
Abstract
Theileria-infected macrophages display many features of cancer cells such as heightened invasive capacity; however, the tumor-like phenotype is reversible by killing the parasite. Moreover, virulent macrophages can be attenuated by multiple in vitro passages and so provide a powerful model to elucidate mechanisms related to transformed macrophage virulence. Here, we demonstrate that in two independent Theileria-transformed macrophage cell lines Grb2 expression is down-regulated concomitant with loss of tumor virulence. Using peptidimer-c to ablate SH2 and SH3 interactions of Grb2 we identify TGF-receptor II and the p85 subunit of PI3-K, as Grb2 partners in virulent macrophages. Ablation of Grb2 interactions reduces PI3-K recruitment to TGF-RII and decreases PIP3 production and dampens JNK phosphorylation and AP-1-driven transcriptional activity down to levels characteristic of attenuated macrophages. Loss of TGF-R>PI3-K>JNK>AP-1 signaling negatively impacts on virulence traits such as reduced JAM-L/ITG4A and Fos-B/MMP9 expression that contribute to virulent macrophage adhesion and invasiveness.
Similar content being viewed by others


Introduction
Theileria annulata is an intracellular protozoan and member of the phylum Apicomplexa. Theileria parasites (T. annulata and T. parva) are unusual eukaryotes in that they possess the property of being able to transform their host cells. Transformed leukocytes display many characteristics of tumors such as uncontrolled proliferation independent of exogenous growth factors, resistance to apoptosis and an increased ability to disseminate and invade organs in vivo1,2. Moreover, virulence of transformed macrophages is reversed upon drug-induced parasite death3,4 and attenuated by multiple in vitro passages5.
In regions endemic for tropical theileriosis indigenous breeds of cattle (ex: Bos indicus also known as Sahiwals) are more resistant to disease, exhibit less symptoms and recover from an infectious dose, which is often fatal to non-indigenous breeds (ex: B. taurus also known as Holstein-Friesian—HF)6. Nonetheless, no great difference in infection-induced expression levels of pro-inflammatory factors such as IL-67, GM-CSF8 and TNF9 was detected between disease-resistant and susceptible cattle. By contrast, induction of TGF-β2 by T. annulata infection is greater in macrophages isolated from disease-susceptible (HF) compared to disease-resistant (Sahiwals) cattle and this correlates with the higher invasive capacity of HF-infected macrophages10. Upon attenuation in vitro, the HF cell line displayed both reduced TGF-β2 expression and invasiveness, which was re-established by exogenous TGF-β210. The ensembles of these observations are consistent with a pro-metastasis role of TGF-β2 in virulent Theileria-infected macrophages.
Transforming growth factor-β (TGF-β) is a family of cytokines known to regulate cell growth, differentiation and motility11. TGF-β1 and TGF-β3 can bind with high affinity to the TGF-β type II receptor (TGF-RII), which leads to the recruitment of the type I receptor (TGF-RI). The constitutively active TGF-RII serine/threonine kinase phosphorylates and activates the TGF-RI receptor kinase, which leads to the recruitment and activation of Smad2 and Smad3. Activated Smad2 and Smad3 then bind to Smad4 and the entire complex translocates to the nucleus to activate downstream target genes12. While type II receptor binds with low affinity to TGF-β2, type III receptor (TGF-RIII) binds with high affinity to all three isoforms and is needed to present TGF-β2 to TGF-RII13. Besides the canonical Smad pathway, many other pathways such as JNK14, Ras14, PI3-K15 and PKA16 are also regulated by TGF-β. This could explain why TGF-β signaling pathways play such a complex role in mediating cellular processes.
Phosphatidylinositol-4, 5-bisphosphate 3-kinases (PI3-K) is a family of signal transducing enzymes that are also involved in cell growth, proliferation, motility and survival17. The PI3-K family is divided into 3 classes based on their primary protein structure, regulation and in vitro lipid substrate specificity18. Class I PI3-kinases are made up of a p110 catalytic subunit and a p85 regulator subunit, which harbors an SH3 domain. PI3-kinases are capable of phosphorylating the 3-position hydroxyl group of the inositol ring of phosphatidylinositol to form phosphatidylinositol biphosphate (PIP2) and phosphatidylinositol triphosphate (PIP3) and other 3-phosphorylated phosphoinositides. This group of signaling kinases contains phosphoinositide-binding domains that are recruited for binding to cell membranes. Both we and others have previously demonstrated that T. parva and T. annulata-infected B-lymphocytes rely on constitutively activated PI3-K for their continued proliferation8,19,20.
PI3-K also acts as an upstream regulator of the c-Jun NH2-terminal kinase (JNK)8,21. JNK is constitutively activated in Theileria-infected cells22,23. Proteins of the Jun and Fos families combine to form the heterodimeric AP-1 transcription factor, which enters the nucleus and binds to specific DNA sequences to modulate target gene expression. AP-1 activity is induced by stimuli such as growth factors and cytokines that bind to specific cell surface receptors24. AP-1 is a key transcription factor induced in virulent Theileria-transformed leukocytes23 and AP-1-driven transcription underpins the hyper-dissemination virulence phenotype25.
The Growth factor receptor-bound protein 2 (Grb2) is an adaptor used to recruit accessory proteins in signal transduction pathways26. Grb2 contains three domains; one Src Homology 2 (SH2) and two Src Homology 3 (SH3) domains at the N- and C-termini27,28. The SH2 domain participates in signal transduction in tyrosine kinase pathways by recognizing specific amino acids motifs in partner proteins, whereas SH3 domains recognize proline-rich sequences within specific partners. The two SH3 domains form a direct complex with the proline-rich regions of the partner proteins, while the SH2 domain binds to tyrosine-phosphorylated motifs in receptors29.
We have previously demonstrated that heightened PI3-K activity is essential for the proliferation of Theileria-transformed B cells8 and have shown that one mechanism by which PI3-K mediates proliferation of infected B lymphocytes is through induction of a granulocyte-monocyte colony-stimulating factor (GM-CSF)-dependent autocrine loop8. Moreover, we have shown that the level of TGF-β2 is augmented in virulent macrophages and this led us to test the role of TGF-β2 in activation of the PI3-K signaling pathway10. We now describe how loss of Grb2 expression ablates PI3-K recruitment to TGF-RII and how a TGF-β2/Grb2/PI3-K/AP-1 pathway contributes to the hyper-invasive virulence phenotype of Theileria-transformed macrophages.
Results
Grb2 is a TGF-β2-target in Theileria-transformed macrophages
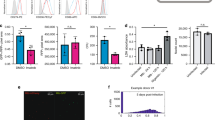
Grb2 is implicated in many cellular functions including adhesion. We have previously shown that Theileria-infected Ode macrophages that display a heightened capacity of adhesion typical of virulent disease-causing infected macrophages compared to live vaccine lines used to treat tropical theileriosis that are attenuated for this virulence trait30. So, we checked the expression of Grb2 in virulent and attenuated macrophages by qPCR analysis (Fig. 1A). In Fig. 1A one can observe an attenuation-associated 50% decrease in Grb2 expression and the decrease was restored by stimulating attenuated macrophages with exogenous TGF-β2. When virulent macrophages are treated with peptidimer-c transcription of Grb2 is reduced by approximately 80%. Furthermore, as TGF-β2 is also down regulated in attenuated Jed4 macrophages isolated from an infected animal in Tunisia30 it follows that Grb2 expression is also diminished (Fig. 1A, right panel). Changes in transcription of Grb2 were reflected at the protein level (Fig. 1B), as an approximate 40% decline in Grb2 protein was observed in attenuated macrophages and protein levels are restored by exogenous TGF-β2. Taken together; the results show that TGF-β2-signaling is responsible for high levels of Grb2 in virulent Theileria-infected macrophages of different geographical origins and ablation of Grb2 accompanies loss of Theileria-transformed macrophage virulence.

Grb2 is a TGF-β2-target gene in Theileria-infected macrophages.
(A) Total RNA was extracted from virulent (V) and attenuated (A) macrophages. Relative mRNA levels of the different genes were determined by qPCR and normalized to GAPDH transcripts. Left panel. The transcription level of Grb2 is higher in virulent macrophages compared to attenuated ones. The treatment with peptidimer-c decreases the expression level of Grb2 in virulent macrophages, whereas adding exogenous TGF-β2 to attenuated macrophages restores Grb2 expression. Right panel. Levels of Grb2 expression in Jed4 macrophages. (B) Western blot analysis with anti-Grb2 antibody using a whole cell lysates (10 μg of protein) derived from virulent and attenuated macrophages treated with TGF-β2 or not. Grb2 is diminished in attenuated macrophages and stimulation by TGF-β2 of attenuated macrophages restores protein levels compared to virulent Ode. Grb2 protein levels were normalized to β-actin protein levels. *p < 0.05 compared to virulent macrophages, #p < 0.05 compared to attenuated macrophages, §p < 0.05 compared to virulent macrophages treated with PEN.
Grb2 associates with TGF-RII and the p85 subunit of PI3-K
We examined whether Grb2 is an upstream player in the PI3-K signaling pathway essential for Theileria-infected leukocyte proliferation by using peptidimer-c to see if the PI3-K levels were decreased after ablation of SH3 interactions, as peptidimer-c specifically recognizes SH3 domains of Grb229,31. Firstly, we examined if there was an association between Grb2 and the p85 subunit and if there was a difference in their interaction between the virulent and attenuated cell lines would treatment of virulent macrophages with peptidimer-c ablate the interaction to levels observed for attenuated macrophages. To detect interactions between Grb2 and p85 pull-downs using GST-Grb2 beads were performed (Fig. 2A). The pull-down demonstrated an interaction between Grb2 and p85 in Theileria-infected macrophages and revealed a dampened association in attenuated macrophages. The association between Grb2 and p85 was ablated by peptidimer-c that competes for Grb2 SH3 domain binding to partners (Fig. 2A, left panel). Thus, the interaction with p85 occurs via the SH3 domains of Grb2 and this can be blocked by peptidimer-c.

Grb2 associates with TGF-RII and the p85 subunit of PI3-K.
(A) Left panel. Pull-down assay performed with TGF-RII receptor expressed by Theileria-transformed macrophages incubated with GST-Grb2 beads. Bottom two rows demonstrate equivalent amounts of actin and TGF-RII in the cell extracts mixed with the GST-Grb2 beads. The top row demonstrates more TGF-RII is bound to GST-Grb2 in virulent macrophages and binding is not diminished by PEN treatment. TGF-RII binding is ablated when Grb2 SH3-interactions are dampened by treatment with peptidimer-c. Right panel. Immunoprecipitation analysis with anti-p85 antibody using whole cell lysates derived from virulent macrophages (V) incubated with GST-Grb2 beads overnight. Bottom two rows indicate that equivalent amounts of cell extract containing p85α and actin were mixed with GST-Grb2 beads. Top row shows the amount of p85α bound to GST-Grb2 beads in virulent and attenuated macrophages. Treatment of virulent macrophages with PEN did not diminish the amount of p85α bound to GST-Grb2. Ablation of SH3 domain interactions of Grb2 with peptidimer-c in virulent macrophages diminishes Grb2 binding of p85α to attenuated levels. (B) Ablating Grb2 partner-interactions with peptidimer-c reduces PI3-K activity to attenuated levels. PI3-K activity in virulent macrophages is decreased when virulent macrophages are treated with 1 μM of peptidimer-c and adding exogenous TGF-β2 to attenuated macrophages increases PI3-K activity. *p < 0.05 compared to virulent macrophages, #p < 0.05 compared to attenuated macrophages, §p < 0.05 compared to virulent macrophages treated with PEN.
Grb2 is known to associate with TGF-RII following receptor phosphorylation at Tyr284 by Src32. GST-Grb2 pull-downs were therefore also used to demonstrate an interaction between Grb2 and TGF-RII in Theileria-infected macrophages. Again, the association was diminished upon treatment of virulent macrophages with peptidimer-c (Fig. 2A, right panel). Taken all together, one suggests that Grb2 recruits the regulatory p85 subunit of PI3-K to TGF-RII in Theileria-infected macrophages.
p85 recruitment to TGF-RII by Grb2 stimulates PI3-K activity
Having demonstrated that the Grb2 and p85 interaction is ablated by peptidimer-c it follows that peptidimer-c treatment should dampen PI3-K activity, as p85 recruits the catalytic p110 subunit proximal to the plasma membrane, where it converts PIP2 to PIP3. The level of PIP3 therefore is a reflection of PI3-K activity. Treating virulent macrophages with peptidimer-c reduced PI3-K activity to that of attenuated macrophages (Fig. 2B). Importantly, TGF-β2 stimulation of attenuated macrophages restores host cell PIP3 to virulence levels demonstrating that Grb2 recruitment of p85 to TGF-RII augments PI3-K activity.
Recruitment of p85 to TGF-RII by Grb2 promotes JNK phosphorylation and AP-1 activation
Activator Protein 1 (AP-1) is constitutively induced via PI3-K-signaling in Theileria-infected leukocytes8. AP-1 activity is required for the survival of T. parva-transformed B cells23 and for dissemination of T. annulata-transformed macrophages25. It follows ablation of the Grb2-p85-TGF-RII association and consequent drop in PI3-K activity should be reflected in decreased JNK phosphorylation and AP-1-mediated transcription and this was confirmed by western blot analysis of JNK phosphorylation and by measuring AP-1-driven luciferase activity (Fig. 3). Treatment of virulent macrophages with peptidimer-c ablates JNK phosphorylation and when virulent macrophages are treated with peptidimer-c the loss of PI3-K activity is comparable to the loss observed with the specific PI3-K inhibitor Wortmannin (Fig. 3).

JNK phosphorylation and AP-1 activity are affected by ablation of Grb2 interactions.
(A) Top panel. JNK phosphorylation is dampened in virulent macrophages treated with peptidimer-c compared to non-treated virulent macrophages. Treatment with the penetrating peptide PEN had no effect. (B) Bottom panel. AP-1 luciferase activity in virulent macrophages is decreased upon treatment with 1μM of peptidimer-c. Virulent macrophages were also treated with the PI3-K inhibitor Wortmannin. When attenuated macrophages were stimulated with 5 ng/ml of TGF-β2, AP-1 luciferase activity returned to virulent levels. Luciferase and β-galactosidase activities were measured as described in Methods and for each independent transfection relative luciferase activity was normalized to the corresponding β-galactosidase activity. *p < 0.05 compared to virulent macrophages, §p < 0.05 compared to virulent macrophages treated with PEN.
Ablation of SH3 interactions of Grb2 dampens AP-1 transactivation and impacts on cellular adhesion and invasion
Our focus turned to JAM-L, as it can bind to CAR and recruit PI3-K33. Since we have demonstrated above that Grb2 activates PI3-K/AP-1 signaling and knowing that Grb2 can regulate cellular adhesion34, we asked if Grb2 regulates adhesion of Theileria-infected macrophages through JAM-L, as anti-JAM-L antibodies dampen adhesion of Theileria-infected macrophages30. So we used peptidimer-c to ablate the SH3 domain interactions of Grb2 and looked at JAM-L expression and adhesion capacity in Theileria-infected macrophages. By qPCR there was an almost 60% decrease in jam-l expression indicating that upon attenuation transcriptional jam-l declines (Fig. 4A). Treatment of virulent macrophages with peptidimer-c, or a specific inhibitor of PI3-K (Wortmannin) also decreased jam-l transcription.

JAM-L is a downstream adhesion effector of Grb2/PI3K/AP-1 signaling.
Total RNA was extracted from virulent, engineered (delta169) virulent and attenuated Jed4 macrophages. Relative mRNA levels for jam-l were determined by qPCR and normalised to GAPDH transcripts. (A) Jam-l is transcriptionally up regulated in virulent compared to attenuated macrophages. Inhibition of Grb2 interactions and PI3-K signaling in virulent macrophages decreases the level of jam-l. (B) The level of jam-l transcription in engineered (delta169) Jed4 macrophages is reduced to that of attenuated Jed4 macrophages. (C) The protein expression levels of Fos-B and MMP9 in virulent macrophages is downregulated upon treatment with peptidimer-c. Fos-B and MMP9 protein levels were normalized to β-actin protein level. *p < 0.05 compared to virulent macrophages, §p < 0.05 compared to virulent macrophages treated with PEN.
To link jam-l expression to AP-1-mediated transcription, we exploited a Theileria-transformed macrophage cell line of Tunisian origin, where AP-1-driven transcription had been handicapped by engineered expression of a dominant-negative mutant (Δ169) of c-Jun25. In the engineered cell line (virulent delta169) jam-l mRNA levels drop to those typical of attenuated macrophages demonstrating that expression of jam-l is AP-1-dependent (Fig. 4B). Since jam-l transcription decreases upon attenuation and peptidimer-c treatment ablates jam-l expression in virulent macrophages, we examined the ability of peptidimer-c to dampen transformed macrophage adhesion.
The Matrix MetalloProteinase (MMP) 9 plays a central role in tumor progression and metastasis by stimulating cell migration, tumor invasion and angiogenesis35. Consistently, MMP9 functions as a mediator of metastasis of Theileria-transformed leukocytes36,37. Moreover, mmp9 expression is driven by the transcription factor AP-1 and differences in mmp9 transcription between virulent and attenuated Theileria-transformed macrophages is due to differential recognition of the AP-1 motif in the mmp9 promoter38. Significantly, a fast migrating AP-1 family member failed to bind to the mmp9 promoter in attenuated macrophages38. By its size the fast migrating species could be Fos-B, a member of a family of transcription factors made up of Fos, Fos-B, Fos-L1 and Fos-L2 that are leucine zipper proteins, which dimerize with members of the Jun family to form different AP-1 heterodimers39. As ablation of SH3 domain interactions of Grb2 diminishes transactivation of AP-1 (Fig. 3), we examined the expression levels of both Fos-B and MMP9 and both were diminished following peptidimer-c treatment of virulent macrophages (Fig. 4).
Ablation of p85 recruitment by Grb2 decreases adhesion and invasiveness of Theileria-infected macrophages
In order to confirm the role of Grb2 in promoting adhesion of virulent Theileria-infected macrophages, we performed a dynamic in vitro adhesion assay. Peptidimer-c treatment reduces adhesion of virulent macrophages to attenuated levels (Fig. 5A). Wortmannin treatment had a similar affect consistent with adhesion being regulated by a Grb2>PI3-K signaling pathway. In parallel, we performed in vitro invasion (Matrigel chamber) assays. The capacity of virulent macrophages to traverse Matrigel is significantly decreased when virulent macrophages are treated with peptidimer-c (Fig. 5B). No effect was observed when treated with the penetrating peptide PEN used as a negative control.

Ablation of SH3 domain interactions of Grb2 diminishes adhesion to fibronectin and Matrigel traversal.
(A) Adhesion to fibronectin decreases to attenuated levels when virulent macrophages are treated with peptidimer-c, or Wortmannin. Virulent macrophages were treated with 1 μM of peptidimer-c and/or 100 nM of Wortmannin. Adhesion to fibronectin was then measured. (B) Matrigel traversal of virulent Theileria-transformed macrophages. Blockade of SH3 domain interactions of Grb2 by peptidimer-c treatment decreased Matrigel traversal of virulent macrophages. No effect was observed upon treatment with PEN. *p < 0.05 compared to virulent macrophages, §p < 0.05 compared to virulent macrophages treated with PEN.
Discussion
We targeted Grb2 because it is an adaptor protein that facilitates formation of large signaling complexes that play pivotal roles in receptor-mediated signal transduction, an example is the association of Grb2 with TGF-RII following receptor phosphorylation on Tyr284 by Src32. Additionally, there’s a significant difference in TGF-β2 expression between the virulent and attenuated Theileria-infected macrophages10. For these reasons we dissected a possible Grb2/TGF-RII association by ablating Grb2 SH3 interactions by peptidimer-c treatment26. We found that not only does Grb2 interact with TGF-RII, but that Grb2 is also a TGF-β2-target gene in Theileria-transformed macrophages. As we have shown that ablating Grb2 SH3 interactions dampens AP-1-driven luciferase it suggests that Grb2 is also an AP-1-target gene and possibly even a CREB-target gene30. Multiple binding partners have been described for Grb2 including the regulatory p85 subunit of PI3-K40. Firstly, we found that TGF-β2 stimulation increased PI3-K activity, whilst ablation of SH3 interactions of Grb2 decreased its activity. Then, we confirmed using GST-Grb2 pull downs that TGF-β2 stimulation of PI3-K activity was facilitated by Grb2; the decrease in association between Grb2 and the p85 subunit in peptidimer-c treated cells confirmed that Grb2 is a key adaptor in the TGF-β2 > PI3-K signaling cascade.
The survival of Theileria-transformed cells depends on the constitutive activation of different transcription factors including AP-123. In Theileria-infected macrophages there is a significant variation in PI3-K levels between virulent and attenuated cells, where PI3-K activity is augmented in aggressive, invasive virulent macrophages. However, PI3-K activity is decreased by ablation of Grb2 SH3 domain interactions by peptidimer-c treatment that also reduces AP-1 activity to levels equivalent to Wortmannin inhibition of PI3-K. In contrast, addition of TGF-β2 to attenuated macrophages restores PI3-K activity. All of the above is consistent with the notion that TGF-β2 stimulation of attenuated macrophages restores Grb2 expression, which results in recruitment of the p85 regulatory subunit of PI3-K proximal to TGF-RII and activation of PI3-K signaling and activation of AP-1 in Theileria-transformed macrophages.
JAM-L is implicated in adhesion of Theileria-infected macrophages30 and in another cellular system clustering of JAM-L activates PI3-K signaling33 suggesting a possible interaction occurs between JAM-L and PI3-K in Theileria-infected macrophages. Ablation of SH3 interactions of Grb2 by peptidimer-c, or inhibition of PI3-K signaling by Wortmannin both decreased mRNA levels of JAM-L. Furthermore, as JAM-L is an AP-1-target gene30 it suggests that PI3-K increases expression of JAM-L through AP-1 transactivation.
AP-1 complexes of altered mobilities can be observed in both T. annulata and T. parva-infected leukocytes, suggesting that parasite infection induces AP-1 heterodimers of different composition41. Previous examination of the same Theileria-transformed macrophages (Ode) as used here, revealed loss of binding to the mmp9 promoter of a fast-migrating AP-1 family member and reduced MMP9 expression in attenuated Ode macrophages38. Here, using specific anti-Fos-B antibodies we identified the fast migrating AP-1 family member and treatment with peptidimer-c ablated Fos-B levels and AP-1 transactivation and consequently MMP9 expression in virulent Theileria-transformed macrophages. Virulent Ode macrophages treated with peptidimer-c resemble attenuated Ode macrophages generated by multiple in vitro passages.
In summary, we propose a scheme that explains how changes in expression of TGF-β2 orchestrate the switch from a virulent to an attenuated phenotype via Grb2 recruitment of PI3-K to TGF-RII that results in activation of AP-1 to regulate both adhesion and invasiveness of transformed macrophage (Fig. 6). The ensembles of our results indicate that peptidimer-c could eventually see use as a treatment for tropical theileriosis and perhaps even have applications in treatment of a wide range of cancers. Finally, as attenuated Theileria-infected macrophages display heightened oxidative stress that underpins their loss of adhesion/invasiveness42 it will be interesting to examine if loss of TGF-β2-signaling contributes to impaired regulation of oxidative stress in attenuated macrophages42. One possibility is that Grb2 binds catalase43 and its recruitment augments the conversion of H2O2 into H2O, so enabling Theileria-transformed leukocytes to better resists oxidative stress stemming for infection induced uncontrolled host cell proliferation44. Thus, future experiments will address the possible role of activation of the TGF-β2/Grb2/PI3-K/AP-1 pathway to regulation of oxidative stress in Theileria-transformed macrophages.

Pathway showing how TGF-β2 induction of Grb2 recruits PI3-K to TGF-RII to activate AP-1-driven gene transcription that underpins heightened adhesion and invasiveness of Theileria-transformed virulent macrophages.
Materials and Methods
Theileria annulata-infected cell lines and cell culture
T. annulata-infected macrophages used in this study are the Ode and Jed4 virulent and attenuated vaccine lines25,45. Virulent (early passage) Ode corresponds to passage 62 and attenuated (late passage) Ode to passage 309, whereas virulent and attenuated Jed4 correspond to passages 18 and 327, respectively. All cells were incubated at 37 °C with 5% CO2 in Roswell Park Memorial Institute medium (RPMI) supplemented with 10% Fetal Bovine Serum (FBS), 2 mM L-Glutamine, 100 U penicillin, 0.1 mg/ml streptomycin and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES).
Ablation of the interaction of Grb2 and its partners
The SH3 interactions of Grb2 with its partners were ablated using a specific Grb2-SH3 competitive peptide (peptidimer-c)29,31. A concentration 1 μM was used and incubated for 1 h at 37 °C in 5% CO2.
Activation of TGF-β-signaling pathway
The TGF-β signaling pathway was activated by using recombinant bovine TGF-β2 (rbo-TGF-β2) (NIBSC, Potters Bar. UK). A concentration of 2.5 ng/ml was added to the cell culture for 24 h.
Total RNA extraction and reversed transcription
Total RNA of Theileria-infected macrophages was isolated using the RNeasy mini kit (Qiagen), according to the manufacturer’s instructions. The quality and quantity of RNA was measured by Nanodrop spectrophotometer. For the reverse transcription, 1 μg isolated RNA was diluted by water to a final volume of 12 μL, warmed at 65 °C for 10 min, then incubated on ice for 2 min. Afterwards, 8 μl of reaction solution (0.5 μL random hexamer, 4 μL 5× RT buffer, 1.5 μL 10 mM dNTP, 1 μL 200U/μLRT-MMLV (Promega) and 1 μL 40 U/μL RNase inhibitor (Promega) was added to get a final reaction volume of 20 μL and incubated at 37 °C for 2 h. The resultant RNA was stored at −20 °C.
Quantitative polymerase chain reaction (qPCR)
mRNA expression levels were measured by qPCR on Light Cycler 480 (Roche) using SYBR Green detection (Thermo). GAPDH was used as internal control to normalize for mRNA levels. The detection of a single product was verified by dissociation curve analysis and relative quantities of mRNA calculated using the method described46. For list of primers used, see below (Table 1).
Western blot analysis
Cells were washed with cold PBS and then lysed on ice for 30 min in lysis buffer (50 μL RIPA buffer 2X, 50 μL PBS). Cells were centrifuged at 13000 rpm for 15 min at 4 °C to eliminate cellular debris. Protein concentration was determined by using Bradford method (by reading optical density at 595 nm). Cell extract was mixed with 5X Laemmli and denatured at 95 °C for 5 min. 10 μg of protein were separated by migration through a denaturing 10% SDS-PAGE gel and electro-transferred onto a nitrocellulose membrane (Protan). The membrane was blocked by 5% non-fat milk-TBST (for antibody of interest), 3% non-fat milk-PBST (for anti-Actin antibody) for 1 h at RT. Antibodies used were as follows: mouse monoclonal antibody anti-Grb-2 (Santa Cruz Biotechnology #8034) diluted 1/1000 in 5% BSA-TBST, rabbit polyclonal antibody anti-JNK (Santa Cruz Biotechnology #571) diluted 1/1000 in 5% BSA-TBST, rabbit polyclonal antibody anti-p85 (Santa Cruz Biotechnology) diluted 1/1000 in 5% BSA-TBST and mouse monoclonal antibody anti- Phospho-SAPK/JNK (Thr183/Tyr185) (Cell signaling #9255) diluted 1/1000 in 5% BSA-TBST all incubated overnight at 4 °C. Goat polyclonal antibody anti-Actin (Santa Cruz biotechnology, I-19) diluted by 1/2000 in 3% BSA-PBST was incubated for 1 h at RT. After washing, membranes were incubated with peroxidase-conjugated secondary antibody (mouse anti-IgG, rabbit anti-IgG and goat anti-IgG (Santa Cruz biotechnology) diluted by 1/5000 for 1 h at RT. After washing, membranes were developed using Super Signal detection kit (Thermo).
GST-Grb2 pull downs
Cells were washed with cold PBS and then lysed on ice for 30 min in lysis buffer (50 μL RIPA buffer 2X, 50 μL PBS). The cells were centrifuged at 13000 rpm for 15 min at 4 °C to eliminate cellular debris. 50 μl of cell lysate was collected and 10 μl of GST-Grb2 beads and incubated over night with gentle rocking at 4 °C. Cell extract was mixed with 2X Laemmli and denatured at 95 °C for 5 min. Proteins were separated by migration through a denaturing 8% SDS-PAGE gel and electro-transferred onto a nitrocellulose membrane (Protan). The membrane was blocked by 5% non-fat milk-TBST for 1 h at RT. The proteins were analyzed using a rabbit polyclonal primary antibody anti-TGFβ RII (Santa Cruz biotechnology #sc-220-R) diluted by 1/200 5% BSA-TBST and a rabbit polyclonal primary antibody anti-p85 both incubated overnight at 4 °C. After washing, membranes were incubated with peroxidase-conjugated secondary antibody rabbit anti-IgG (Santa Cruz biotechnology #2005) diluted by 1/5000 in 5% non-fat milk-TBST for 1 h at RT. After washing, membranes were developed using Super Signal detection kit (Thermo).
AP-1-luciferase assay
Ode macrophages were transfected by electroporation using the Amaxa Nucleofector, kit V and program T -017 (Amaxa Lonza protocol). 5 × 105 cells/ml were washed once with PBS at room temperature and then cells were suspended in 100 μl of Nucleofector solution V (Amaxa). Cells were co-transfected with 2 μg of luciferase reporter plasmid (3X-TRE) and/or reporter plasmid (β-galactosidase: β-gal). After transfection, cells were suspended in fresh complete medium and incubated at 37 °C, 5% CO2 for 24 h. Measurements of luciferase and β-galactosidase activities were performed using the Dual Light Assay system (Life Technologies) and luminometer Centro LB 960 (Berthold) according to the manufacturer’s instructions.
PI3-Kinase activity
Cells were grown to 80–90% confluence in a 6-well plate, pre-treated with peptidimer-c (1 μM for 1 h) and TGF-β2 (5 ng/ml for 30 min) and incubated at 37 °C. Extraction of PIP3 from cells followed the protocol in the PIP3 Mass ELISA kit (Echelon #k-2500 s). PIP3 levels were estimated using the PI3-K activity/inhibitor assay kit (Millipore #17–493) according to the manufacturer’s instructions. Briefly, the kit contains recombinant protein GRP-1, which binds to the glutathione plate and captures either PIP3 generated by the PI3-kinase reaction, or the biotinylated-PIP3 tracer included in the kit. The captured biotinylated-PIP3 is detected using streptavidin-HRP conjugate and following colorimetric development the optical density was measured at 405 nm.
Dynamic monitoring of cell adhesion: xCELLigence (Roche)
Fibronectin (0.7 μg/ml) was added to wells on 96X E-plate then the plate incubated for 30 min at 37 °C. The protein-coated plates were washed with PBS and incubated with 0.5% BSA solution in PBS for 1 h at 4 °C. The wells of the treated plates were washed with PBS. 50 μL of the cell suspension was transferred to wells on E-plates. The extent of cell adhesion and spreading, was monitored every 5 min for a period of 2 h. The xCELLigence System monitors cellular events in real time without the incorporation of labels. The System measures electrical impedance across interdigitated micro-electrodes integrated on the bottom of tissue culture E-Plates.
Matrigel chambers assay
The invasive capacity of Ode macrophages was assessed in vitro using Matrigel migration chambers, as described47. Culture coat 96-well medium BME cell invasion assay was obtained from Culturex instructions (3482-096-K). After 24 h of incubation at 37 °C, each well of the top chamber was washed once in buffer. The top chamber was placed back on the receiver plate. 100 μL of cell dissociation solution/Calcein AM were added to the bottom chamber of each well, incubated at 37 °C for 1 h to fluorescently label cells and dissociate them from the membrane before reading at 485 nm excitation, 520 nm emission using the same parameters as the standard curve.
Statistical analysis
All experiments were repeated at least three times and the results were expressed as mean ± standard error (SE). The statistical analysis were calculated using Student’s t-test. Differences were considered significant when P < 0.05.
Additional Information
How to cite this article: Haidar, M. et al. TGF-β2 induces Grb2 to recruit PI3-K to TGF-RII that activates JNK/AP-1-signaling and augments invasiveness of Theileria-transformed macrophages. Sci. Rep. 5, 15688; doi: 10.1038/srep15688 (2015).
References
Dobbelaere, D., Fernandez, P., Machado, J., Botteron, C. & Heussler, V. Interference by the intracellular parasite Theileria parva with T-cell signal transduction pathways induces transformation and protection against apoptosis. Vet Immunol Immunopathol 72, 95–100 (1999).
Dobbelaere, D. & Heussler, V. Transformation of leukocytes by Theileria parva and T. annulata. Annu Rev Microbiol 53, 1–42, doi: 10.1146/annurev.micro.53.1.1 (1999).
McHardy, N., Wekesa, L. S., Hudson, A. T. & Randall, A. W. Antitheilerial activity of BW720C (buparvaquone): a comparison with parvaquone. Res Vet Sci 39, 29–33 (1985).
Dobbelaere, D. A., Coquerelle, T. M., Roditi, I. J., Eichhorn, M. & Williams, R. O. Theileria parva infection induces autocrine growth of bovine lymphocytes. Proc Natl Acad Sci USA 85, 4730–4734 (1988).
Somerville, R. P., Adamson, R. E., Brown, C. G. & Hall, F. R. Metastasis of Theileria annulata macroschizont-infected cells in scid mice is mediated by matrix metalloproteinases. Parasitology 116(Pt 3), 223–228 (1998).
Glass, E. J. et al. Bos taurus and Bos indicus (Sahiwal) calves respond differently to infection with Theileria annulata and produce markedly different levels of acute phase proteins. Int J Parasitol 35, 337–347, doi: 10.1016/j.ijpara.2004.12.006 (2005).
Campbel, J. D. & Spooner, R. L. Macrophages behaving badly: infected cells and subversion of immune responses to Theileria annulata. Parasitology today 15, 10–16 (1999).
Baumgartner, M. et al. Constitutive PI3-K activity is essential for proliferation, but not survival, of Theileria parva-transformed B cells. Cellular microbiology 2, 329–339 (2000).
Guergnon, J. et al. A tumour necrosis factor alpha autocrine loop contributes to proliferation and nuclear factor-kappaB activation of Theileria parva-transformed B cells. Cellular microbiology 5, 709–716 (2003).
Chaussepied, M. et al. TGF-b2 induction regulates invasiveness of Theileria-transformed leukocytes and disease susceptibility. PLoS Pathog 6, e1001197, doi: 10.1371/journal.ppat.1001197 (2010).
Massague, J. TGF-beta signal transduction. Annu Rev Biochem 67, 753–791, doi: 10.1146/annurev.biochem.67.1.753 (1998).
Itoh, S. & ten Dijke, P. Negative regulation of TGF-beta receptor/Smad signal transduction. Curr Opin Cell Biol 19, 176–184, doi: 10.1016/j.ceb.2007.02.015 (2007).
Lopez-Casillas, F., Wrana, J. L. & Massague, J. Betaglycan presents ligand to the TGF beta signaling receptor. Cell 73, 1435–1444 (1993).
Zhang, Y. E. Non-Smad pathways in TGF-beta signaling. Cell research 19, 128–139, doi: 10.1038/cr.2008.328 (2009).
Miyazono, K. Transforming growth factor-beta signaling in epithelial-mesenchymal transition and progression of cancer. Proc Jpn Acad Ser B Phys Biol Sci 85, 314–323 (2009).
Zhang, L. et al. A transforming growth factor beta-induced Smad3/Smad4 complex directly activates protein kinase A. Mol Cell Biol 24, 2169–2180 (2004).
Katso, R. et al. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis and cancer. Annu Rev Cell Dev Biol 17, 615–675, doi: 10.1146/annurev.cellbio.17.1.615 (2001).
Leevers, S. J., Vanhaesebroeck, B. & Waterfield, M. D. Signalling through phosphoinositide 3-kinases: the lipids take centre stage. Curr Opin Cell Biol 11, 219–225 (1999).
Guergnon, J. et al. A PKA survival pathway inhibited by DPT-PKI, a new specific cell permeable PKA inhibitor, is induced by T. annulata in parasitized B-lymphocytes. Apoptosis: an international journal on programmed cell death 11, 1263–1273, doi: 10.1007/s10495-006-7702-6 (2006).
Heussler, V. T. et al. The Akt/PKB pathway is constitutively activated in Theileria-transformed leucocytes, but does not directly control constitutive NF-kappaB activation. Cellular microbiology 3, 537–550 (2001).
Ding, J. et al. PI3K/Akt/JNK/c-Jun signaling pathway is a mediator for arsenite-induced cyclin D1 expression and cell growth in human bronchial epithelial cells. Curr Cancer Drug Targets 9, 500–509 (2009).
Galley, Y. et al. Jun NH2-terminal kinase is constitutively activated in T cells transformed by the intracellular parasite Theileria parva. Proc Natl Acad Sci USA 94, 5119–5124 (1997).
Chaussepied, M. et al. Upregulation of Jun and Fos family members and permanent JNK activity lead to constitutive AP-1 activation in Theileria-transformed leukocytes. Molecular and biochemical parasitology 94, 215–226 (1998).
Yang, J. T. et al. Overlapping and independent functions of fibronectin receptor integrins in early mesodermal development. Dev Biol 215, 264–277, doi: 10.1006/dbio.1999.9451 (1999).
Echebli, N. et al. Engineering attenuated virulence of a Theileria annulata-infected macrophage. PLoS Negl Trop Dis 8, e3183 (2014).
Vidal, M. et al. Differential interactions of the growth factor receptor-bound protein 2N-SH3 domain with son of sevenless and dynamin. Potential role in the Ras-dependent signaling pathway. The Journal of biological chemistry 273, 5343–5348 (1998).
Lowenstein, E. J. et al. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 70, 431–442 (1992).
Chardin, P., Cussac, D., Maignan, S. & Ducruix, A. The Grb2 adaptor. FEBS Lett 369, 47–51 (1995).
Ye, Y. B. et al. The cytotoxicity of a Grb2-SH3 inhibitor in Bcr-Abl positive K562 cells. Biochem Pharmacol 75, 2080–2091, doi: 10.1016/j.bcp.2007.12.021 (2008).
Haidar, M., Echebli, N., Ding, Y., Kamau, E. & Langsley, G. TGF-beta2 promotes transcription of COX2 and EP4 leading to a PGE2-driven auto-stimulatory loop that enhances virulence of Theileria-transformed macrophages. Infect Immun, doi: 10.1128/IAI.02975-14 (2015).
Cussac, D. et al. A Sos-derived peptidimer blocks the Ras signaling pathway by binding both Grb2 SH3 domains and displays antiproliferative activity. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 13, 31–38 (1999).
Galliher-Beckley, A. J. & Schiemann, W. P. Grb2 binding to Tyr284 in TbetaR-II is essential for mammary tumor growth and metastasis stimulated by TGF-beta. Carcinogenesis 29, 244–251, doi: 10.1093/carcin/bgm245 (2008).
Verdino, P., Witherden, D. A., Havran, W. L. & Wilson, I. A. The molecular interaction of CAR and JAML recruits the central cell signal transducer PI3K. Science 329, 1210–1214, doi: 10.1126/science.1187996 (2010).
Cheng, S. Y., Sun, G., Schlaepfer, D. D. & Pallen, C. J. Grb2 promotes integrin-induced focal adhesion kinase (FAK) autophosphorylation and directs the phosphorylation of protein tyrosine phosphatase alpha by the Src-FAK kinase complex. Mol Cell Biol 34, 348–361, doi: 10.1128/MCB.00825-13 (2014).
Sugiura, Y., Shimada, H., Seeger, R. C., Laug, W. E. & DeClerck, Y. A. Matrix metalloproteinases-2 and -9 are expressed in human neuroblastoma: contribution of stromal cells to their production and correlation with metastasis. Cancer research 58, 2209–2216 (1998).
Hall, R. et al. Mechanism(s) of attenuation of Theileria annulata vaccine cell lines. Tropical medicine & international health : TM & IH 4, A78–84 (1999).
Adamson, R. & Hall, R. A role for matrix metalloproteinases in the pathology and attenuation of Theileria annulata infections. Parasitology today 13, 390–393 (1997).
Adamson, R., Logan, M., Kinnaird, J., Langsley, G. & Hall, R. Loss of matrix metalloproteinase 9 activity in Theileria annulata-attenuated cells is at the transcriptional level and is associated with differentially expressed AP-1 species. Molecular and biochemical parasitology 106, 51–61 (2000).
Jochum, W., Passegue, E. & Wagner, E. F. AP-1 in mouse development and tumorigenesis. Oncogene 20, 2401–2412, doi: 10.1038/sj.onc.1204389 (2001).
Wang, J., Auger, K. R., Jarvis, L., Shi, Y. & Roberts, T. M. Direct association of Grb2 with the p85 subunit of phosphatidylinositol 3-kinase. The Journal of biological chemistry 270, 12774–12780 (1995).
Forsyth, L. M. et al. Tissue damage in cattle infected with Theileria annulata accompanied by metastasis of cytokine-producing, schizont-infected mononuclear phagocytes. Journal of comparative pathology 120, 39–57, doi: 10.1053/jcpa.1998.0256 (1999).
Metheni, M. et al. The level of H(2)O(2) type oxidative stress regulates virulence of Theileria-transformed leukocytes. Cellular microbiology 16, 269–279, doi: 10.1111/cmi.12218 (2014).
Yano, S., Arroyo, N. & Yano, N. Catalase binds Grb2 in tumor cells when stimulated with serum or ligands for integrin receptors. Free Radic Biol Med 36, 1542–1554, doi: 10.1016/j.freeradbiomed.2004.04.006 (2004).
Metheni, M., Lombes, A., Bouillaud, F., Batteux, F. & Langsley, G. HIF-1alpha induction, proliferation and glycolysis of Theileria-infected leukocytes. Cellular microbiology 17, 467–472, doi: 10.1111/cmi.12421 (2015).
Singh, S. et al. Impact of field vaccination with a Theileria annulata schizont cell culture vaccine on the epidemiology of tropical theileriosis. Vet Parasitol 101, 91–100 (2001).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic acids research 29, e45 (2001).
Lizundia, R. et al. c-Jun NH2-terminal kinase/c-Jun signaling promotes survival and metastasis of B lymphocytes transformed by Theileria. Cancer research 66, 6105–6110, doi: 10.1158/0008-5472.CAN-05-3861 (2006).
Acknowledgements
MH was supported by a PhD fellowship from the National Council for Scientific Research, CNRS, Beirut, Lebanon; JW by an Erasmus student exchange fellowship with the Chemistry Department at Leeds University, England. GL acknowledges support from ANR grant (11 BSV3 01602), Labex ParaFrap (ANR-11-LABX-0024), INSERM and the CNRS.
Author information
Authors and Affiliations
Contributions
M.H. and J.W. performed the biochemical experiments; J.W. and W.L. peptide synthesis of peptidimer-c; G.N. bioinformatics of putative pathways activated by TGF-β stimulation; G.L. and M.V. conceived the study and M.H. and G.L. prepared figures and wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Haidar, M., Whitworth, J., Noé, G. et al. TGF-β2 induces Grb2 to recruit PI3-K to TGF-RII that activates JNK/AP-1-signaling and augments invasiveness of Theileria-transformed macrophages. Sci Rep 5, 15688 (2015). https://doi.org/10.1038/srep15688
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep15688
This article is cited by
-
Theileria parasites subvert E2F signaling to stimulate leukocyte proliferation
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
