161, 1559–1562 (2000).
Blackwood, K. S. et al. Viability testing of material derived from Mycobacterium tuberculosis prior to removal from a Containment Level-III Laboratory as part of a Laboratory Risk Assessment Program. BMC Infectious Diseases 5, 4 (2005).
Lanigan, M. D., Vaughan, J. A., Shiell, B. J., Beddome, G. J. & Michalski, W. P. Mycobacterial proteome extraction: comparison of disruption methods. Proteomics 4, 1094–1100 (2004).
Yáñez, M. A. et al. Determination of mycobacterial antigens in sputum by enzyme immunoassay. J. Clin. Microbiol. 23, 822–825 (1986).
Vandeventer, P. E. et al. Mechanical Disruption of Lysis-Resistant Bacterial Cells by Use of a Miniature, Low-Power, Disposable Device. J. Clin. Microbiol. 49, 2533–2539 (2011).
Piersimoni, C., Gherardi, G., Nista, D. & Bornigia, S. Impact of a Chemistry-Based DNA Extraction Method on Performance of a Commercial Amplification Assay for Detection of Mycobacterium tuberculosis Complex. J. Clin. Microbiol. 47, 282–283 (2009).
Doebler, R. W. et al. Continuous-Flow, Rapid Lysis Devices for Biodefense Nucleic Acid Diagnostic Systems. Journal of Laboratory Automation 14, 119–125 (2009).
Vandeventer, P. E., Mejia, J., Nadim, A., Johal, M. S. & Niemz, A. DNA Adsorption to and Elution from Silica Surfaces: Influence of Amino Acid Buffers. J. Phys. Chem. B 10742–10749 (2013).
Vandeventer, P. E. et al. Multiphasic DNA Adsorption to Silica Surfaces under Varying Buffer, pH and Ionic Strength Conditions. J. Phys. Chem. B 116, 5661–5670 (2012).
Roskos, K. et al. Simple System for Isothermal DNA Amplification Coupled to Lateral Flow Detection. PLoS ONE 8, e69355 (2013).
Lu, H.-W., Roskos, K., Hickerson, A. I., Carey, T. & Niemz, A. System for portable nucleic acid testing in low resource settings. Proc SPIE 8615, 86150I–86150I–12 (2013).
Acknowledgements
We thank Kimberlee Musser for advice related to the Halse et al.12 qPCR method and for supplying the internal control plasmid. We further thank Meg Savlov and Sergio Steele at Public Health - Seattle & King County for the collection of clinical sputum samples.
Author information
Authors and Affiliations
Contributions
T.M.F., K.M.W., A.L.B., R.D., G.A.C. and A.N. planned the experiments, T.M.F., K.M.W., A.L.B., D.O. and I.T. executed the experiments, M.N. oversaw the collection of clinical sputum specimens, T.M.F. and K.M.W. performed the majority of the data analysis, T.M.F., A.N., G.A.C. and K.M.W. wrote the manuscript, all authors were involved in manuscript review and editing.
Ethics declarations
Competing interests
T.M.F. is an employee and R.D. is president and co-founder of Claremont BioSolutions, the company commercializing the PureLyse technology. Both have equity interests in the company. K.M.W., A.L.B., D.O., M.N., I.T., G.A.C. and A.N. declare no potential conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ferguson, T., Weigel, K., Lakey Becker, A. et al. Pilot study of a rapid and minimally instrumented sputum sample preparation method for molecular diagnosis of tuberculosis. Sci Rep 6, 19541 (2016). https://doi.org/10.1038/srep19541
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep19541
This article is cited by
-
Tuberculosis: current scenario, drug targets, and future prospects
Medicinal Chemistry Research (2021)
-
Closed-type pre-treatment device for point-of-care testing of sputum
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
