
Abstract
Orchids make up about 10% of all seed plant species, have great economical value and are of specific scientific interest because of their renowned flowers and ecological adaptations. Here, we report the first draft genome sequence of a lithophytic orchid, Dendrobium catenatum. We predict 28,910 protein-coding genes and find evidence of a whole genome duplication shared with Phalaenopsis. We observed the expansion of many resistance-related genes, suggesting a powerful immune system responsible for adaptation to a wide range of ecological niches. We also discovered extensive duplication of genes involved in glucomannan synthase activities, likely related to the synthesis of medicinal polysaccharides. Expansion of MADS-box gene clades ANR1, StMADS11, and MIKC*, involved in the regulation of development and growth, suggests that these expansions are associated with the astonishing diversity of plant architecture in the genus Dendrobium. On the contrary, members of the type I MADS box gene family are missing, which might explain the loss of the endospermous seed. The findings reported here will be important for future studies into polysaccharide synthesis, adaptations to diverse environments and flower architecture of Orchidaceae.
Similar content being viewed by others

Introduction
Orchids, constituting approximately 10% of all seed plant species, have enormous value for commercial horticulture and are of specific scientific interest because of their spectacular flowers, ecological adaptations1,2 and secondary metabolites3,4,5,6. Dendrobium is the third largest genus of Orchidaceae and contains approximately 1,450 species, characterised by a fleshy stem with abundant polysaccharides and growing in diverse habitats3,4,5,6. A draft genome sequence of Dendrobium officinale Kimura & Migo has been reported before but the highly fragmented assembly and the presence of multiple peaks in K-mer analyses, suggesting that its sequence is likely derived from an artificial hybrid7, seriously complicate correct interpretation of the genome. To complement the lack of a high quality, well assembled genome sequence for Dendrobium, we here present the genome of D. catenatum Lindl., a lithophytic orchid found in subtropical and temperate regions2 and commonly used as a health food in many Asian countries3,4,5. Analysis of the D. catenatum genome sequence offers insights into flower development and polysaccharide synthesis, as well as its wide distribution.
Results
Genome sequencing and genome characteristics
Dendrobium catenatum (Supplementary Note 1) has thirty-eight (2N = 2X = 38) small chromosomes of approximately 2 μm (Supplementary Note 2 and Supplementary Fig. 1). To sequence its complete genome, we generated a total of 222.51 Gb of raw reads, with multiple insert libraries ranging in size from 180 bp to 20 Kb (Supplementary Table 1). A K-mer analysis estimated the genome size of D. catenatum at 1.11 Gb (Supplementary Fig. 2). Assembly was done with SOAPdenovo28 and Platanus9, but completeness and N50 length of scaffolds were much better with the latter tool (Supplementary Fig. 3), the results of which were used in subsequent analyses. The total length of its assembly was 1.01 Gb (Supplementary Table 2). Mapping all of the paired-end reads to the assembly revealed that 97% of the sequence had a coverage depth greater than five (Supplementary Fig. 4). Further quality analysis indicated that 93% of the set of eukaryotic core genes (CEGMA)10 were present and 97% were partially represented, suggesting near completeness of the euchromatin component. In addition, 93%–95% of the RNA seq data set could be mapped onto the assembled sequence (Supplementary Tables 3 and 4). These results suggest that our genome assembly is of high quality.
A total of 789 Mb of repetitive elements occupying more than 78.1% of the D. catenatum genome were annotated using a method combining structural and homology information. Retrotransposable elements, known to be the dominant form of repeats in angiosperm genomes, constituted a large part of the D. catenatum genome and included the most abundant subtypes, such as LTR/Copia (27.36%), LTR/Gypsy (18.49%), LINE/L1 (8.44%) and LINE/RTE (5.68%), among others. In addition, the percentage of de novo predicted repeats was notably larger than that obtained for repeats based on Repbase11, indicating that D. catenatum has many unique repeats compared to other sequenced plant genomes (Supplementary Note 3 and Supplementary Table 5). Among these elements, long terminal repeats (LTRs) were the most dominant type, accounting for approximately 46% of the genome. After calculating their times of insertion, we discovered that a burst of LTR activity occurred during the last five million years (Supplementary Fig. 5) and therefore, we deduced that these LTRs were inserted into the genome after D. catenatum diverged from Phalaenopsis species (which is estimated to have occurred 22.6–59.6 million years ago, Fig. 1). We annotated 28,910 protein-coding genes (Supplementary Note 4), of which 22,394 (74.9%) were supported by transcriptome data (Supplementary Fig. 6 and Supplementary Table 6). Notably, we found that D. catenatum has, on average, longer genes than most other sequenced plant species, although similar to that of the butterfly orchid Phalaenopsis equestris (Shauer) Rchb. f.12, due to both species having longer average intron lengths (Supplementary Fig. 7 and Supplementary Table 7). Therefore, this feature might be a unique characteristic of Orchidaceae. In addition, we identified 49 microRNAs, 310 transfer RNAs, 248 ribosomal RNAs and 144 small nuclear RNAs in the D. catenatum genome (Supplementary Table 8).

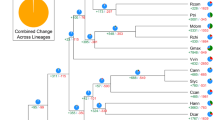
Phylogenetic position and Ks distributions for D. catenatum.
(a) Phylogenetic tree showing the topology and divergence times for 12 plant species, including D. catenatum. Estimated divergence times are indicated by light blue boxes at internodes. Numbers at nodes indicate bootstrap values. The brown bar indicates the orchid-specific whole-genome duplication (WGD), while the green bar indicates a more ancient monocot-specific WGD (Thanks Li-Jun Chen for taking the images of species). (b) Distribution of synonymous substitutions per synonymous site (Ks) for the whole D. catenatum paranome. (c) Distribution of synonymous substitutions per synonymous site (Ks) for orthologous genes found in syntenic regions. Two consistent peaks highlighted by the dashed rectangles are considered to reflect the most recent and older WGD events.
We determined the expansion and contraction of orthologous protein families among species using CAFÉ2.213, which is based on a probabilistic graphical model. For each species, expanded and contracted (compared with their ancestors) gene families were compared with D. catenatum to identify gene families that were uniquely expanded or contracted in D. catenatum (Supplementary Note 5). Seven hundred and fifty-six gene families were found to be expanded in D. catenatum (30 of these significantly) and 804 families contracted (of which four significantly; Supplementary Fig. 8). For the significantly expanded gene families, we conducted GO enrichment analysis and found enrichment for the GO terms ‘DNA metabolic process’, ‘cellular macromolecule metabolic process’, ‘RNA-directed DNA polymerase activity’, ‘primary metabolic process’ and ‘ribonuclease H activity’ (Supplementary Table 9).
We identified 5,758,781 heterozygous single nucleotide polymorphisms (SNPs) in the D. catenatum genome. The heterozygous SNP rate for the whole genome was estimated at 6.28 × 10–3, whereas the SNP rate in exons was as low as 4.98 × 10–3 (Supplementary Fig. 9). Of the 139,830 SNPs that were found in exons, 69,459 caused non-synonymous mutations, affecting 18,404 genes and this suggested that D. catenatum is a high heterozygosity genome. We conducted a Gene Ontology (GO)14 and KEGG15 enrichment analyses of the affected genes and found enrichment of the KEGG pathways ‘Biosynthesis of secondary metabolites’, ‘Plant hormone signal transduction’, ‘Metabolic pathways’ and ‘Isoflavonoid biosynthesis’(Supplementary Table 10) and the GO terms ‘ATP binding’, ‘protein tyrosine kinase activity’ and ‘transition metal ion binding’ (Supplementary Table 11).
Genome evolution
We constructed a highly supported phylogenetic tree and estimated the divergence times of 12 plants based on genes extracted from a total of 677 single-copy families (Fig. 1a). As expected, we found that the D. catenatum is most closely related to P. equestris from which it separated approximately 38 million years ago.
Both the distribution of synonymous substitutions per synonymous site (Ks) across all paralogous genes (regardless of gene order, Fig. 1b) and for duplicated genes lying in synteny blocks (Fig. 1c) show two obvious peaks at Ks values between 0.7–0.9 and 1.5–1.8, suggestive of two rounds of whole-genome duplication (WGDs) in the D. catenatum lineage (Supplementary Note 6). Dating of the WGDs suggests that the most recent WGD appeared near to the Cretaceous–Paleogene (K/Pg) boundary16 and is shared with the WGD event documented recently for P. equestris12. Since it has been suggested that WGDs might facilitate species diversification17,18, it would be interesting to see whether the WGD has also been shared with the species-rich subfamily Orchidoideae (3630 species), which diverged from the Epidendroideae (about 20,000 species, amongst which D. catenatum and P. equestris) about 59 million years ago19 and with the subfamilies Apostasioideae, Vanilloideae and Cypripedioideae, which only include 17, 185 and 180 species, respectively20. Cypripedioideae and the ancestor of Orchidoideae and Epidendroideae subfamilies are assumed to have diverged from each other about 68 million years ago19. Unfortunately, whole genome sequences, or extensive transcriptome data sets from members of these other subfamilies are not yet available. The older peak in the Ks age distribution probably points to one or more older WGD events that have occurred in the monocot lineage, as already previously suggested21.
Gene family evolution
We have also zoomed in on some specific gene families.
Terpene synthase genes
As secondary metabolites, most plant terpenes and their corresponding synthases have evolved selectively to increase fitness by adaptation to specific ecological niches22. Plant terpene synthase (TPS) genes can be divided into seven subfamilies (a, b, c, d, e/f, g and h)22. The TPS genes of D. catenatum and P. equestris all fall into known angiosperm TPS clades, TPS-a, TPS-b, TPS-e/f, TPS-c and TPS-g (Fig. 2). The genome of D. catenatum encodes 39 members of TPS, whereas there are only 21 in the genome of P. equestris. Notably, rapid expansion by tandem gene duplication is particularly common in the TPS-a subfamily of these two orchids. Furthermore, the specific placement of TPS-a genes for D. catenatum and P. equestris (Fig. 2; Supplementary Tables 12 and 13) implies that the expansion of this gene family has occurred in the ancestor of the Epidendroideae subfamily, or at least prior to the divergence of D. catenatum and P. equestris and might have contributed to species radiation in this subfamily, containing over 20,000 species. Indeed, although this needs to be further investigated, a previous study has suggested that the expansion of the TPS-a subfamily might be linked to the radiation of the flowering plants23.

Phylogeny of putative full-length TPSs from D. catenatum (green), P. equestris (purple), O. sativa (blue) and A. thaliana (cyan).
See text for details.
Disease resistance genes
Plant disease resistance genes (R genes) play a key role in recognizing proteins expressed by specific avirulence genes of pathogens21 and form various subfamilies, such as the TIR-domain-containing (for example, TOLL/INTERLEUKIN LIKE RECEPTOR/RESISTANCE PROTEIN) (TIR-NB-LRR), the non-TIR-domain containing (NB-LRR) and the non-TIR coiled-coil domain-containing (CC-NB-LRR) R-protein subfamilies24. The genomes of D. catenatum and P. equestris possess 157 and 79 R genes, respectively (Supplementary Table 14). Although further investigation is required, the dramatic expansion of its R genes suggests that D. catenatum may possess a more powerful disease immune system than P. equestris.
Heat-shock proteins
As molecular chaperones, heat-shock proteins (Hsp) are ubiquitous in plant cells. Hsp genes are not only associated with stress caused by heat shock and other abiotic factors, but have recently also been found to be associated with response to biotic stress25,26. Hsp genes function to manage the stress-induced denaturation of other proteins and can be classified into seven major families based on their molecular weight: small Hsps, Hsp20, Hsp40, Hsp60, Hsp70, Hsp90 and Hsp110. Of those, plants mainly contain Hsp20, Hsp70 and Hsp90 subfamilies. The genome of D. catenatum contains 20 members of Hsp70, whereas there are only 9 in that of P. equestris. Interestingly, in particular Hsp70 genes encoding proteins localizing in the cytoplasm have more members in D. catenatum than in P. equestris (11 vs. 3) (Supplementary Fig. 10). Due to the fact that D. catenatum is found in subtropical and temperate regions (Supplementary Fig. 11), can grow in wet and dry environments4,27, can tolerate both low and high temperatures4,27 and has a much wider distribution than P. equestris (Supplementary Fig. 11), it is interesting to speculate that the additional Hsp70 genes in the D. catenatum genome might have helped in the adaptation to a much wider variety of environments. However, more future work will be necessary to prove or disprove this hypothesis.
Evolution of polysaccharide synthase gene families
The fleshy stem of D. catenatum contains various types of polysaccharides, many of which have medicinal, such as anti-inflammatory, immuno-enhancing, antioxidant and anti-glycation activities4,5,28,29. Among those, particularly glucomannan (GM) and galactoglucomannan (GGM) are two major medicinal polysaccharides in D. catenatum30. Genes involved in GM and GGM biosynthesis were identified through their homology with genes in the Arabidopsis genome. A biosynthetic pathway was proposed and the tissue-specific expression patterns of GM and GGM biosynthesis genes were examined31,32 (Fig. 3 and Supplementary Note 7). The result suggests that the downstream genes of the biosynthesis pathway are highly expressed in stem tissues where high levels of GM and GGM accumulate. Therefore, we focussed on the analysis of these genes in the D. catenatum genome.

Proposed biosynthetic pathway of GM and GGM in Dendrobium stem.
The biosynthetic pathway was modified according to the pathways proposed in Amorphophallus konjac31,32. GM or GGM biosynthesis is supposed to be generated from sucrose, mainly produced by photosynthesis in the leaf tissue (Supplementary Note 7). The enzymes indicated in red are highly expressed in the stem. Only abbreviations of gene names are shown: Csl, Cellulose synthase like gene; FRK, fructokinase; Fru, fructose; Fru-6-P, Fructose-6-phosphate; GGP, GDP-glucose-pyrophosphorylase; GMPP, GDP-mannose pyrophosphorylase; GT, glycosyltransferase; HXK, hexokinase; INV, invertase; MSR, mannan synthesis-related; PGI, phosphoglucose isomerase; PGM, phosphoglucomutase; PMI, phosphomannose isomerase; PMM, phosphomannomutase; RWA, Reduced Wall Acetylation proten; SuS, sucrose synthase; UGE, UDP-galactose epimerase; UGP, UDP-glucose pyrophosphorylase.
Since GM or GGM polysaccharides of D. catenatum are easily extracted with water, they may not be tightly bound to the cell wall and probably act as storage polysaccharides in specialized mucilage cells rather than being structural polysaccharides28,30. Konjac glucomannan (KGM) is water-soluble and accumulates in storage tissues33. It also has several bioactivities, such as reducing plasma cholesterol, removing free radicals and inhibiting tumor genesis and metastasis34. In addition, the backbone structure of KGM is similar to that of GM. Therefore, we included konjac EST sequences responsible for GM synthesis to search for D. catenatum orthologs32.
Previous studies showed that CslA (Cellulose synthase-like A) genes of glycosyltransferase (GT) family 2 are involved in GM backbone synthesis35,36. We found 13 CslA genes in the D. catenatum genome, compared to only 6 copies in the P. equestris genome. This expansion of CslA genes in the D. catenatum genome is mainly due to tandem duplication (three arrays: Dca006365, Dca006366; Dca007032, Dca007033, Dca007034; Dca013434, Dca013437). Interestingly, these genes were grouped in the same clade with AkCslA3, a konjac GM synthase (Supplementary Fig. 12). In addition, two of these genes (Dca006366 and Dca007032) were significantly higher expressed in stem than in other tissues (root, leaf and flower, Supplementary Fig. 13). Therefore, these expanded CslA genes may act as GM or GGM synthases in D. catenatum. Although CslD genes were reported to synthesize mannan rather than GM in A. thaliana, a recent study showed that konjac CslD may also be involved in the synthesis of GM31,36. Based on our phylogenetic analysis, two D. catenatum genes (Dca018361 and Dca000653) cluster with the konjac CslD EST clones (Supplementary Fig. 14). These two genes were highly expressed in stem and leaf, respectively (Supplementary Fig. 15) and suggest their potential roles in the synthesis of GM or GGM.
Arabidopsis CslD5 was reported to play an important role in osmotic stress tolerance37. In addition, GM present in the pseudobulb of an epiphytic CAM orchid, Cattleya forbesii Lindl. × Laelia tenebrosa Rolfe, has been associated with drought tolerance38. The large accumulation of starch, fructan and GM in storage organs of geophytes is critical for their survival in detrimental conditions39. Because Dendrobium species accumulate high amounts of GM and GGM in their stems and/or leaves, it would be interesting to know whether this is also related to adaptation to environmental stresses, such as drought, a common condition experienced by epiphytic or lithophytic Dendrobium species in their natural environment40. The online microarray data from Arabidopsis eFP Browser seems to support this because A. thaliana CslA (CslA7 and CslA10) and CslD (CslD2 and CslD3) genes are induced by drought, osmotic, salt or cold stress (Supplementary Fig. 16).
Storage of carbohydrates in geophytes can serve as carbon and energy sources for the maintenance under adverse environments and for growth under favorable conditions39. In addition, soluble sugars, such as glucose and sucrose, can act as osmolytes under osmotic stress41. Accumulation of these metabolites is enhanced in response to environmental stresses and has been shown to contribute to drought and freezing tolerance41,42. In Easter lily bulbs, when stored at –1.0 °C, large amounts of sucrose, mannose, fructose and oligosaccharides accumulated, suggesting that not only starch but also GM was degraded to soluble sugars during frozen storage43. Therefore, degradation of GM and GGM to monomers in Dendrobium stems might also be induced by stress and play a role in increasing tolerance for drought, cold, salt and osmosis. GM or GGM were reported to be hydrolysed by glycosyl hydrolase families 5 (GH5) enzymes44,45,46. We thus performed a phylogenetic analysis of GH5 genes and analysed their tissue-specific gene expression. As the data show, several DcaGH5 genes were expressed at higher levels in the stems (Supplementary Fig. 17). Among these genes, Dca014977 clusters with LeMAN4, HvMAN1 and AtMAN1, which have been demonstrated to possess hydrolytic activities to GM and GGM44,45,46 (Supplementary Fig. 18). Interestingly, the expression of AtMAN1 and OsMAN4, the rice GH5 gene that grouped with HvMAN1, was significantly induced by cold, osmotic, salt or drought stress (Supplementary Fig. 19). All together, these results strongly suggest that the biological functions of GM or GGM in storage organs of D. catenatum are related to environmental stress tolerance.
A genome-wide analysis of 12 previously sequenced plant genomes and subsequent KEGG enrichment analysis of the 629 D. catenatum specific gene families (Supplementary Figs 20 and 21; Supplementary Table 15) showed that the functional pathways of these unique families were significantly enriched in ‘Tyrosine metabolism’, ‘Fatty acid metabolism’ and ‘Glycolysis/Gluconeogenesis’ (Supplementary Table 16). Intriguingly, the D. catenatum specific genes implicated in the ‘Glycolysis/Gluconeogenesis’ pathway could help to shape and maintain the physiological mechanism that synthesises and stores polysaccharides in the stem.
Evolution of MADS-box genes
Given that orchids are a unique model system for flower development12, we characterised their MADS-box genes, which hold diverse functions in many important processes during plant development, in greater detail. An investigation of the D. catenatum genome revealed 63 putative functional MADS-box genes and 12 pseudogenes (Table 1). As earlier reported for P. equestris, there seem to be fewer MADS box genes present in orchids than in most other angiosperms, such as rice (Oryza sativa; 75 genes) and A. thaliana (108 genes). D. catenatum has 35 type II MADS-box genes (Table 1), compared with 29 in P. equestris. Phylogenetic analysis (Supplementary Fig. 22) shows that most type II MADS-box genes have been duplicated in D. catenatum, except for those in the B-PI clade. Among these clades, ANR1 (with three members), StMADS11 (three members), MIKC* (three members) and Bs (two members) contain more members than in P. equestris (two members in ANR1 and one member in other three clades, respectively) (Supplementary Fig. 23). The ANR1 MADS-box gene in Arabidopsis is a key gene involved in regulating lateral development in response to external nitrate supply47. Genes in the StMADS11 clade have functions in controlling flowering time and inflorescence architecture48,49. Genes in the Bs clade can regulate seed development and fruit size50. Recent evidence indicated that the closely related MIKC* MADS-domain proteins are important for the functioning of the A. thaliana male gametophyte51. However, genes corresponding to the FLC, AGL12 and AGL15 clades could not be found in the D. catenatum genome nor in the P. equestris genome. FLC genes have recently been found in cereals, although they have proved difficult to identify because they diverged extensively within a relatively short period52. However, AGL12 clade genes are present in the genomes of rice and A. thaliana, while AGL15 clade genes are only present in A. thaliana. Therefore, we hypothesise that orthologues of FLC, AGL12 and AGL15 might have been lost in orchids.
Only 28 putative functional MADS-box type I genes and one pseudogene were found in D. catenatum (Supplementary Table 17), suggesting that the D. catenatum type I MADS-box genes have experienced a lower birth rate compared with those of type II MADS-box genes. Tandem gene duplication events seem to have contributed to the increase in type I Mα MADS-box genes (Supplementary Fig. 23), indicating that these genes have mainly been duplicated by recent, small-scale duplications53. We found that type I Mα MADS-box genes DcMADS30 and DcMADS31 and DcMADS57 and DcMADS58 are located side by side in scaffold12110 and scaffold5677, respectively. In addition, three type I Mα MADS-box genes DcMADS47, DcMADS48 and DcMADS50 were also found in the same scaffold7526. Interestingly, the D. catenatum genome does not contain any type I Mβ MADS-box genes, although these genes do exist in Arabidopsis, poplar and rice. Interactions among type I MADS-box genes are important for the initiation of endosperm development54. The failed development of endosperm in orchids might be related to the smaller number of type I MADS box genes in the D. catenatum genome.
In conclusion, Dendrobium represents a fascinating groups of orchids because of their fleshy stem, various flower architectures and synthesis of many kinds of different polysaccharides and the D. catenatum genome sequence forms an important resource for further exploring orchid gene and genome evolution.
Method
Sample preparation and sequencing
For genome sequencing, we collected leaves, stems and flowers from an individual of wild D. catenatum (voucher specimen : CHINA.Yunnan: Guangnan county, on rock in evergreen broad-leaf forest, alt. 1350 m,10 March, 2010, Z.J. Liu 4979, NOCC) and extracted genomic DNA using a modified CTAB protocol. Sequencing libraries with insert sizes ranging from 180 bp to 20 Kb (Supplementary Table 1) were constructed using a library construction kit (Illumina, San Diego, CA). These libraries were then sequenced using Illumina HiSeq 2000 platform. The raw reads generated were filtered according to the sequencing quality, presence of adaptor contamination and duplication. Thus, only high-quality reads were used for genome assembly.
Genome size estimation
To estimate the genome size of D. catenatum, we used reads from pair-end libraries to determine the distribution of K-mer values. According to the Lander–Waterman theory55, the genome size can be determined by the total number of K-mers that were divided by the peak value of the K-mer distribution. Given the high heterozygosity in the D. catenatum genome, we found two peaks in the distribution (Supplementary Fig. 2). Using the second peak as the expected K-mer depth and the formula Genome size = Total K-mer/Expected K-mer depth, the size of the haploid genome was estimated to be 1.11 Gb (haploid).
Sequence assembly
Initially, we used SOAPdenovo28 to assemble the genome, which produced an assembly of 1.27 Gb with an N50 scaffold size of 80.56 Kb and a corresponding N50 contig size of 6.64 Kb (Supplementary Table 18). These figures suggest high fragmentation and redundancy. Therefore, to generate a better assembly for further analyses, Platanus9, which can effectively manage high-throughput data from heterozygous samples, was used for whole genome shotgun assembly. We subsequently used GapCloser (http://soap.genomics.cn) to fill gaps remaining after the Platanus built-in gap-filling module had been applied. The final assembly was 1.01 Gb in length, approximately 91% of the estimated genome size, with an N50 scaffold size of 391 Kb and a corresponding N50 contig size of 33.1 Kb (Supplementary Table 2).
Gene and non-coding RNA gene prediction
MAKER56 was used to generate a consensus gene set based on de novo prediction, homology annotation with CEGMA10 and other sequenced monocots and RNA-seq gene prediction. These results were integrated into a final set of 28,910 protein-coding genes for annotation (Supplementary Table 19). We then generated functional assignments of the D. catenatum genes by aligning their CDS (protein-coding sequences) to sequences available in the public protein databases including KEGG15, SwissProt57, TrEMBL57 and InterProScan58 (Supplementary Table 20). tRNA genes were searched for by tRNAscan-SE59. For rRNA identification, we downloaded the Arabidopsis rRNA sequences from NCBI and aligned them against the D. catenatum genome to identify possible rRNAs. Additionally, other types of non-coding RNAs, including miRNA and snRNA, were identified by utilizing INFERNAL60 to search from the Rfam database.
Single nucleotide polymorphisms
We used the BWA program14 to remap the pair-end (500 bp) clean reads to the assembled scaffolds. After merging the BAM results, sorting the alignments by the leftmost coordinates and removing potential PCR duplicates, we used SAMtools15 ‘mpileup’ to identify single nucleotide polymorphisms (SNPs) and short INDELs. We rejected SNPs and InDels within reads with depths lower (<5 folds) or higher (>80 folds) than expected. Filtering was achieved using the vcfutils.pl varFilter tool in the SAMtools package, with parameters -Q 10 -d 5 -D 86. We estimated heterozygosity rates as the density of heterozygous SNPs from the whole genome, gene intervals, introns and exons.
Gene family identification
We downloaded genome and annotation data from Amborella (Am.) trichopoda (http://amborella.huck.psu.edu, version 1.0), Arabidopsis (A.) thaliana (TAIR 10), Brachypodium distachyon (purple false brome; Phytozome v9.0), Musaceae acuminata (http://ensemblgenomes.org, release-21), Oryza sativa (Nipponbare, IRGSP-1.0), Phoenix (Ph.) dactylifera (http://qatar-weill.cornell.edu/research/datepalmGenome), Phalaenopsis equestris (ftp://ftp.genomics.org.cn/from_BGISZ/20130120/), Populus (Po.) trichocarpa (http://ensemblgenomes.org, release-21), Sorghum (S.) bicolor (sorghum; Phytozome v9.0), Spirodela (Sp.) polyrhiza (common duckweed; http://www.spirodelagenome.org) and Vitis vinifera (Phytozome v9.0), Zea mays (http://www.plantgdb.org/ZmGDB), Phyllostachys (Phy.) heterocycla (http://www.bamboogdb.org). We chose the longest transcript to represent each gene and removed gene models with an open reading frame (ORF) shorter than 150 bp. These protein sets were aligned and clustered using OrthoMCL61.
Phylogenomic dating
We conducted phylogenomic dating with PAML McMcTree62. The McMc process was run for 1,500,000 iterations, with a sample frequency of 150 after a burn-in of 500,000 iterations. Other parameters used the default settings of McMcTree. Two independent runs were performed to check convergence. The following constraints were used for time calibrations:
-
1
140–150 million years ago (MYA) for the monocot – dicot split63,
-
2
94 MYA as the lower boundary for the Vitis – Eurosid split52,
-
3
130 MYA as the lower boundary for the Alismatales – Acorales and core monocots (Commelinids, Asparagales, Liliales, etc.) split64, and
-
4
200 MYA as the upper boundary for basal angiosperms65.
Based on these divergence time ranges and the inferred phylogenetic tree, the divergence times between the 12 species were estimated using McMcTree software.
Identification of resistance genes
HMMER V3.0 was used to align the protein sequences of D. catenatum against the hidden Markov model of the Pfam NBS (NB-ARC). The TIR and LRR domains were detected by using the Pfam_Scan (−E 0.01 –domE 0.01). MARCOIL66 and paircoil267 were utilized for identification of the CC motif.
Identification of polysaccharide-related genes
We collected polysaccharide-related genes of Arabidopsis first by using the CAZY database and other information resources. Then, we performed TBLASTN search against all coding sequences (CDS) datasets of each plant species. These CDS datasets were downloaded from Phytozome (poplar, Selaginella and Physcomitrella), ConGenIE (Norway spruce), QATAR-WEILL.CORNELL (dates palm), RAP-DB (rice) and TAIR (Arabidopsis). In case of Amo. Konjac, RNA-seq data in NCBI SRA (accession number SRX098311) was downloaded and assembled by CLC genomic workbench software. Homologous genes from these species with BLAST E-values less than 1e-5 were then used as BLASTX queries against all protein sequences in Arabidopsis. If the top-hit genes of this BLASTX results belonged to polysaccharide-related genes defined previously, the queries were defined as orthologues in each species. The phylogenetic trees of collected orthologs were constructed by ClustalW68.
Evolution of MADS box genes in D. catenatum
The MADS-box domain is comprised of 60 amino acids, which we identified for all the potential MADS-box sequences of D. catenatum. Next, we aligned all the MADS-box genes with ClustalW. An un-rooted neighbour-joining phylogenetic tree was constructed in MEGA569 with default parameters. Confidence on the tree branches was evaluated by bootstrap analysis (1000 replicates).
Associated references and supplementary information are available in the online version of the paper.
Additional Information
Accession codes: Genome sequences have been submitted to the National Center for Biotechnology Information (NCBI). Whole genome assemblies have been deposited in DDBJ/EMBL/GenBank under the accession codes JSDN00000000 (URL: http://www.ncbi.nlm.nih.gov/bioproject/262478).
How to cite this article: Zhang, G.-Q. et al. The Dendrobium catenatum Lindl. genome sequence provides insights into polysaccharide synthase, floral development and adaptive evolution. Sci. Rep. 6, 19029; doi: 10.1038/srep19029 (2016).
References
Roberst, D. L. D. & Orchids, K. W. Curr Biol 18, 325–329 (2008).
Pridgeon A. M., Cribb, J. P., Chase W. M. & Rasmussen F. N. In Genera Orchidacearum, Vol. 6, Epidendroideae (Part Three), 3–544 (Oxford University Press, Oxford, UK, 2013).
Leitch, I. J. et al. Genome size diversity in orchids: consequences and evolution. Ann Bot 104, 469–481 (2009).
Ng, T. B. et al. Review of research on Dendrobium, a prized folk medicine. Appl Microbiol Biot 93, 1795–1803 (2012).
Pan, L. H. et al. Comparison of hypoglycemic and antioxidative effects of polysaccharides from four different Dendrobium species. Int J Biol Macromol 64, 420–427 (2014).
Dressler, R. L. In Phylogeny and Classification of the Orchid Family. 7–278 (Cambridge University Press, Australia, 1993).
Yan, L. et al. The genome of Dendrobium officinale illuminates biology of the important traditional Chinese orchid herb. Mol Plant 10.1016/j.molp.2014.12.011 (2014).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1, 18 (2012).
Kajitani, R. et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res 24, 1384–1395 (2014).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Cai, J. et al. The genome sequence of the orchid Phalaenopsis equestris. Nat Genet 47, 65–72 (2015).
De Bie, T., Cristianini, N., Demuth, J. P. & Hahn, M. W. CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271 (2006).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29 (2000).
Ogata, H. et al. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 27, 29–34 (1999).
Fawcett, J. A., Maere, S. & Van de Peer, Y. Plants with double genomes might have had a better chance to survive the Cretaceous-Tertiary extinction event. Proc Natl Acad Sci USA 106, 5737–5742 (2009).
Van de Peer, Y., Maere, S. & Meyer, A. The evolutionary significance of ancient genome duplications. Nat Rev Genet 10, 725–732 (2009).
Oderda, G. M. et al. Creating a path to the summit by thinking off the map: report of the 2008-2009 Academic Affairs Committee. Am J Pharm Educ 73 Suppl, S7 (2009).
Gustafsson, A. L., Verola, C. F. & Antonelli, A. Reassessing the temporal evolution of orchids with new fossils and a Bayesian relaxed clock, with implications for the diversification of the rare South American genus Hoffmannseggella (Orchidaceae: Epidendroideae). BMC Evol Biol 10, 177 (2010).
The Angiosperm Phylogeny, G. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot J Linn Soc 161, 105–121 (2009).
Salse, J. In silico archeogenomics unveils modern plant genome organisation, regulation and evolution. Curr Opin Plant Biol 15, 122–130 (2012).
Chen, F., Tholl, D., Bohlmann, J. & Pichersky, E. The family of terpene synthases in plants: a mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J 66, 212–229 (2011).
Amborella Genome Project. The Amborella genome and the evolution of flowering plants. Science 342, 1241089 (2013).
McHale, L., Tan, X., Koehl, P. & Michelmore, R. W. Plant NBS-LRR proteins: adaptable guards. Genome Biol 7, 212 (2006).
Wang, W., Vinocur, B., Shoseyov, O. & Altman, A. Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci 9(5), 244–252 (2004).
Rensing, S. A. et al. The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science 319, 64–69 (2008).
Duan, J. & Duan, Y. P. In Cultivative Technology of Dendrobium catenatum. 1–151 (Fujian Science and Technology Press, Fuzhou, China, 2013).
Wang, J. H., Zha, X. Q., Luo, J. P. & Yang, X. F. An acetylated galactomannoglucan from the stems of Dendrobium nobile Lindl. Carbohyd Res 345, 1023–1027 (2010).
Hsieh, Y. S. et al. Structure and bioactivity of the polysaccharides in medicinal plant Dendrobium huoshanense. Bioorg Med Chem 16(11), 6054–6068 (2008).
Xing, X. et al. A review of isolation process, structural characteristics and bioactivities of water-soluble polysaccharides from Dendrobium plants. Bioact Carbohydrates Dietary Fibre 1, 131–147 (2013).
Diao, Y. et al. De novo transcriptome and small RNA analyses of two amorphophallus species. PLoS ONE 9, e95428 (2014).
Gille, S. et al. Deep sequencing of voodoo lily (Amorphophallus konjac): an approach to identify relevant genes involved in the synthesis of the hemicellulose glucomannan. Planta 234, 515–526 (2011).
Chua, M., Hocking, T. J., Chan, K. & Baldwin, T. C. Temporal and spatial regulation of glucomannan deposition and mobilization in corms of Amorphophallus konjac (Araceae). Am J Bot 100, 337–345 (2013).
Yao-ling, L., Rong-hua, D., Ni, C., Juan, P. & Jie, P. Review of Konjac Glucomannan: Isolation, Structure, Chain Conformation and Bioactivities. J Single Mol Res 1, 7–14 (2013).
Dhugga, K. S. et al. Guar seed beta-mannan synthase is a member of the cellulose synthase super gene family. Science 303, 363–366 (2004).
Verhertbruggen, Y., Yin, L., Oikawa, A. & Scheller, H. V. Mannan synthase activity in the CSLD family. Plant Signal Behav 6, 1620–1623 (2011).
Zhu, J. et al. A cellulose synthase-like protein is required for osmotic stress tolerance in Arabidopsis. Plant J 63(1), 128–140 (2010).
Stancatoa, G. C., Mazzafera, P. & Buckeridge, M. S. Effect of a drought period on the mobilization of non-structural carbohydrates, photosynthetic efficiency and water status in an epiphytic orchid. Plant Physiol Bioch 39, 1009–1016 (2001).
Ranwala, A. P. & Miller, W. B. Analysis of nonstructural carbohydrates in storage organs of 30 ornamental geophytes by high-performance anion-exchange chromatography with pulsed amperometric detection. New Phytol 180(2), 421–433 (2008).
Fan, H. H. et al. Effects of exogenous nitric oxide on antioxidation and DNA methylation of Dendrobium huoshanense grown under drought stress. Plant Cell Tiss Org 109, 307–314 (2012).
Rosa, M. et al. Soluble sugars–metabolism, sensing and abiotic stress: a complex network in the life of plants. Plant Signal Behav 4(5), 388–393 (2009).
Mattana, M. et al. Overexpression of Osmyb4 enhances compatible solute accumulation and increases stress tolerance of Arabidopsis thaliana. Physiol Plantarum 125, 212–223 (2005).
Miller, W. B. & Langhans, R. W. Low temperature alters carbohydrate metabolism in Easter lily bulbs. HortScience 25(4), 463–465 (1990).
Hrmova, M. et al. Hydrolysis of (1,4)-β-D-mannans in barley (Hordeum vulgare L.) is mediated by the concerted action of (1,4)- β-D-mannan endohydrolase and β-D-mannosidase. Biochem J 399(1), 77–90 (2006).
Schroder, R., Wegrzyn, T. F., Sharma, N. N. & Atkinson, R. G. LeMAN4 endo-beta-mannanase from ripe tomato fruit can act as a mannan transglycosylase or hydrolase. Planta 224(5), 1091–1102 (2006).
Wang, Y., Vilaplana, F., Brumer, H. & Aspeborg, H. Enzymatic characterization of a glycoside hydrolase family 5 subfamily 7 (GH5_7) mannanase from Arabidopsis thaliana. Planta 239(3), 653–665 (2014).
Zhang, H. & Forde, B. G. An Arabidopsis MADS box gene that controls nutrient-induced changes in root architecture. Science 279, 407–409 (1998).
Torti, S. & Fornara, F. AGL24 acts in concert with SOC1 and FUL during Arabidopsis floral transition. Plant Signal Behav 7, 1251–1254 (2012).
Liu, C. et al. A conserved genetic pathway determines inflorescence architecture in Arabidopsis and rice. Dev Cell 24, 612–622 (2013).
Prasad, K. & Ambrose, B. A. Shaping up the fruit: control of fruit size by an Arabidopsis B-sister MADS-box gene. Plant Signal Behav 5, 899–902 (2010).
Kwantes, M., Liebsch, D. & Verelst, W. How MIKC* MADS-Box Genes Originated and Evidence for Their Conserved Function Throughout the Evolution of Vascular Plant Gametophytes. Mol Biol Evol 29, 293–302 (2012).
Ruelens, P. et al. FLOWERING LOCUS C in monocots and the tandem origin of angiosperm-specific MADS-box genes. Nat Commun. 4, 2280 (2013).
Parenicova, L. et al. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: new openings to the MADS world. Plant Cell 15, 1538–1551 (2003).
Masiero, S., Colombo, L., Grini, P. E., Schnittger, A. & Kater, M. M. The emerging importance of type I MADS box transcription factors for plant reproduction. Plant Cell 23, 865–872 (2011).
Lander, E. S. & Waterman, M. S. Genomic mapping by fingerprinting random clones: a mathematical analysis. Genomics 2, 231–239 (1988).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 31, 365–370 (2003).
Zdobnov, E. M. & Apweiler, R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25, 955–964 (1997).
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337 (2009).
Li, L., Stoeckert, C. J., Jr. & Roos, D. S. OrthoMCL: identification of or tholog groups for eukaryotic genomes. Genome Res 13, 2178–2189 (2003).
Yang, Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13, 555–556 (1997).
Gaut, B. S., Morton, B. R., McCaig, B. C. & Clegg, M. T. Substitution rate comparisons between grasses and palms: synonymous rate differences at the nuclear gene Adh parallel rate differences at the plastid gene rbcL. Proc Natl Acad Sci USA 93, 10274–10279 (1996).
Hsiao, Y. Y. et al. Transcriptomic analysis of floral organs from Phalaenopsis orchid by using oligonucleotide microarray. Gene 518, 91–100 (2013).
Li, H. et al. Rice MADS6 interacts with the floral homeotic genes SUPERWOMAN1, MADS3, MADS58, MADS13 and DROOPING LEAF in specifying floral organ identities and meristem fate. Plant Cell 23, 2536–2552 (2011).
Delorenzi, M. & Speed, T. An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics 18(4), 617–625 (2002).
McDonnell, A. V., Jiang, T., Keating, A. E. & Berger, B. Paircoil2: improved prediction of coiled coils from sequence. Bioinformatics 22(3), 356–358 (2006).
Thompson, J. D., Gibson, T. J. & Higgins, D. G. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics 10.1002/0471250953.bi0203s00 (2002).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol Biol Evol 28, 2731–2739 (2011).
Potato Genome Sequencing, C. et al. Genome sequence and analysis of the tuber crop potato. Nature 475, 189–195 (2011).
Leseberg, C. H., Li, A., Kang, H., Duvall, M. & Mao, L. Genome-wide analysis of the MADS-box gene family in Populus trichocarpa. Gene 378, 84–94 (2006).
Duan, W. et al. Genome-wide analysis of the MADS-box gene family in Brassica rapa (Chinese cabbage). Mol Genet Genomics 290, 239–255 (2015).
Wei, B. et al. Genome-wide analysis of the MADS-box gene family in Brachypodium distachyon. PLoS ONE 9, e84781 (2014).
Arora, R. et al. MADS-box gene family in rice: genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genomics 8, 242 (2007).
Acknowledgements
The authors acknowledge support from the 948 programme from the State Forestry Administration P. R. China (no. 2011–4–53), the Funds for Forestry Science and Technology Innovation Project of Guangdong, China (no. 2011KJCX009; no. 2013KJCX014–05), the Funds for Environmental Project of Shenzhen, China (no. 2013-02), the Funds for Technology Research and Development Project of Shenzhen, China (no. CXZZ20120830103025851), the Funds for the Development of Strategic Emerging Industries of Shenzhen, China (no. NY20130205008) to Z.-J. L. The authors appreciate technical help of Dr. Saqib Muhummad (AIST) for assembling Amorphophallus (Amo.) konjac RNA-seq data. Y.V.d.P. acknowledges the Ghent University Multidisciplinary Research Partnership ‘Bioinformatics: From Nucleotides to Networks’ and support from the European Union Seventh Framework Programme (FP7/2007-2013) under European Research Council Advanced Grant Agreement 322739–DOUBLE-UP.
Author information
Authors and Affiliations
Contributions
Z.-J.L. and G.-Q.Z. managed the project. Z.-J.L., G.-Q.Z., C.B., Y.V.d.P., Y.-B.L., Q.X., K.-W.L., L.-S.Z., F.C., W.-C.T., Z.-W.W. and Y.-Y.H. planned and coordinated the project and wrote the manuscript. Z.-J.L., Y.S., Y.-Y.S., Y.-Q.Z., L.-J.C., H.D., S.-C.N., J.H., M.W., G.-H.L., X.-J.X., H.H., Y.Y. and H.-J.Y. collected and grew the plant material. Q.X., Z.-J.L., W.-C.T., K.-W.L., L.-J.C., Y.S., Y.-Y.S., M.L. and Y.Y. prepared samples. C.B., G.-Q.Z., Z.-J.L., Y.-Q.Z. and K.-W.L. sequenced and processed the RAW data. Z.-W.W., S.-L.Z., X.Z., C.D., C.B. and G.-Q.Z. annotated the genome. Z.-J.L., Z.-W.W., S.-L.Z., X.Z., L.-S.Z., F.C. and C.D. analyzed gene family. Z.-J.L., Z.-W.W., K.-W.L., G.-Q.Z. and C.B. conducted genome evolution analysis. C.B., L.-S.Z., F.C., Z.-J.L., G.-Q.Z., K.Y., N.M., C.-M.Y., M.O.-T. and Q.X. conducted secondary metabolites and R gene analysis. W.-C.T., Y.-Y.H., Z.-J.L., S.-B.C., L.-S.Z., F.C., W.-L.W., Y.-Y.C. and K.-W.L. conducted the MADS-box gene analysis. C.B., H.D., L.-S.Z., Z.-J.L., G.-Q.Z., Y.-Q.Z. and K.-W.L. conducted transcriptome sequencing and analysis.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhang, GQ., Xu, Q., Bian, C. et al. The Dendrobium catenatum Lindl. genome sequence provides insights into polysaccharide synthase, floral development and adaptive evolution. Sci Rep 6, 19029 (2016). https://doi.org/10.1038/srep19029
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep19029
This article is cited by
-
Uncovering the involvement of DoDELLA1-interacting proteins in development by characterizing the DoDELLA gene family in Dendrobium officinale
BMC Plant Biology (2023)
-
Genome-wide identification, characterization and transcriptional profile of the SWEET gene family in Dendrobium officinale
BMC Genomics (2023)
-
Genome-wide identification of Aux/IAA and ARF gene families reveal their potential roles in flower opening of Dendrobium officinale
BMC Genomics (2023)
-
Genome-wide analysis of the NAC gene family and functional verification of the DcNAC043s in Dendrobium catenatum
Plant Growth Regulation (2023)
-
Identification and transcriptome data analysis of ARF family genes in five Orchidaceae species
Plant Molecular Biology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
