
Abstract
Mutagenesis is an important tool to study gene regulation, model disease-causing mutations and for functional characterisation of proteins. Most of the current methods for mutagenesis involve multiple step procedures. One of the most accurate methods for genetically altering DNA is recombineering, which uses bacteria expressing viral recombination proteins. Recently, the use of in vitro seamless assembly systems using purified enzymes for multiple-fragment cloning as well as mutagenesis is gaining ground. Although these in vitro isothermal reactions are useful when cloning multiple fragments, for site-directed mutagenesis it is unnecessary. Moreover, the use of purified enzymes in vitro is not only expensive but also more inaccurate than the high-fidelity recombination inside bacteria. Here we present a single-step method, named REPLACR-mutagenesis (Recombineering of Ends of linearised PLAsmids after PCR), for creating mutations (deletions, substitutions and additions) in plasmids by in vivo recombineering. REPLACR-mutagenesis only involves transformation of PCR products in bacteria expressing Red/ET recombineering proteins. Modifications in a variety of plasmids up to bacterial artificial chromosomes (BACs; 144 kb deletion) have been achieved by this method. The presented method is more robust, involves fewer steps and is cost-efficient.
Similar content being viewed by others
Introduction
Site-directed mutagenesis (SDM), also known as directed mutagenesis, is used to generate mutations, add or delete domains in cDNAs or gene promoters in order to study the resulting translation product for protein engineering and/or functional characterisation. SDM is also used to model mutations found in clinical samples for functional studies at the cellular and molecular level.
There are a vast number of techniques and commercial kits to generate point mutations, short additions or deletions, mainly based on PCR such as overlap extension and megaprimer PCR1,2,3,4,5,6,7. Yet most involve several steps and are limited by the size of insertion or deletion due to the use of complementary primer pairs. In most cases, mutagenesis is accompanied with ligation of the resulting linear PCR fragment to form a circular vector, which is inefficient and can result in unwanted additions or deletions due to the activities of the polymerase used.
Homologous recombination (HR) is a process where a pair of homologous DNA fragments exchanges nucleotides, often as means to repair DNA breaks. HR was first used to modify genetic material in yeast8. Later on, the bacteriophage enzymes that perform HR independently of the bacterial system were discovered and used to modify plasmids, bacterial artificial chromosomes (BACs) and bacterial genomes9,10,11,12, a method now called recombineering (recombination-mediated genetic engineering)13. These enzymes allow genetic modifications by recognising complementary DNA strands for strand invasion or annealing14,15. One limitation for this seamless cloning technique is the need of selection of positive clones, something we, among many others, have solved by using selection/counter-selection systems16,17,18,19.
Recently, a series of methods based on the assembly of overlapping DNA fragments using in vitro reactions have been developed, such as Gibson assembly20, GeneArt seamless cloning (Life technologies), In-Fusion HD cloning (Clontech), ligase-independent cloning (LIC) and the variations of the latter (SLIC and SLICE)21,22,23. These in vitro methods can join several fragments of DNA with 15–25 nucleotide homology at both termini. Yet, there are limitations for these assemblies such as single-stranded DNA secondary structures and cloning of primer dimers24. A common feature of these methods is the use of purified DNA-modifying enzymes (exonucleases, recombinases, polymerases and ligases), which not only makes these methods highly expensive but also dependent on selective buffers and reaction conditions. In addition, in vitro DNA termini-joining can also yield many non-specific products, which is in stark contrast to in vivo recombineering that is highly selective and robust due to the presence of endogenous proofreading replication and repair pathways.
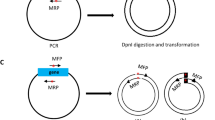
Although in vitro methods are highly advantageous for multiple fragment cloning, they are, however, completely unnecessary for generating mutations at a single locus (substitutions, additions or deletions) in plasmids. Here we present a single-step method where, by in vivo recombineering, we are able to generate site-directed modifications (point mutations, deletions, additions and substitutions) in plasmid vectors in a cost-effective manner with high efficiency and accuracy. Since the method is primarily focused on generating mutations in plasmids and the only step needed is PCR followed by recombineering of the linearised ends of the plasmid, we named the method as REPLACR-mutagenesis (Recombineering of Ends of linearised PLAsmids after PCR; Fig. 1).

Principle of REPLACR-mutagenesis and primer design strategy for sequence substitution, addition or deletion.
(a) Primers containing the desired mutation are designed to target a specific region in the original vector. A high-fidelity polymerase is used to generate a linear PCR product such that both the ends contain overlapping sequences for recombination. Bacteria expressing the recombineering proteins (Redγ, β, α and RecA) are transformed with the PCR product. Recombination takes places inside the bacteria thereby yielding a circular plasmid containing the desired mutation. Bacterial colonies are then screened for the correct clone by PCR and sequencing. (b) Forward and reverse primers contain the desired addition/substitution as a part of homology regions needed for recombination, besides containing a 3′ extension for effective template binding. The homology region (17 bp or more) for substitutions also contains the desired nucleotide change. (c) For generating deletion mutants, forward primer contains the sequence adjoining the sequence to be deleted. The reverse primer however contains a sequence homologous to the forward primer and the adjoining sequence in the vector.
Results
Optimal homology for REPLACR-mutagenesis
We rationalised that the linear ends of a PCR product could be circularised by recombineering if they had enough homology. Thus, we first amplified a plasmid by PCR such that the resulting linear PCR products had a varying number of homologous nucleotides at their ends. To determine the optimal length of homology needed for highest recombination efficiency, we tested 23 bp primer pairs with homology ranging from 2 bp to 23 bp (See Supplementary Table S1 for primer sequences). The primers targeted a ScaI restriction site (AGTACT) in the wild-type (WT) human luteinizing hormone/chorionic gonadotropin receptor (LHCGR) plasmid25 by the addition of two nucleotides (AT) in the middle of a ScaI sequence, such that the resulting site in the plasmid was not amenable to ScaI digestion. The number of correct clones found over the LHCGR_WT background was used to calculate the efficiency with respect to the homology at the ends of the PCR products (Fig. 2a). A 2 bp homology at the ends of PCR products is insufficient for recombination, thereby resulting in no positive clones. From 5 bp homology onwards, there was an increase in efficiency of REPLACR-mutagenesis up to 17 bp, with the maximum efficiency of 84%. Since the primers were only 23 bp long, a 20 bp and 23 bp homology among the primers favoured the formation of primer dimers thereby resulting in undesired PCR products. The bacterial colonies containing either the mutated plasmids or LHCGR_WT background were screened by colony PCR and subsequent ScaI restriction digestion of the PCR products. Supplementary Figure S1 shows the ScaI restriction digestion patterns of PCR products obtained for one representative experiment and the results are summarised in Supplementary Table S2, detailing the number of colonies obtained, screened, correct ones found and the associated efficiencies. The colony PCR conditions are mentioned in Supplementary Table S3.

Efficiency of REPLACR-mutagenesis.
(a) The effect of homology at the ends of PCR products is plotted against the achieved efficiencies with REPLACR-mutagenesis. The efficiency of mutagenesis increases with increasing homology, where a 2 bp homology is insufficient to yield any correct products while a 17 bp homology gives the highest efficiency at 84%. (b) The same PCR products with 14 bp and 17 bp homology were used with two commercial kits (Gibson Assembly and GeneArt Seamless cloning) and the achieved efficiencies were compared with REPLACR-mutagenesis. REPLACR-mutagenesis is least efficient among the three methods when the PCR products have only 14 bp homology, however with the recommended 17 bp homology at the ends of PCR products, comparable efficiencies for all the methods can be observed. The data is presented as mean ± standard error of mean (SEM) of three independent repeats.
Using oligonucleotides that span 22–24 nucleotides of primer-template binding plus a 5′-tail containing the homology arms can further increase the total length of the homology. As mentioned later in the manuscript, different mutants were made where longer primers with more more than 17 bp homology (20, 23 and 30 bp homology) were used, however, the associated efficiencies were very similar to the 84% efficiency as achieved by 17 bp homology (See Supplementary Figure S2). Thus, a 17 bp homology with 3′ overhangs for both primers is sufficient for efficient REPLACR-mutagenesis.
In addition, when three PCR products with 11 bp, 14 bp and 17 bp homology were transformed in non-recombineering bacteria (E.coli DH-10β), no colonies were obtained because linear PCR products cannot recombine own their own in the absence of enzymes needed for recombination.
Efficiency comparison with commercial kits (Gibson Assembly and GeneArt seamless cloning)
Since the PCR products with 14 bp and 17 bp homology at their termini gave highest efficiencies with REPLACR-mutagenesis (Fig. 2a), we used the same PCR products with two commercially available kits, namely Gibson assembly and GeneArt seamless cloning. The efficiencies of the Gibson assembly and GeneArt seamless cloning were similarly determined by ScaI digestion of the colony PCR products (see Supplementary Fig. S3). Although PCR products with 14 bp homology gave higher efficiencies with Gibson assembly and GeneArt, but for PCR products with recommended 17 bp homology, efficiencies all the methods were comparable (Fig. 2b). Moreover, we consistently found GeneArt cloning to result in smaller number of overall colonies as compared with REPLACR-mutagenesis or Gibson Assembly (see Supplementary Table S4).
Substitutions
All substitutions were targeted to plasmids encoding the human LHCGR, follicle-stimulating hormone receptor (FSHR) or beta-2 adrenergic receptor (β2AR). We generated many single nucleotide substitutions, namely, LHCGR_Asn291Ser, LHCGR_Val454Ile, FSHR_Ala444Thr, FSHR_ Gly70Ala, β2AR_Asp79Asn, β2AR_Asp130Asn and β2AR_Cys341Gly and a double nucleotide substitution, β2AR_Tyr350Ala (See Supplementary Figure S4). The primers used for creating substitutions and verifying via DNA sequencing are mentioned in Supplementary Tables S5 and S6, respectively.
Deletions
Because the method is not limited to point substitutions, we also tested whether we could delete nucleotides. First, we deleted a single nucleotide in the LHCGR (1850delG), which results in a frame-shift of the C-terminal tail of the receptor; the functional tests for this mutant receptor have been reported elsewhere26. This deletion was achieved as verified by sequencing (see Supplementary Figure S5). As deletion of a nucleotide worked as efficiently as nucleotide substitutions, we proceeded to generate larger editing of the DNA sequence of some plasmids.
The deletion of 12 nucleotides of the signal peptide of the LHCGR gene (LHCGR-Lys12-Leu15del) was achieved using the same method. Once again, we produced hundreds of colonies where most of them were correct as verified by sequencing (see Supplementary Figure S5).
Finally, we used REPLACR-mutagenesis to delete an entire 144 kb DNA sequence from a human LHCGR BAC clone (RPCI-11-186L7), a one step “BAC-shaving” as compared with the multistep systems to-date27. The primer sequences used for the deletion are mentioned in Supplementary Table S5. The resulting deletion was verified by sequencing (Supplementary Fig. S5). The primers used for sequencing are specified in Supplementary Table S6. PCR conditions for deletion are mentioned in Supplementary Table S7. In addition, since the chloramphenicol resistance gene and origin of replication were not affected during PCR, the resulting plasmid could propagate in chloramphenicol containing Luria-Bertani (LB) medium, demonstrating the integrity of the backbone during PCR.
Additions
In order to generate mutants with additional nucleotides, the LHCGR_Leu10-Gln17Dup, a 27-nucleotide duplication, was generated. This construct produced many colonies but most of them were negative, probably due to a preference for near-end recombination and the presence of 2 identical sequences (duplication). The correct colonies were obtained by generating forward and reverse strands separately in two separate PCR reactions and thereafter the two products were mixed together. The PCR products were heated up to 95 °C and slowly allowed to cool for annealing of DNA strands, followed by DpnI digestion before transformation in Red/ET bacteria, as previously described28. The sequence was verified by sequencing (Supplementary Figure S6). Neither Gibson assembly nor GeneArt produced any correct colonies for this duplication.
In order to show that there is no limit on the number of additional nucleotides to be incorporated as far as they have homology arms, we inserted a 45-nucleotide nuclear localization signal to a plasmid containing Cryptochrome Circadian Clock 2 (CRY2) gene29. Moreover, a 60-nucleotide addition of a flexible domain was also achieved using REPLACR-mutagenesis (Supplementary Figure S6). The primer sequences used to generate the mutations are mentioned in Supplementary Tables S5. The sequencing primers used to verify the additions of 45 and 60 nucleotides were TTGCTCGTTGGCATCAGAAGG and AGCTGCTGCTAATGCAGGAT, respectively. We did not try longer additions as the cost for larger primers become higher than using synthetic DNA “blocks” and Gibson assembly.
Modifications of larger plasmids
A 24 kb Wnt1 targeting vector was used to introduce a 2 bp addition. The two bases (TG) were introduced in one the three MfeI restriction sites (CAATTG) present in the original vector such that the resulting sequence (CAATGTTG) leaves only two MfeI restriction sites in the mutated plasmid. MfeI restriction digestion of the original Wnt1 vector with three MfeI restriction sites results in three bands (15476, 6379, 1844 bp; lane 2 in Fig. 3) whereas the modified Wnt1 vector with a mutated MfeI site results in two bands (15476 and 8225 bp; lane 4 in Fig. 3), as expected. The mutated region was verified by sequencing (Supplementary Figure S7). The primers for creating the mutation and sequence verification by DNA sequencing are stated in Supplementary Tables S5 and S6, respectively. The PCR conditions are mentioned in Supplementary Table S8. The mutated plasmid could replicate in kanamycin conditioned LB-agar plates, similar to the original plasmid, demonstrating the integrity of the antibiotic resistance gene as well as the origin of replication. Overall, the sequencing of the mutated site, the expected restriction digestion pattern and the ability of the plasmid to propagate in bacteria cultured in the appropriate antibiotic containing medium demonstrates the integrity of a larger and complex plasmid mutated with REPLACR-mutagenesis.

Modifications in larger plasmids.
The 24 kb Wnt1 targeting vector (undigested; lane 1) contains three MfeI restriction sites, which upon MfeI digestion gives three bands (15476, 6379, 1844 bp; lane 2). One of the MfeI sites is mutated by REPLACR-mutagenesis, such that the resulting plasmid (lane 3) upon MfeI digestion gives only two bands (15476 and 8225 bp; lane 4).
Modifying plasmid with similar incompatibility to the recombineering plasmid
A potential limitation of REPLACR-mutagenesis could be the modification of plasmids with origin of replication incompatible with the Red/ET plasmid (pSC101). To test this, recombineering bacteria were prepared with Red/ET plasmid carrying tetracycline resistance gene. We have previously modified a similar Red/ET plasmid (pSC101 BADgbaRecA) containing hygromycin rather than tetracycline resistance gene16. A fragment of the temperature sensitive repressor (RepA) was deleted (944 bp) and was verified by sequencing (Supplementary Figure S8). The primers for mutagenesis, sequencing and the PCR conditions are specified in Supplementary Tables S5, S6 and S9, respectively. The original plasmid can only be grown at 30 °C due to active repressor while the mutated plasmid with an inactive repressor could now be grown at 37 °C. This shows the utility of the REPLACR-mutagenesis to modify plasmids with similar incompatibility to the recombineering plasmid. This is possible because the electrocompetent bacteria are already expressing the recombination enzymes during their preparation and the original recombineering plasmid cannot replicate at 37 °C, thereby eliminating any selective pressure. However, we found very few colonies after REPLACR-mutagenesis and the recombination efficiency of 33% as only one out three colonies were positive. This suggests that the incompatibility of the origins of replication has a negative effect, which result in low efficiencies, but it is possible to achieve mutations even in such circumstances.
Discussion
REPLACR-mutagenesis presents a quick and robust in vivo recombineering based mutagenesis protocol. The only step needed is the transformation of PCR products in bacteria expressing viral recombination proteins. An effective primer design is thus crucial for this method. The general primer design strategy, specific for additions, deletions and substitutions is summarised in Fig. 1. Primers should be designed such that the resulting PCR products contain a homology at their termini of around 17bp. As seen in Fig. 2a, a 17 bp homology at the ends of PCR products gave the highest efficiency of 84% over the background. For 23 bp primer pairs, up to 17 bp homology (with 6 additional nucleotides at 3′ end) resulted in expected PCR products whereas a 20 bp and 23 bp homology among primers resulted in undesired PCR products. Therefore, besides containing the homology regions, the primers should include a 3′ extension (6 nucleotides or more), such that primer-template binding is favoured over primer-primer self-complementarity. For creating substitutions, the homology region also contains the substituted nucleotide(s), with an extended 3′ end. For additions, a 20 bp region for primer template binding is sufficient and the additions can be made on the 5′ end of either one or both primers. The added regions should contain a 17 bp homology for maximum efficiency. However, for creating deletions, one of the primers should contain the adjoining sequences between the region to be deleted and the other primer containing a 17 bp homology to the 5′ end of the first primer and an additional 3′ sequence in the other direction. It is however possible to design longer primers, with more than 17 bp homology but there is no significant increase in efficiency (see Supplementary Figure S2). In addition, longer primers also increase the cost as well as the chances of formation of secondary structures, thereby decreasing the PCR efficiency in some cases.
The bacteria used for recombination contain Red/ET plasmid, which during their preparation are made to express viral recombination proteins under arabinose promoter, by addition of L-Arabinose and subsequently frozen until use. The replication of Red/ET plasmid is temperature sensitive and only replicates at 30 °C30. Thus, after transforming the PCR products, the bacteria are grown at 37 °C and the resulting bacterial colonies only contain the desired mutated plasmid and not the original Red/ET plasmid. In addition, it is also possible to modify plasmids with incompatibility as the Red/ET plasmid because the electrocompetent bacteria are already expressing the recombination proteins to circularise the PCR products. However, the plasmid to be mutated should have a different antibiotic resistance gene than the Red/ET plasmid.
The method was used to successfully generate a variety of point substitutions, deletions ranging from one nucleotide deletion to as large as 144 kb deletion (shaving) in human LHCGR BAC in one single step, which would otherwise require multiple steps via traditional restriction digestion based deletion techniques or those involving selection/counter-selection cassettes17,27,31,32. We were also able to add nucleotides ranging from one to 60 nucleotides. The limitation of adding nucleotides by longer primers is the cost of primers themselves. The method was thus used to generate mutations in small plasmids (6–10 kb) to as large as a 24 kb Wnt1 targeting vector.
Traditional PCR based mutagenesis methods typically require a variety of steps and the application of many enzymes such as kinases for phosphorylation of 3′ ends and ligases to form circular plasmids. Similarly, recombination-based mutagenesis and cloning methods (Gibson Assembly and GeneArt seamless cloning) also require the application of expensive enzymes and multiple steps. In both GeneArt and Gibson assembly, PCR of the template DNA is followed by DNA purification, in vitro recombination and subsequent transformation into bacteria. Our method reduces the number of steps needed for creating mutations to just one-step since the PCR product is directly transformed in the bacteria and there is no need for an additional in vitro incubation of PCR product with recombineering enzymes. In addition, it is considerably cheaper since the electrocompetent bacteria have to be prepared only once for a large number of mutagenesis experiments. Moreover, REPLACR-mutagenesis is as efficient as the commercially available Gibson assembly and GeneArt seamless cloning kits. Finally, a median efficiency of 75% was found for all the mutations made using our method (see Table 1).
The method bears similarity in primer design to PCR-based mutagenesis methods like quikchange site-directed mutagenesis (Agilent Technologies), where nicks in circular PCR products are repaired by bacterial endogenous DNA repair machinery7. However, most PCR products are linear and the number of circular PCR products with nicks that can be repaired by bacterial endogenous repair systems is very low and henceforth the method becomes inefficient particularly for mutagenesis of more than one nucleotide. Thus, the expression of viral recombination proteins in bacteria as proposed in our method greatly enhances the efficiency of the recombination at the ends of linear PCR products as well as the number of bacterial colonies obtained. Moreover, REPLACR-mutagenesis is capable of complex additions and deletions in fewer steps than what would be needed by other PCR-based mutagenesis methods such as overlap extension PCR mutagenesis. Although there have been reports of using in vivo recombineering-based methods for mutagenesis in one single transformation step such as “en passant mutagenesis” but the bacterial colonies have to be grown and selected by colony PCR twice in different conditions, thereby prolonging the experiment33. The presented method however, involves direct screening of bacterial colonies obtained after the transformation step.
Although in the case of the large duplications we experienced problems during the PCR step, we solved it by generating single complementary strands of the vector and then re-joining them in an isothermal reaction, as previously described28, before transformation into Red/ET electrocompetent bacteria. The generation of complimentary DNA strands using two PCR reactions with only one primer each is a known way to PCR the DNA regions with tandem repeats. One of the limiting factors in REPLACR-mutagenesis is the PCR itself; since most high-fidelity polymerases are recommended for PCR products up to 20–25 kb, though there have been some improvements in development of better polymerases. Nevertheless, most plasmids containing the cDNA of genes are smaller than 10–12 kb and hence the utility of the method is sufficient for most routine mutagenesis experiments.
In conclusion, REPLACR-mutagenesis provides a cost-effective way involving fewer steps to produce mutations with high accuracy due to the nature of the Red/ET recombineering system.
Methods
Materials
KOD-Xtreme hot-start DNA polymerase was purchased from Merck Millipore. Three previously described plasmids carrying cDNA of the human LHCGR, FSHR and β2AR were used for all the substitutions25,34,35. Restriction endonucleases DpnI and MfeI were purchased from New England Biolabs (NEB) and ScaI was purchased from Promega. Wnt1 targeting vector (24 kb) was purchased from the KOMP Repository (University of California Davis and Children’s Hospital Oakland Research Institute, USA). pCRY2FL(deltaNLS)-mCherryN1 was a gift from Chandra Tucker (Addgene plasmid # 26871)29. The Red/ET plasmid, pSC101BADgbaRecA[tet], was purchased from Genebridges (Dresden, Germany).
Preparation of Electrocompetent bacteria for recombineering
Red/ET recombineering system (Red γ, β, α and RecA) containing recombineering plasmid (pSC101BADgbaRecA[tet]; hereafter called as Red/ET) was purchased from GeneBridges. Electrocompetent cells were prepared as described in the manuals from Genebridges using L-arabinose to induce the phage recombinases. Recombineering was performed in electrocompetent HS996 E.coli cells harbouring the Red/ET plasmid using standard recombination procedures14. Briefly, four microcentrifuge tubes with 1ml Luria-Bertani (LB) medium supplemented with either tetracycline (3 μg/ml) or hygromycin (15 μg/ml) were inoculated with bacteria harbouring the Red/ET plasmid and cultured overnight at 30 °C in a table-top thermomixer (Eppendorf). Next day, the 4 ml bacterial culture was transferred to a 250 ml LB culture with the appropriate antibiotic and was cultured for a further 3 h (250 rpm at 30 °C). Thereafter, L-arabinose was added to a final concentration of 0.35% and the temperature increased to 37 °C to induce the expression of the recombinases for 1 h. Bacteria were then collected by centrifugation 6000 X g for 15 min at 4 °C and were then resuspended in ice-cold water. This procedure was repeated once before resuspension in 10% glycerol, followed by centrifugation as above and resuspension of the bacteria in the remaining 10% glycerol (about 1 ml). Bacteria were then aliquoted into ice-cold microcentrifuge tubes (50 μl per tube) and snap-frozen in liquid nitrogen before storage at −80 °C until further use.
PCR
PCR primers were designed to generate addition(s), substitution(s) or deletion(s) of specific regions in the wild-type (WT) LHCGR, FSHR, β2AR or CRY2 plasmids25,29,35,36 (Fig. 1). Primer sequences specific for the mutation are listed in Supplementary Table S5. PCR was performed using a high-fidelity polymerase (KOD-Xtreme, Millipore) (see Supplementary Tables S7-S12 for PCR conditions). PCR products were purified by ethanol precipitation followed by DpnI digestion of the template plasmid. DpnI-digested PCR products were ethanol-precipitated and subsequently used for bacterial transformation. DpnI digestion can also be directly performed on the PCR products (1–2 μl) without purification and the DpnI digested products can then be transformed in recombineering bacteria.
Site-directed mutagenesis
Mutagenesis involves only one-step: transformation of the PCR products generated using mutagenesis primers in recombineering bacteria (see Fig. 1 for principle and primer design).
Recombineering
For generating mutants, one tube of electrocompetent cells was used per sample. Cells were first thawed on ice and the PCR product (100 ng) was then added, followed by electroporation in a 1-mm cuvette. Electroporation was performed at 1.35 kV, 25 μF, 200 ohms using an Eppendorf electroporator (2510). After electroporation, bacteria were incubated in 1 ml LB medium at 37 °C, shaking for 1–2 h before plating on LB-agar plates conditioned with the appropriate antibiotic(s).
Analysis of correct clones was performed first by PCR by primers flanking the targeted area using Biotools DNA polymerase and buffer. The general conditions included an initial denaturation at 96 °C for 2 min, followed by 30 cycles with 95 °C for 45 s, 57 °C for 45 s and 72 °C for 1–3 min, depending on the length of the product. Products were analysed by gel electrophoresis and then by sequencing (Turku Centre for Biotechnology, Finland). Sequencing was performed in both directions to ensure accuracy of the mutated modified sequences (See Supplementary Table S6 for sequencing primers).
Determining optimal homology length needed for REPLACR-mutagenesis
Primers were designed for the addition of two nucleotides (AT) in the middle of a ScaI restriction site (AGTACT) in WT human LHCGR plasmid, thereby disrupting the restriction site25. The homology between forward and reverse primers was varied from 2bp to 23 bp (See Supplementary Table S1 for primer sequences). PCR products were subjected to REPLACR-mutagenesis protocol as mentioned above (Fig. 1). The resulting bacterial colonies were analysed by colony PCR (forward primer: AGGGTCCTGATTTGGCTGAT, reverse primer: TGGCATGTCTTAATCGCAGC; see Supplementary Table S3 for PCR conditions). The expected PCR product for the mutated plasmids should be 366 bp and not amenable to ScaI digestion whereas the LHCGR_WT background PCR product should be 364 bp and following ScaI digestion to yield 190 bp and 174 bp products (indistinguishable as a single band; see Supplementary Fig. S1).
Gibson Assembly and GeneArt seamless cloning
The same PCR products with 14 bp and 17 bp homology, as used above with REPLACR-mutagenesis, were subjected to recombination by Gibson Assembly cloning (NEB) and GeneArt seamless cloning (Life technologies) kits following the manufacturers’ protocol and using 100 ng of the linear PCR product. The resulting bacterial colonies were subjected to colony PCR (Supplementary Table S3) and analysed by ScaI restriction digestion of the colony PCR products (see Supplementary Fig. S3 and Table S4).
Additional Information
How to cite this article: Trehan, A. et al. REPLACR-mutagenesis, a one-step method for site-directed mutagenesis by recombineering. Sci. Rep. 6, 19121; doi: 10.1038/srep19121 (2016).
References
Ho, S. N., Hunt, H. D., Horton, R. M., Pullen, J. K. & Pease, L. R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77, 51–59 (1989).
Smith, M. In vitro mutagenesis. Annu. Rev. Genet. 19, 423–462, 10.1146/annurev.ge.19.120185.002231 (1985).
Ling, M. M. & Robinson, B. H. Approaches to DNA mutagenesis: an overview. Anal. Biochem. 254, 157–178, 10.1006/abio.1997.2428 (1997).
Weiner, M. P. et al. Site-directed mutagenesis of double-stranded DNA by the polymerase chain reaction. Gene 151, 119–123 (1994).
Barik, S. Site-directed mutagenesis in vitro by megaprimer PCR. Methods Mol. Biol. 57, 203–215, 10.1385/0-89603-332-5:203 (1996).
Wei, D., Li, M., Zhang, X. & Xing, L. An improvement of the site-directed mutagenesis method by combination of megaprimer, one-side PCR and DpnI treatment. Anal. Biochem. 331, 401–403, 10.1016/j.ab.2004.04.019 (2004).
Tseng, W. C., Lin, J. W., Wei, T. Y. & Fang, T. Y. A novel megaprimed and ligase-free, PCR-based, site-directed mutagenesis method. Anal. Biochem. 375, 376–378, 10.1016/j.ab.2007.12.013 (2008).
Baudin, A., Ozier-Kalogeropoulos, O., Denouel, A., Lacroute, F. & Cullin, C. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res. 21, 3329–3330 (1993).
Murphy, K. C. Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J. Bacteriol. 180, 2063–2071 (1998).
Testa, G. et al. BAC engineering for the generation of ES cell-targeting constructs and mouse transgenes. Methods Mol. Biol. 256, 123–139, 10.1385/1-59259-753-X:123 (2004).
Muyrers, J. P., Zhang, Y., Testa, G. & Stewart, A. F. Rapid modification of bacterial artificial chromosomes by ET-recombination. Nucleic Acids Res. 27, 1555–1557 (1999).
Zhang, Y., Buchholz, F., Muyrers, J. P. & Stewart, A. F. A new logic for DNA engineering using recombination in Escherichia coli. Nat. Genet. 20, 123–128, 10.1038/2417 (1998).
Muyrers, J. P., Zhang, Y. & Stewart, A. F. Techniques: Recombinogenic engineering–new options for cloning and manipulating DNA. Trends Biochem. Sci. 26, 325–331 (2001).
Zhang, Y., Muyrers, J. P., Testa, G. & Stewart, A. F. DNA cloning by homologous recombination in Escherichia coli. Nat. Biotechnol. 18, 1314–1317, 10.1038/82449 (2000).
Maresca, M. et al. Single-stranded heteroduplex intermediates in lambda Red homologous recombination. BMC Mol. Biol. 11, 54, 10.1186/1471-2199-11-54 (2010).
Rivero-Muller, A., Lajic, S. & Huhtaniemi, I. Assisted large fragment insertion by Red/ET-recombination (ALFIRE)–an alternative and enhanced method for large fragment recombineering. Nucleic Acids Res. 35, e78, 10.1093/nar/gkm250 (2007).
Wang, H. et al. Improved seamless mutagenesis by recombineering using ccdB for counterselection. Nucleic Acids Res. 42, e37, 10.1093/nar/gkt1339 (2014).
Wang, S., Zhao, Y., Leiby, M. & Zhu, J. A new positive/negative selection scheme for precise BAC recombineering. Mol. Biotechnol. 42, 110–116, 10.1007/s12033-009-9142-3 (2009).
Wong, Q. N. et al. Efficient and seamless DNA recombineering using a thymidylate synthase A selection system in Escherichia coli. Nucleic Acids Res. 33, e59, 10.1093/nar/gni059 (2005).
Gibson, D. G. Synthesis of DNA fragments in yeast by one-step assembly of overlapping oligonucleotides. Nucleic Acids Res. 37, 6984–6990, 10.1093/nar/gkp687 (2009).
Aslanidis, C. & de Jong, P. J. Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 18, 6069–6074 (1990).
Zhang, Y., Werling, U. & Edelmann, W. SLiCE: a novel bacterial cell extract-based DNA cloning method. Nucleic Acids Res. 40, e55, 10.1093/nar/gkr1288 (2012).
Li, M. Z. & Elledge, S. J. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nature methods 4, 251–256, 10.1038/nmeth1010 (2007).
Stevenson, J., Krycer, J. R., Phan, L. & Brown, A. J. A practical comparison of ligation-independent cloning techniques. PLoS One 8, e83888, 10.1371/journal.pone.0083888 (2013).
Rivero-Muller, A. et al. Rescue of defective G protein-coupled receptor function in vivo by intermolecular cooperation. Proc. Natl. Acad. Sci. USA 107, 2319–2324, 10.1073/pnas.0906695106 (2010).
Rivero-Muller, A. et al. A novel inactivating mutation of the LH/chorionic gonadotrophin receptor with impaired membrane trafficking leading to Leydig cell hypoplasia type 1. Eur. J. Endocrinol. 172, K27–36, 10.1530/EJE-14-1095 (2015).
Testa, G. et al. Engineering the mouse genome with bacterial artificial chromosomes to create multipurpose alleles. Nat. Biotechnol. 21, 443–447, 10.1038/nbt804 (2003).
Edelheit, O., Hanukoglu, A. & Hanukoglu, I. Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol. 9, 61, 10.1186/1472-6750-9-61 (2009).
Kennedy, M. J. et al. Rapid blue-light-mediated induction of protein interactions in living cells. Nature methods 7, 973–975, 10.1038/nmeth.1524 (2010).
Miller, C. A., Ingmer, H. & Cohen, S. N. Boundaries of the pSC101 minimal replicon are conditional. J. Bacteriol. 177, 4865–4871 (1995).
Li, X. T., Thomason, L. C., Sawitzke, J. A., Costantino, N. & Court, D. L. Positive and negative selection using the tetA-sacB cassette: recombineering and P1 transduction in Escherichia coli. Nucleic Acids Res. 41, e204, 10.1093/nar/gkt1075 (2013).
Zhang, Y., Muyrers, J. P., Rientjes, J. & Stewart, A. F. Phage annealing proteins promote oligonucleotide-directed mutagenesis in Escherichia coli and mouse ES cells. BMC Mol. Biol. 4, 1 (2003).
Tischer, B. K., Smith, G. A. & Osterrieder, N. En passant mutagenesis: a two step markerless red recombination system. Methods Mol. Biol. 634, 421–430, 10.1007/978-1-60761-652-8_30 (2010).
Jean-Alphonse, F. et al. Spatially restricted G protein-coupled receptor activity via divergent endocytic compartments. J. Biol. Chem. 289, 3960–3977, 10.1074/jbc.M113.526350 (2014).
Li, X. & Heyer, W. D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 18, 99–113, 10.1038/cr.2008.1 (2008).
Singh, M. et al. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat. Biotechnol. 28, 585–593, 10.1038/nbt.1640 (2010).
Acknowledgements
This work has been supported by the Academy of Finland, Turku Doctoral Programme of Biomedical Sciences (TuBS), Turku Doctoral Programme of Molecular Medicine (TuDMM) and the Ahokas and Sigrid Jusélius Foundations.
Author information
Authors and Affiliations
Contributions
A.R.M. devised the concept. All authors participated in the design of the research. A.T., M.K., J.Cz. and A.R.M. conducted the experiments. A.T. and A.R.M. prepared the manuscript and all the co-authors have approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Trehan, A., Kiełbus, M., Czapinski, J. et al. REPLACR-mutagenesis, a one-step method for site-directed mutagenesis by recombineering. Sci Rep 6, 19121 (2016). https://doi.org/10.1038/srep19121
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep19121
This article is cited by
-
State-of-art engineering approaches for ameliorated production of microbial lipid
Systems Microbiology and Biomanufacturing (2024)
-
A simplified Gibson assembly method for site directed mutagenesis by re-use of standard, and entirely complementary, mutagenesis primers
BMC Biotechnology (2022)
-
A high-efficiency method for site-directed mutagenesis of large plasmids based on large DNA fragment amplification and recombinational ligation
Scientific Reports (2021)
-
Efficient strategy for introducing large and multiple changes in plasmid DNA
Scientific Reports (2018)
-
A restriction-free method for gene reconstitution using two single-primer PCRs in parallel to generate compatible cohesive ends
BMC Biotechnology (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
