
Abstract
Incarvillea sinensis is a Bignoniaceae plant used to treat rheumatism and relieve pain in traditional Chinese medicine. As a major component of I. sinensis, incarvillateine has shown analgesic activity in mice formalin tests. Using a series of animal models, this study further evaluated the effects of incarvillateine against acute, inflammatory and neuropathic pain. Incarvillateine (10 or 20 mg/kg, i.p.) dose-dependently attenuated acetic acid-induced writhing, but did not affect thermal threshold in the hot plate test. In a Complete Freund’s Adjuvant model, incarvillateine inhibited both thermal hyperalgesia and paw edema and increased interleukin-1β levels. Additionally, incarvillateine attenuated mechanical allodynia induced by spared nerve injury or paclitaxel, whereas normal mechanical sensation was not affected. Incarvillateine did not affect locomotor activity and time on the rotarod at analgesic doses and no tolerance was observed after 7 consecutive daily doses. Moreover, incarvillateine-induced antinociception was attenuated by theophylline, 1,3-dipropyl-8-cyclopentylxanthine and 3,7-dimethyl-1-propargylxanthine, but not naloxone, indicating that the effects of incarvillateine on chronic pain were related to the adenosine system, but not opioid system. These results indicate that incarvillateine is a novel analgesic compound that is effective against inflammatory and neuropathic pain and that its effects are associated with activation of the adenosine system.
Similar content being viewed by others
Introduction
Pain is the most common reason people seek medical care and treatment of pain remains challenging in the clinic. In particular, chronic inflammatory and neuropathic pain can persist and significantly affect the health and quality of life of patients. Various drug classes, including antidepressants, anti-epileptics, opioids and topical anesthetics, are used to treat pain in the clinic1. However, their clinical applications are limited by either poor therapeutic action and/or marked side effects2. Therefore, there is a constant demand for novel compounds that will effectively inhibit pathological pain with limited influence on normal nociception and other physiological functions.
Among various therapeutic targets, the adenosine receptor system is promising for the treatment of pain3. Adenosine receptors, namely A1, A2A, A2B and A3 receptors, are widely distributed in the spinal cord and brain areas involved in pain transmission, as well as peripheral sensory afferents or adjacent cells4,5,6. Administration of adenosine or adenosine receptor agonists inhibits pain-related behaviors in animal and human models7,8,9,10. Compared to classical analgesics, such as opioids, adenosine receptor agonists are particularly effective in chronic pain that arises from inflammation and neuropathy11,12. However, direct-acting agonists may exert CNS side-effects, including general sedation and locomotor suppression, which raises serious concerns when considering the development of agents affecting the adenosine system13,14.
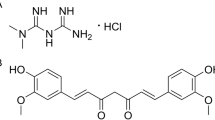
Incarvillea sinensis is a Bignoniaceae plant that has been widely used as a herbal medicine for more than 1400 years in China. In traditional Chinese medicine, I. sinensis is used to treat rheumatism, bruises and wounds and is effective in attenuating pain and inflammation15. Since the 1990 s, photochemists and pharmacologists have investigated the active components in I. sinensis, isolating more than 13 monoterpene alkaloids and 7 macrocyclic spermine alkaloids16,17,18,19,20,21,22,23,24. Incarvillateine (Fig. 1) is considered the major active component, with a characteristic dimeric structure and five contiguous stereocenters on the bicyclic piperidine moiety16. Previous reports showed that incarvillateine attenuates formalin-induced pain in mice with a higher potency than morphine25. Moreover, the antinociceptive effect of incarvillateine in this model can be attenuated by non-selective adenosine receptor antagonist, non-selective opioid receptor antagonist and μ and κ opioid receptor antagonists25,26. Nevertheless, no other study has addressed the pharmacological effects of incarvillateine. The difficult isolation procedure and small quantity yield also pose hurdles for further research and development of incarvillateine.

Chemical structure of incarvillateine.
For more than a decade, efforts have been made to artificially synthesize incarvillateine and its total synthesis was achieved in 200427,28,29. We also developed a method for concise, total, enantioselective synthesis of incarvillateine in a rapid, efficient and economic manner30. Taking advantage of these improvements, we investigated the pharmacological effects of incarvillateine in a set of experimental pain models, with a focus on its effects on chronic pathological pain. Furthermore, we explored possible mechanisms underlying its antinociceptive effects. Our results showed that incarvillateine is a novel analgesic for inflammatory and neuropathic pain, with limited influence on normal pain and motor function. The effects of incarvillateine against pathological pain may be closely related to the activation of the adenosine receptor system, but not the opioid receptor system.
Results
Effect of incarvillateine in the writhing and hot plate tests
The antinociceptive effects of incarvillateine on acute pain were evaluated using the mice writhing and hot plate tests. In the writhing test, intraperitoneal (i.p.) injection of acetic acid induced significant nociceptive responses in vehicle-treated mice. Administration of incarvillateine 30 min before acetic acid injection resulted in a dose-dependent antinociceptive effect [F(3, 35) = 11.33, p < 0.001], producing 61.4 and 78.5% inhibition of the pain behavior at doses of 10 and 20 mg/kg, respectively. The classical NSAID, aspirin DL-lysine (100 mg/kg, i.p.), also produced 85.4% inhibition of pain (Fig. 2A). In the 55 °C hot plate test, incarvillateine did not affect the pain threshold significantly, whereas morphine (10 mg/kg, s.c.) produced a potent antinociceptive effect up to 90 min after administration (Fig. 2B).

Effect of incarvillateine on pain behaviors in the mice acetic acid writhing (A) and hot plate (B) tests. In the writhing test, mice were intraperitoneally administered vehicle (Veh), incarvillateine (INCA, 10 or 20 mg/kg), or aspirin DL-lysine (Asp, 100 mg/kg) 30 min prior to acetic acid injection. In the hot plate test, mice were intraperitoneally administered vehicle (Veh), incarvillateine (INCA, 10 or 20 mg/kg), or subcutaneously administered morphine (Mor, 10 mg/kg) and tested before and at different times after drug administration. Each data point represents the mean ± S.E.M., n = 10. ***P < 0.001, compared with vehicle-treated mice.
Incarvillateine reduced Complete Freund’s adjuvant (CFA)-induced inflammation and pain
Injection of CFA in the paw is an established model of inflammation that induces thermal hyperalgesia from 24 to 72 h31. Consistent with previous reports, thermal withdrawal latency was decreased 48 h after CFA injection. Administration of incarvillateine dose-dependently attenuated CFA-induced thermal hyperalgesia [F(3, 140) = 51.71, p < 0.001], with significant effects observed at both 10 and 20 mg/kg 30 min after administration. At 20 mg/kg, the effect of incarvillateine lasted for more than 150 min. Indomethacin, a positive control, also significantly attenuated CFA-induced decreases in withdrawal latency (Fig. 3A).

Effects of incarvillateine on CFA-induced inflammation and pain.
(A) At 48 h after CFA injection, mice were intraperitoneally administered vehicle (Veh), incarvillateine (INCA, 10 or 20 mg/kg), or indomethacin (Indo, 30 mg/kg) and the thermal withdrawal latency was measured before and at different times after drug administration. (B) Paw edema was measured 30 min after intraperitoneal administration of vehicle (Veh) and incarvillateine (INCA, 10, 20 mg/kg). (C) IL-1β levels in inflamed hindpaw tissue were determined by enzyme-linked immunosorbent assay (ELISA) immediately after paw edema measurements. Each data point represents the mean ± S.E.M., n = 7–13. *P < 0.05, **P < 0.01, ***P < 0.001, compared with vehicle-treated mice. ###P < 0.001, compared with control mice that received saline instead of CFA.
In addition to attenuating thermal hyperalgesia, incarvillateine also attenuated CFA-induced paw edema. As shown in Fig. 3B, intraplantar injection of CFA resulted in a significant increase in paw thickness. Incarvillateine (20 mg/kg) produced a mild, but significant, inhibition of paw edema [F(3,45) = 16.91, p < 0.001]. Consistent with this observation, the CFA-induced increase in tissue interleukin (IL)-1β was significantly inhibited by 20 mg/kg incarvillateine [F(3,27) = 89.06, p < 0.001; Fig. 3C].
Incarvillateine reduced spared nerve injury (SNI)-induced neuropathic pain
As shown in Fig. 4A, SNI surgery, which reflects neurotrauma in the clinic, induced significant mechanical allodynia. Compared to sham-operated mice, the mean mechanical sensitivity in SNI mice decreased from 0.88 to < 0.07 g. Vehicle administration did not affect the mechanical withdrawal threshold, whereas 20 mg/kg incarvillateine produced a significant antinociceptive effect for more than 90 min [F(4, 264) = 78.29, p < 0.001]. Gabapentin, a positive control, also reversed SNI-induced decreases in mechanical threshold. To investigate possible tolerance in incarvillateine-induced antinociception, a repeated dose study was conducted in the same model. As shown in Fig. 4B, no significant reduction in the antinociception was observed in SNI mice treated with 20 mg/kg incarvillateine once daily for 7 consecutive days (p > 0.05). Moreover, 100 mg/kg gabapentin, but not 20 mg/kg incarvillateine, affected mechanical sensitivity in normal mice (Fig. 4C).

Effects of incarvillateine on SNI-induced neuropathic pain.
(A) After 5–7 days of recovery from SNI surgery, mice were intraperitoneally administered vehicle (Veh), incarvillateine (INCA, 10 or 20 mg/kg), or gabapentin (Gaba, 100 mg/kg). The mechanical withdrawal threshold was measured using von Frey filaments before and at different times after drug administration. (B) Mice were intraperitoneally administered with incarvillateine (INCA, 20 mg/kg) once daily for 7 consecutive days and the mechanical threshold was measured 30 min after administration on day 1, 3, 5 and 7. (C) Control mice naïve to SNI injury were intraperitoneally administered incarvillateine (INCA, 20 mg/kg) or gabapentin (Gaba, 100 mg/kg) and the mechanical threshold was measured 30 min after administration. Each data point represents the mean ± S.E.M., n = 9–10. *P < 0.05, ***P < 0.001, compared with vehicle-treated mice.
Incarvillateine reduced paclitaxel-induced neuropathic pain
The antiallodynic effects of incarvillateine were also investigated in paclitaxel-induced painful neuropathy, another experimental model of neuropathic pain. As expected, daily administration of 2 mg/kg paclitaxel for 5 consecutive days induced a serious decrease in mechanical withdrawal threshold and there was no significant difference among groups before drug administration. Vehicle administration did not affect the mechanical withdrawal threshold, whereas incarvillateine produced a rapid, significant and dose-dependent antinociceptive effect [F(3, 144) = 48.16, p < 0.001; Fig. 5]. At 20 mg/kg, the effect of incarvillateine lasted for more than 60 min. Gabapentin also reversed paclitaxel-induced decreases in mechanical threshold.

Effects of incarvillateine on paclitaxel-induced neuropathic pain.
After 5 days paclitaxel treatment, mice were intraperitoneally administered vehicle (Veh), incarvillateine (INCA, 10 or 20 mg/kg), or gabapentin (Gaba, 100 mg/kg). The mechanical withdrawal threshold was measured using von Frey filaments before and at different times after drug administration. Each data point represents the mean ± S.E.M., n = 10. ***P < 0.001, compared with vehicle-treated mice.
Effects of incarvillateine on motor performance
The possible effects of incarvillateine on motor performance were assessed by evaluating spontaneous locomotor activity and the rotarod test. Administration of 10 or 20 mg/kg incarvillateine did not affect total distance traveled [F(2, 27) = 0.37, p > 0.05; Fig. 6A] or time on the rotarod [F(2, 54) = 0.09, p > 0.05; Fig. 6B], indicating that the antinociceptive effects of incarvillateine were not due to inhibition of motor function.

Effects of incarvillateine on motor performance.
(A) Mice were intraperitoneally administered vehicle (Veh) or incarvillateine (INCA, 10 or 20 mg/kg) and locomotor activity was measured for 120 min. (B) Mice were intraperitoneally administered vehicle (Veh) or incarvillateine (INCA, 10 or 20 mg/kg) and the time on the rotarod was recorded 30 and 90 min after drug administration. Each data point represents the mean ± S.E.M., n = 10.
Role of opioid and adenosine receptor systems in the antinociceptive effects of incarvillateine
To investigate whether the antinociceptive effects of incarvillateine were mediated via activation of the opioid and/or adenosine system, the nonselective opioid antagonist naloxone, nonselective adenosine antagonist theophylline and 3 adenosine subtype-targeting antagonists were administered before incarvillateine administration. As shown in Fig. 7A, SNI surgery induced a decrease in mechanical withdrawal threshold, with mean values less than 0.06 g. There were no significant differences among the mice intracerebroventricularly (i.c.v.) administered vehicle, naloxone, or theophylline alone. Incarvillateine produced a significant antinociceptive effect in vehicle-pretreated SNI mice 30 min after administration. Pretreatment with naloxone did not alter the effect of incarvillateine. In contrast, theophylline significantly inhibited the effect of incarvillateine [F(5, 52) = 19.83, p < 0.001], indicating that adenosine receptors may be involved in incarvillateine-induced antinociception. Furthermore, the selective A1 antagonist 1,3-dipropyl-8-cyclopentylxanthine (DPCPX) and the relative preferential A2 antagonist 3,7-dimethyl-1-propargylxanthine (DMPX), but not the selective A2A antagonist SCH-58261, attenuated incarvillateine-induced antinociception [F(7, 62) = 40.84, p < 0.001; Fig. 7B].

Effects of opioid and adenosine antagonists on incarvillateine-induced antinociception.
(A) SNI mice were intracerebroventricularly administered 5 μl ACSF, the nonselective opioid antagonist naloxone (Nal, 10 nmol), or the nonselective adenosine antagonist theophylline (Theo, 10 nmol) 10 min prior to incarvillateine (INCA, 20 mg/kg) administration. The mechanical threshold was measured 30 min after incarvillateine administration. (B) SNI mice were intraperitoneally administered vehicle (Veh), the selective A1 antagonist DPCPX (0.1 mg/kg), the relatively preferential A2 antagonist DMPX (1 mg/kg), or the selective A2 A antagonist SCH58261 (0.5 mg/kg) 15 min prior to incarvillateine (INCA, 20 mg/kg) administration. The mechanical threshold was measured 30 min after incarvillateine administration. (C) CFA mice were intracerebroventricularly administered 5 μl ACSF, the nonselective opioid antagonist naloxone (10 nmol), or the nonselective adenosine antagonist theophylline (10 nmol) 10 min prior to incarvillateine (INCA, 20 mg/kg) administration. The thermal withdrawal latency was measured 30 min after incarvillateine administration. Each data point represents the mean ± S.E.M., n = 8–10. **P < 0.01, ***P < 0.001, compared with vehicle-treated mice. ###P < 0.001, compared with incarvillateine-treated mice.
Similar results were observed in the CFA-induced thermal pain model. Incarvillateine significantly increased the withdrawal latency in vehicle-pretreated CFA mice 30 min after administration. Pretreatment with theophylline, but not naloxone, significantly inhibited the antinociceptive effect of incarvillateine [F(3, 32) = 20.79, p < 0.001; Fig. 7C].
Discussion
The goal of this study was to provide a systematic description of the pharmacological profile of incarvillateine, the proposed major active component of I. sinensis. This study extends a previous report, which evaluated incarvillateine in the formalin test, to several other pain models, including intractable inflammatory and neuropathic pain. Our results indicate that incarvillateine is a potent antiallodynic and antihyperalgesic compound, with a particular efficacy in pathological pain conditions and a mechanism involving adenosine receptor system.
The enormous clinical demands and shortage of effective therapy options has intensified research efforts to develop novel pathological pain therapies. A current research goal is to identify novel chemical structures with better or additional efficacy and/or greater safety relative to currently available treatments32. Active compounds from natural products and traditional herbal medicines are of interest. Incarvillateine has attracted attention due to its unique structure and potent analgesic activity in a mouse formalin model16,25. The structure of incarvillateine is dimeric and possesses five contiguous stereocenters on the bicyclic piperidine moiety. Through analysis of basic constructive units, such as incarvilline and ferulic acid and synthesized structure-related alkaloids, the monoterpene alkaloid and cyclobutane moieties are hypothesized to regulate the antinociceptive effects of incarvillateine. In particular, the cyclobutane ring seems indispensable, as incarvilline, incarvine C and related chemicals lacking the cyclobutane ring showed no or weak activity33,34. To our knowledge, incarvillateine does not belong to any structural groups that have been reported to show efficacy against pathological pain.
Previous reports indicate that incarvillateine affects behavioral manifestation in the mouse formalin test, a model addressing direct nociceptor stimulation in the first phase and inflammatory pain in the second phase35. In this study, the effects of incarvillateine on pain perception were evaluated in detail with a series of experimental models. The hot plate test represents acute phasic thermal pain, whereas tonic pain was measured in chemical, inflammatory and nerve-injury-induced pain models. Among these, the tonic pain models are considered to be more relevant to clinical pathological pain36, with each model reflecting one specific etiological factor. Incarvillateine has a broad spectrum of antinociceptive action against pathological pain, with a potency much higher than the reference drug gabapentin. However, incarvillateine did not affect acute phasic pain at the doses effective in reducing allodynia and hyperalgesia. The differential activity of incarvillateine in physiological and pathological pain may be correlated with adenosine receptor plasticity under pathological conditions10 and is of particular interest, as in most clinical cases the normal response to acute physiological pain is preferred while treating pathological pain. In addition, we also observed a mild anti-inflammatory effect following incarvillateine administration. Because inflammation and inflammatory mediators play an important role in pain pathology37, the anti-inflammatory effect, particularly the inhibition of IL-1 production, may be contributing factors in incarvillateine-induced antinociception.
To reveal the mechanism of incarvillateine in pathological pain, in vivo antagonist experiments were performed with specific antagonists. It was reported that the antinociceptive effects of many compounds involve activation of the opioid system by promoting endogenous peptide release or directly activating receptors and that these effects were diminished by corresponding antagonists38,39,40. In this study, naloxone, a nonselective opioid antagonist, did not affect the antinociceptive effects of incarvillateine in SNI and CFA injured mice. Although the results from these two models are consistent, they appear to be in contrast to a previous report, which suggested a concomitant involvement of the opioid and adenosine receptor systems in incarvillateine-induced antinociception in the formalin test25. As opioid agonists are likely to be effective in the hot plate test, induce locomotor hyperactivity and tend to develop tolerance, the lack of these characteristics in our studies did not support the involvement of the opioid receptor system in incarvillateine antinociception. Moreover, the difference in pain models used may account for the different conclusions. Nevertheless, the present results excluded the opioid mechanism of incarvillateine, at least under chronic pathological pain conditions.
Adenosine receptors, especially the A1 receptor, are considered attractive targets for the development of analgesics. Endogenous adenosine and selective A1 agonists have been well-documented to possess analgesic effects41. The activation of the adenosine system is involved as a mechanism not only in some analgesic compounds, but also in some therapies, such as Zusanli acupuncture, which triggers adenosine release and activates the A1 receptor42. Selective A1 agonists are also effective against neuropathic pain induced by SNI7,43. The attenuation of incarvillateine antinociception by the nonselective antagonist theophylline clearly suggests the involvement of the adenosine system. Further, receptor-subtype-preferential antagonists were used to distinguish whether the effects of incarvillateine were mediated through A1 or A2 receptors. The three antagonists used were DPCPX, DMPX and SCH58261, targeting A1, A2 and A2A respectively. The antinociceptive effects of incarvillateine were significantly attenuated by DPCPX and DMPX, but not SCH58261. It should be noted that, although DMPX is frequently used as an A2A antagonist, its selectivity for the rat A2A was only 3 fold in relative to A144. Therefore, DMPX-induced antagonism of incarvillateine antinociception may also be due to A1 receptor blockade. Thus, we believe that the A1 receptor is the major target mediating incarvillateine antinociception. The lack of motor dysfunction also supports this hypothesis, as motor function is primarily regulated by the A2 receptor45. Although these findings underlined the involvement of the adenosine system and point to the A1 receptor as a mediator of incarvillateine action, further work is needed to address the specific mechanisms, including binding and activating properties and the regulatory effects of incarvillateine on different adenosine receptor subtypes, as well as its influence on endogenous adenosine release or metabolism. Moreover, the possible role of incarvillateine biotransformation cannot be neglected.
In conclusion, the present study demonstrated that incarvillateine, a monomer from I. sinensis, is effective against pain induced by chemical substances, inflammation and neural injury, without any significant alterations in acute thermal and mechanical nociception, as well as motor functions. Its antinociceptive effects are related to the adenosine, but not the opioid, receptor system. Of note, incarvillateine produces neither stimulant nor sedative effect on mice and does not cause lethality under the antinociceptive doses studied, after acute and even 7 days of treatment, suggesting a low toxicity potential. Therefore, incarvillateine may be a novel leading molecule for development as a non-opioid analgesic.
Materials and Methods
Animals
All experimental animals were supplied by Beijing Animal Center (Beijing, China) and maintained on a 12 h light/dark cycle. Male Kunming mice (18–22 g) were used for the writhing test and locomotor activity measurements and male C57BL/6 mice (8–10 weeks old) were used for other procedures. Animals had free access to food and water. All experimental procedures were conducted in accordance with the guidelines for the use of experimental animals approved by the local ethics committee and the Institutional Review Committee on Animal Care and Use.
Drugs and reagents
Incarvillateine hydrochlorate was synthesized in-house as previously described30 and the purity was greater than 95%. It was dissolved in 1% DMSO in saline and intraperitoneally administered (10 ml/kg body weight). Naloxone, theophylline, DPCPX, DMPX and SCH58261 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Naloxone and theophylline were dissolved in artificial cerebrospinal fluid (ACSF; 147 mM NaCl, 2.7 mM KCl, 1.2 mM CaCl2, 0.85 mM MgCl2, 1.0 mM Na2HPO4, pH 7.4) and i.c.v. administered (5 μl/animal). DPCPX, DMPX and SCH58261 were dissolved in 1% DMSO in saline and i.p. administered (10 ml/kg body weight).
Acetic acid writhing test
The antinociceptive effects of incarvillateine were initially assessed using the acetic acid-induced writhing test as described previously46. Briefly, mice were i.p. injected with 0.6% acetic acid (0.2 ml) and the number of writhes, characterized by a wave of contraction in the abdomen followed by extension of the hind limbs, was recorded for a period of 15 min, starting at 5 min after injection. Incarvillateine was administered 30 min before acetic acid administration.
Hot plate test
The hot plate test was performed as described previously46. Mice were individually placed on the surface of a hot plate (HUGO SACHS Elektronik-Harvard Apparatus GmbH, March-Hugstetten, Germany) maintained at 55 ± 0.5 °C. The latency was recorded from starting to the end point of jumping, licking, or shaking hind paws. A cutoff time of 60 s was imposed to prevent the possibility of tissue damage. Mice were tested before and repeatedly at different times after incarvillateine administration.
CFA-induced inflammation and pain
Inflammation and pain were induced over a 48-h period by intraplantar injection of CFA (10 μl, F5881, Sigma-Aldrich) into the left hind paw. Control mice were injected with 10 μl saline. The effects of incarvillateine on hyperalgesia were determined at different times after administration by measuring the thermal sensitivity. To determine the effects of incarvillateine on inflammation, the ventral-dorsal paw thickness was measured with a digital caliper before CFA injection and 30 min after incarvillateine administration and paw edema rate was calculated as a percentage of change from baseline paw thickness. Mice were then sacrificed and the injected paws were cut and homogenized in 0.01 M PBS. After centrifugation at 10000 g/min for 5 min, 100 μl supernatant was used for the determination of IL-1β levels by enzyme-linked immunosorbent assay (ELISA).
SNI-induced neuropathic pain
SNI surgery was performed according to a previously described method47 with minor modifications. Briefly, mice were anesthetized with pentobarbital sodium (40 mg/kg, i.p.) and the sciatic nerve and its three terminal branches (the common peroneal, tibial and sural nerves) were exposed by incising the skin and the biceps femoris muscle on the lateral surface of the thigh. The common peroneal and the tibial nerves were tight-ligated with 5.0 silk and transected distal to the ligation, removing 1–2 mm of the distal nerve stump. During the procedure, any contact or stretching of the sural nerve was avoided. The sham mice underwent the same surgical procedure without ligation or transection. After 5–7 days of recovery, the effects of incarvillateine were determined by measuring mechanical sensitivity with von Frey filaments.
Paclitaxel-induced neuropathic pain
To induce peripheral neuropathy, mice were injected with paclitaxel (2 mg/kg, i.p.) once daily for 5 consecutive days as described previously48. One day after the last paclitaxel administration, the effects of incarvillateine were determined by measuring mechanical sensitivity with von Frey filaments.
Role of opioid and adenosine receptor systems in incarvillateine antinociception
The possible involvement of opioid and adenosine receptor systems in incarvillateine-induced antinociception was examined in the SNI and CFA models. After recovery from intracerebroventricular catheterization and SNI surgeries, the mice were i.c.v. administered ACSF, the nonselective adenosine antagonist theophylline (10 nmol), or the nonselective opioid antagonist naloxone (10 nmol) 10 min before incarvillateine (20 mg/kg, i.p.) administration. To verify the roles of specific adenosine receptor subtypes, the selective A1 adenosine receptor antagonist DPCPX (0.1 mg/kg), the relatively preferential A2 adenosine receptor antagonist DMPX (1.0 mg/kg) and the selective A2A adenosine receptor antagonist SCH58261 (0.5 mg/kg) were i.p. administered 15 min before incarvillateine administration. The changes in mechanical or thermal sensitivity were determined 30 min after incarvillateine administration.
Mechanical sensitivity measurement
Mechanical sensitivity was measured using von Frey filaments according to the up-down method described by Dixon49. Mice were individually placed in a plastic enclosure on an elevated wire mesh floor and allowed to habituate for 30 min before testing. A series of von Frey monofilaments were presented to the sural nerve innervation area in the plantar surface of the injured hind-paw, beginning with the 0.6 g filament. The next weaker filament was applied if a brisk paw withdrawal was observed; otherwise, the next stronger filament was applied. After the first cross of response threshold occurred, four additional stimuli were presented. The 50% response threshold was calculated based on the last six stimuli, using the formula by Dixon49.
Thermal sensitivity measurement
Thermal sensitivity was measured with a plantar test device (Model 390 G; IITC Life Science Inc., Woodland Hills, CA, USA) as previously described50. Mice were placed in Plexiglas chambers on a heated (30 °C) glass surface and allowed to habituate for 30 min before testing. A mobile radiant heat source was then positioned under the plantar surface of the hind paw and the paw withdrawal latency was recorded. A cutoff time of 20 s was imposed to prevent the possibility of tissue damage. For each time point, each mouse was tested three times with an interval of 2 min. The latencies of the three repetitions were averaged. Testing during grooming or exploratory behavior was avoided51.
Locomotor activity measurement
Mice were allowed to habituate the experimental environment for 30 min before testing. On the testing day, mice were administered vehicle or incarvillateine and placed in Plexiglass boxes (40 × 40 × 35 cm). Animal behavior was recorded for 120 min by a ceiling video camera and analyzed using the EthoVision video tracking system (Noldus, Wageningen, The Netherlands).
Rotarod test
Rotarod tests were performed by placing mice on a rod rotating at a speed of 15 rpm. Mice were trained once a day for 3 days and those failed to stay on the rod for 3 min were excluded. On the testing day, the time each mouse stayed on the rod was recorded 30 and 90 min after vehicle or incarvillateine administration.
Statistical analysis
Data are expressed as the mean ± SEM. Multiple comparisons were performed by one-way ANOVA or repeated measures two-way ANOVA followed by Bonferroni post hoc test. The level of statistical significance was defined as p < 0.05.
Additional Information
How to cite this article: Wang, M.-L. et al. Antinociceptive effects of incarvillateine, a monoterpene alkaloid from Incarvillea sinensis and possible involvement of the adenosine system. Sci. Rep. 5, 16107; doi: 10.1038/srep16107 (2015).
References
Finnerup, N. B. et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 14, 162–173 (2015).
Finnerup, N. B., Sindrup, S. H. & Jensen, T. S. The evidence for pharmacological treatment of neuropathic pain. Pain 150, 573–581 (2010).
Zylka, M. J. Pain-relieving prospects for adenosine receptors and ectonucleotidases. Trends Mol. Med. 17,188–196 (2011).
Burnstock, G. & Sawynok, J. Adenosine triphosphate and adenosine receptors and pain in Pharmacology of pain (eds Beaulieu, P. et al.) 303–326, (IASP, 2010).
Dickenson, A. H., Suzuki, R. & Reeve, A. J. Adenosine as a potential analgesic target in inflammatory and neuropathic pains. CNS Drugs 13, 77–85 (2000).
Sawynok, J. & Liu, X. J. Adenosine in the spinal cord and periphery: release and regulation of pain. Prog. Neurobiol. 69, 313–340 (2003).
Curros-Criado, M. M. & Herrero, J. F. The antinociceptive effects of the systemic adenosine A1 receptor agonist CPA in the absence and in the presence of spinal cord sensitization. Pharmacol. Biochem. Behav. 82, 721–726 (2005).
Eisenach, J. C., Rauck, R. L. & Curry, R. Intrathecal, but not intravenous adenosine reduces allodynia in patients with neuropathic pain. Pain 105, 65–70 (2003).
Gan, T. J. & Habib, A. S. Adenosine as a non-opioid analgesic in the perioperative setting. Anesth. Analg. 105, 487–494 (2007).
Suzuki, R., Gale, A. & Dickenson, A. Altered effects of an A1 adenosine receptor agonist on the evoked responses of spinal dorsal horn neurons in a rat model of mononeuropathy. J. Pain 1, 99–110 (2000).
Lavand’homme, P. M. & Eisenach, J. C. Exogenous and endogenous adenosine enhance the spinal antiallodynic effects of morphine in a rat model of neuropathic pain. Pain 80, 31–36 (1999).
Lee, Y. W. & Yaksh, T. L. Pharmacology of the spinal adenosine receptor which mediates the antiallodynic action of intrathecal adenosine agonists. J. Pharmacol. Exp. Ther. 277, 1642–1648 (1996).
Elzein, E. & Zablocki, J. A1 adenosine receptor agonists and their potential therapeutic applications. Expert Opin. Investig. Drugs 17, 1901–1910 (2008).
Jarvis, M. F. Psychomotor aspects of adenosine receptor activation in Purinergic Approaches in Experimental Therapeutics (eds Jacobson, K. A. & Jarvis, M. F. ) 405–421, (Wiley-Liss, 1997).
Chi, Y., Yan, W. & Li, J. Original botanical statistics and commercial study of the Chinese drug touguchao. Zhongguo Zhong Yao Za Zhi 15, 262–265 (1990).
Chi, Y. M., Yan, W. M. & Li, J. S. An alkaloid from Incarvillea sinensis. Phytochemistry 29, 2376–2378 (1990).
Chi, Y. M. et al. A monoterpene alkaloid from Incarvillea sinensis. Phytochemistry 31, 2930–2932 (1992).
Chi, Y. M., Hashimoto, F., Yan, W. M. & Nohara, T. Two alkaloids from Incarvillea sinensis. Phytochemistry 39, 1485–1487 (1995).
Chi, Y. M., Hashimoto, F., Yan, W. M. & Nohara, T. Incarvine A, a monoterpene alkaloid from Incarvillea sinensis. Phytochemistry 40, 353–354 (1995).
Chi, Y. M., Hashimoto, F., Yan, W. M. & Nohara, T. Four novel monoterpene alkaloidal derivatives from Incarvillea sinensis. Phytochemistry 46, 763–769 (1997).
Chi, Y. M., Hashimoto, F., Yan, W. M. & Nohara, T. Five novel macrocyclic spermine alkaloids from Incarvillea sinensis. Tetrahedron Lett. 38, 2713–2716 (1997).
Chi, Y. M. et al. Monoterpene alkaloids from Incarvillea sinensis. VI. absolute stereochemistry of incarvilline and structure of a new alkaloid, hydroxyincarvilline. Chem. Pharm. Bull. 45, 495–498 (1997).
Chi, Y. M. et al. A novel macrocyclic spermine alkaloid from Incarvillea sinensis. J. Asian. Nat. Prod. Res. 9, 115–118 (2007).
Nakamura, M. & Chi, Y. M. Two monoterpene alkaloidal derivatives from Incarvillea sinensis. Phytochemistry 51, 595–597 (1999).
Nakamura, M. et al. Strong antinociceptive effect of incarvillateine, a novel monoterpene alkaloid from Incarvillea sinensis. J. Nat. Prod. 62, 1293–1294 (1999).
Chi, Y. M. et al. Pharmacological study on the novel antinociceptive agent, a novel monoterpene alkaloid from Incarvillea sinensis. Biol. Pharm. Bull. 28, 1989–1991 (2005).
Ichikawa, M., Takahashi, M., Aoyagi, S. & Kibayashi, C. Total Synthesis of (−)-Incarvilline, (+)-Incarvine C and (−)-Incarvillateine. J. Am. Chem. Soc. 126, 16553–16558 (2004).
Ichikawa, M., Aoyagi, S. & Kibayashi, C. Total synthesis of (−)-incarvilline. Tetrahedron Lett. 46, 2327–2329 (2005).
Tsai, A. S., Bergman, R. G. & Ellman, J. A. Asymmetric synthesis of (−)-incarvillateine employing an intramolecular alkylation via Rh-catalyzed olefinic C-H bond activation. J. Am. Chem. Soc. 130, 6316–6317 (2008).
Zhang, F. & Jia, Y. Total synthesis of (−)-incarvilline and (−)-incarvillateine. Tetrahedron 65, 6840–6843 (2009).
Woolf, C. J., Allchorne, A., Safieh-Garabedian, B. & Poole, S. Cytokines, nerve growth factor and inflammatory hyperalgesia: the contribution of tumour necrosis factor alpha. Br. J. Pharmacol. 121, 417–424 (1997).
Gilron, I. & Dickenson, A. H. Emerging drugs for neuropathic pain. Expert Opin. Emerg. Drugs 19, 329–341 (2014).
Nakamura, M. et al. Structure-antinociceptive activity studies of incarvillateine, a monoterpene alkaloid from Incarvillea sinensis. Planta Med. 67, 114–117 (2001).
Chi, Y. M. et al. Anti-inflammatory activities of α-Truxillic acid derivatives and their monomer components. Biol. Pharm. Bull. 28, 1776–1778 (2005).
Tjølsen, A., Berge, O. G., Hunskaar, S., Rosland, J. H. & Hole, K. The formalin test: an evaluation of the method. Pain 51, 5–17 (1992).
Le Bars, D., Gozariu, M. & Cadden, S. W. Animal models of nociception. Pharmacol. Rev. 53, 597–652 (2001).
Schäfers, M., Brinkhoff, J., Neukirchen, S., Marziniak, M. & Sommer, C. Combined epineurial therapy with neutralizing antibodies to tumor necrosis factor-alpha and interleukin-1 receptor has an additive effect in reducing neuropathic pain in mice. Neurosci. Lett. 310, 113–116 (2001).
Guzzo, L. S., Perez, A. C., Romero, T. R., Azevedo, A. O. & Duarte, I. D. Cafestol, a coffee-specific diterpene, induces peripheral antinociception mediated by endogenous opioid peptides. Clin. Exp. Pharmacol. Physiol. 39, 412–416 (2012).
Nakamoto, K. et al. Possible involvement of β-endorphin in docosahexaenoic acid-induced antinociception. Eur. J. Pharmacol. 666, 100–104 (2011).
Takayama, H. Chemistry and pharmacology of analgesic indole alkaloids from the rubiaceous plant, Mitragyna speciosa. Chem. Pharm. Bull. 52, 916–928 (2004).
Maione, S. et al. The antinociceptive effect of 2-chloro-2′-C-methyl-N6-cyclopentyladenosine (2′-Me-CCPA), a highly selective adenosine A1 receptor agonist, in the rat. Pain 131, 281–292 (2007).
Goldman, N. et al. Adenosine A1 receptors mediate local anti-nociceptive effects of acupuncture. Nat. Neurosci. 13, 883–888 (2010).
Luongo, L. et al. 5′-Chloro-5′-deoxy-(±)-ENBA, a potent and selective adenosine A(1) receptor agonist, alleviates neuropathic pain in mice through functional glial and microglial changes without affecting motor or cardiovascular functions. Molecules 17, 13712–13726 (2012).
Yang, M. et al. Characterization of the potency, selectivity and pharmacokinetic profile for six adenosine A2A receptor antagonists. Naunyn Schmiedebergs Arch. Pharmacol. 375, 133–144 (2007).
Karlsten, R., Gordh, T., Jr., Hartvig, P. & Post, C. Effects of intrathecal injection of the adenosine receptor agonists R-phenylisopropyl-adenosine and N-ethylcarboxamideadenosine on nociception and motor function in the rat. Anesth. Analg. 71, 60–64 (1990).
Yu, G. et al. The antinociceptive effects of intracerebroventricular administration of Chicago sky blue 6B, a vesicular glutamate transporter inhibitor. Behav. Pharmacol. 24, 653–658 (2013).
Bourquin, A. F. et al. Assessment and analysis of mechanical allodynia-like behavior induced by spared nerve injury (SNI) in the mouse. Pain 122, 14.e1–14 (2006).
Costa, R. et al. Anti-nociceptive effect of kinin B1 and B2 receptor antagonists on peripheral neuropathy induced by paclitaxel in mice. Br. J. Pharmacol. 164, 681–693 (2011).
Dixon, W. J. Efficient analysis of experimental observations. Annu. Rev. Pharmacol. Toxicol. 20, 441–462 (1980).
Hargreaves, K., Dubner, R., Brown, F., Flores, C. & Joris, J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32, 77–88 (1988).
Callahan, B. L., Gil, A. S., Levesque, A. & Mogil, J. S. Modulation of mechanical and thermal nociceptive sensitivity in the laboratory mouse by behavioral state. J. Pain 9, 174–184 (2008).
Acknowledgements
This study was supported by the National Science and Technology Major Project of China (2011ZX09101-005-01, 2012ZX09301003-001).
Author information
Authors and Affiliations
Contributions
M.L.W., G.Y. and R.B.S. contributed to the research design, data analysis and manuscript writing. M.L.W., S.P.Y., F.Y.Z, Z.T.W. and B.H. conducted the experiments. Y.X.J. and Z.H.G. participated in the discussion of the results.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wang, ML., Yu, G., Yi, SP. et al. Antinociceptive effects of incarvillateine, a monoterpene alkaloid from Incarvillea sinensis and possible involvement of the adenosine system. Sci Rep 5, 16107 (2015). https://doi.org/10.1038/srep16107
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep16107
This article is cited by
-
Nature brings new avenues to the therapy of central nervous system diseases—An overview of possible treatments derived from natural products
Science China Life Sciences (2019)
-
Recent development in antihyperalgesic effect of phytochemicals: anti-inflammatory and neuro-modulatory actions
Inflammation Research (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
