
Abstract
Despite the accumulating knowledge on the development and establishment of the gut microbiota, its role as a reservoir for multidrug resistance is not well understood. This study investigated the prevalence and persistence patterns of an integrase gene (int1), used as a proxy for integrons (which often carry multiple antimicrobial resistance genes), in the fecal microbiota of 147 mothers and their children sampled longitudinally from birth to 2 years. The study showed the int1 gene was detected in 15% of the study population and apparently more persistent than the microbial community structure itself. We found int1 to be persistent throughout the first two years of life, as well as between mothers and their 2-year-old children. Metagenome sequencing revealed integrons in the gut meta-mobilome that were associated with plasmids and multidrug resistance. In conclusion, the persistent nature of integrons in the infant gut microbiota makes it a potential reservoir of mobile multidrug resistance.
Similar content being viewed by others


Introduction
The spread of antibiotic resistance (AR) genes and development of multidrug resistance represent major threats to public health1. Until recently, pathogens have been the prime focus with respect to understanding the spread of multidrug resistance, with the commensal microbiota receiving much less attention. However, recent studies have shown the prevalence of AR genes in the commensal gut microbiota2,3,4,5. Furthermore, the gut microbiota shows a high rate of horizontal gene transfer (HGT), which was indicated to be up to 25-fold greater than that of bacteria in other environments6. Hence, the collective mobile genetic elements (MGEs) in the gut microbiota (i.e. the gut meta-mobilome) represent an important target for both understanding and combating the spread of multidrug resistance5,7.
The gut microbiota forms a complex ecosystem. The gut is assumed sterile at birth8,9 whereas just after birth, it goes through major shifts starting with facultative anaerobic bacteria (Enterococcaceae and Streptococcaceae)10,11. As oxygen levels deplete, strictly anaerobic bacteria (Bifidobacteriales and Bacteroidetes) take over and dominate in the gut12. This progression slows down as the microbiota reaches the adult-like state where an estimated 100–200 species co-exist in close proximity13. Although scientists have started to understand the shifts in the taxonomic composition of the developing microbiota from infancy to adulthood, the knowledge of the meta-mobilome, including the transmission and persistence of multiple antimicrobial resistance genes, is limited.
Antimicrobial resistance genes can be carried in integrons, which are non-mobile elements themselves, but are often found within MGEs like transposons and plasmids14,15. Integrons are platforms for integration, assembly and expression of specific gene cassettes within the MGEs that often encode antimicrobial resistance16. The individual genetic cassettes typically lack their own promoters, but are expressed by a common promoter for all the cassettes within the integron (Fig. 1). There have been 5 classes of integrons (class I–V) classified to date5. The class I integrons are the most widely studied and are found in a broad host range of commensal and pathogenic bacteria17. Class I integrons are found extensively in clinical isolates containing several different AR gene cassettes conferring resistance to antibiotics commonly used against bacterial infections16,18. Up to 8 gene cassettes have been found in a single class I integron16, however hundreds of gene cassettes have been detected in so-called super-integrons19.

Structure of class I integron.
A general representation of a class I integron with resistance gene cassettes at the attachment sites (attC) and a common promoter for the cassettes as Pc and for the integrase as Pint. The following cassettes are a part of the 3′ conserved region and not mobile: sul1 gene encoding resistance to sulfonamides and qacE∆ encoding resistance to quaternary ammonium compounds.
The aim of the current study was to investigate the prevalence and persistence of class I integrons in a large unselected longitudinal cohort of mothers and their children. We used quantitative PCR to identify and study the persistence patterns of integrons. 16S rRNA and metagenome deep sequencing were used to analyze the phylogeny and genetic background of the integrons in the samples and to trace these elements longitudinally.
Materials and Methods
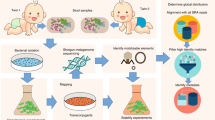
The schematic overview of the workflow is displayed in Fig. 2. The methods were performed in accordance to the approved guidelines and all experimental protocols were approved by Norwegian University of Life Sciences.

Workflow of experimental setup.
Cohort description
IMPACT (Immunology and Microbiology in Prevention of Allergy among Children in Trondheim) study is a controlled non-randomized longitudinal study, which began in 2000. The regional committee for Medical Research Ethics for Central Norway has approved the IMPACT study (ref. 120–2000). This study was granted a license by the Norwegian Data Inspectorate to process personal health data and one of the parents of each child signed a written informed consent form (r. 2003/953-3 KBE/-). Current controlled trials registration number: ISRCTN28090297.
The study involved 720 pairs of pregnant women and their children (up to two years of age). Ninety percent of the children were vaginally delivered and at term. Ninety-seven percent of the infants were breast-fed exclusively for the first six weeks of life. Fecal samples were collected from the pregnant women during the first/second (7–20 weeks) trimester and the third (32–40 weeks) trimester and from the children at 3–10 days, 4 months, 1 and 2 years of age. In the current study, samples from a randomly selected subgroup of 147 mother-child pairs from the IMPACT cohort were analyzed. Information on allergy related hereditary diseases, atopy and antibiotic usage; health and exposure factors for the parent and child is summarized in Supplementary Table S1.
Sample collection
Fecal samples from the subjects of the IMPACT cohort were collected in Cary-Blaire transport and holding medium (BD Diagnostics, Sparks, MD). The samples were frozen at -20 °C within 2 h from collection. The samples were then stored at −80 °C within one month for children and mothers.
DNA purification
Fecal DNA was purified with an automated protocol using DNA extraction kit based on paramagnetic particles (LGC Genomics, UK). In brief, the samples were subjected to mechanical lysis using glass beads and the DNA was purified by eluting from the paramagnetic particles by downstream processes as described by manufacturer. The DNA was stored at −40 °C.
Gene quantification
The abundance of integrons (using the integrase (int1) gene20 as a proxy) in the samples was calculated relative to the 16S rRNA gene by quantitative PCR. Each PCR reaction (25 μl) contained 1× HOT FIREPol PCR mix (Solis BioDyne, Estonia); 200 nM forward and reverse primers; one μl of sample DNA and water. The reaction mix was run on LightCycler 480 (Roche, Germany). Following the thermal cycling the raw fluorescence data was exported into LinRegPCR program. The software performed baseline corrections and calculated the mean PCR efficiency. For the int1 amplicon, we also used High Resolution Melting (HRM) curve analysis, in addition to Sanger sequencing using the BigDye Terminator v.1.1 chemistry (Applied Biosystems) for verification.
The thermal cycling for the 16S rRNA primer-pair (5′-TCCTACGGGAGGCAGCAGT-3′; 5′- GGACTACCAGGGTATCTAATCCTGTT-3′) was an initial denaturation of 95 °C for 15 min followed by 40 cycles of 95 °C for 30 sec and 60 °C for 30 sec. This primer-pair targets conserved regions of the 16S rRNA gene21. The primers flanking the int1 gene (5′-ACGAGCGCAAGGTTTCGGT-3′; 5′-GAAAGGTCTGGTCATACATG-3′) from Sørum et al.11 were used with thermal cycling conditions 95 °C for 15 min and 40 cycles of 95 °C for 30 sec, 53 °C for 30 sec and 72 °C for 30 sec.
Microbial community analyses
Microbial communities were assessed using Illumina sequencing of 16S rRNA gene amplicons (n = 465), with subsets subjected to full metagenome (n = 15) and long-range PCR amplicon (n = 6) analyses. For full metagenomics analysis, samples were selected based on the high relative quantities of int1 gene in the samples. For a long-range PCR, six int1-positive samples were randomly chosen for amplification.
Long-range primers were used to amplify the sequence flanking the region from attI to the 3′ consensus region including the gene cassettes (5′-GGCATCCAAGCAGCAAG-3′; 5′-AAGCAGACTTGACCTGA-3′)11 with the TaKaRa LA PCR kit Ver.2.1. The thermal cycling conditions of 94 °C for 5 min followed by 35 cycles of 98 °C for 10 min, 54 °C for 30 sec and 72 °C for 1 min, with the final extension step at 72 °C for 5 min. The resultant PCR products were analyzed by agarose gel electrophoresis and Illumina sequencing.
For full metagenome, long-range amplicon and metagenome analyses, gDNA was randomly fragmented, tagged, amplified and prepared for sequencing using Nextera XT kit (Illumina, USA).
Portions of the 16S rRNA genes were amplified using PRK341F/PRK806R primers targeting V3-V4 regions22, modified by addition of Illumina-specific adapters. Each PCR reaction (25 μl) contained 1× HOT FIREPol PCR mix (Solis BioDyne, Estonia); 200 nM uniquely tagged forward and reverse primers; 1 μl of sample DNA and water. The thermal cycling conditions were 95 °C for 15 min and 30 cycles of 95 °C for 30 sec, 50 °C for 1 min and 72 °C for 45 sec. PCR products were then pooled, based on their concentrations measured using Quant-iTTM PicoGreen® dsDNA assay kit (Life Technologies, USA), column-purified using E.Z.N.A.® Cycle Pure kit (Omega Bio-tek, USA) and submitted for sequencing.
Sequencing was performed on MiSeq platform (Illumina, USA) using V3 sequencing chemistry with 300 bp paired-end reads. 16S rRNA gene amplicon samples were processed at Norwegian Sequencing Centre (Oslo, Norway), whereas full metagenome samples were sequenced in-house.
Bacterial culturing
For isolation of Bifidobacterium species, 10-fold dilutions of fecal samples in 1% peptone water were anaerobically cultured on Beerens agar at 37 °C. Isolated colonies were then subcultured to purity using the same conditions. DNA was extracted for sequencing of 16S rRNA gene as described above to confirm isolates belonging to Bifidobacterium genus.
Three-fold serial dilutions of fecal samples from the cohort were prepared in distilled water, cultured on lactose agar and in tryptic soya broth with 5% horse blood, incubated at 37 °C for 24 h. The broth was supplemented with 0.1% of both Tween 80 and magnesium chloride to recover damaged Enterobacteriaceae cells.
Data analyses
16S rRNA gene amplicon data were analyzed using QIIME pipeline23. Sequences were first quality filtered (split_libraries.py; sequence length between 200 bp and 1000 bp; minimum average quality score 25; not more than 6 ambiguous bases; and no primer mismatch allowed) and then clustered at 99% homology level using closed-reference uclust search against Greengenes database24 (pick_closed_reference_otus.py). Persistence of operational taxonomic units (OTUs) over time in individuals was assessed using multi-way decomposition PARAFAC analysis of mean-centered abundance data25. This analysis allows detection of the OTUs that bring most of the variation into the system, simultaneously with detecting the time points at which these OTUs are most pronounced. Simpson’s reciprocal diversity index and Bray-Curtis dissimilarity index were used for alpha- and beta-diversity assessment, respectively.
Metagenome data mapping and assembly was performed using Geneious pipeline following authors’ recommendations26. MG-RAST metagenome analyzer was used to analyze the taxonomy and functional classification of the samples27. PATRIC database28 in MG-RAST was used to check the integron abundance in the samples. E-value < 10−5 was used as the cut-off to select integron hits.
Int1 gene persistence was calculated as the ratio of the number of mother-child pairs in who int1 was detected at both time points to the total number of mother-child pairs for who information for both time points was available. The odds ratio for int1 gene detection was calculated by the ratio of int1 persistence to the prevalence of int1 at a later time point.
Fisher exact test, Pearson correlation coefficient and Spearman correlation coefficient were used for pairwise comparisons of int1 and 16S rRNA data (including diversity, OTU abundance and bacterial class abundance data). The significance of the change over time was tested with Friedman’s test - a non-parametric version of ANOVA test which takes into account repeated measurements. The change in int1 gene relative abundance was also compared to the change in log-transformed OTU relative abundances over time in an attempt to identify OTUs that correlated to int1. Regression and classification decision trees were also built in an attempt to identify bacterial classes that correlated to int1. Data analyses were performed using MATLAB® R2014a software (The MathWorks Inc., Natick MA, USA).
Results
Microbiota composition and development
The phylogenetic composition of the microbiota was assessed using deep 16S rRNA gene sequencing. All samples that were amplified with 16S rRNA gene-targeting primers and further amplified with Illumina-adapted primer set were included in the analysis. In total, sequencing data were available for 451 samples. In addition, seven of the samples were analyzed in triplicate to determine technical variation, which was found to be low (Supplementary Fig. S1). The average quality score for the sequence range of 250–299 bp was 25.
On average, 21,277 sequences per sample were generated after quality filtering and assembly. To ensure even amount of sequencing information, 4,000 reads per sample were randomly picked from the full dataset based on the recommendations by Sørensen et al.29. The final dataset after quality filtering and unification of the sequencing information per sample comprised 378 samples, with a total of 8,288 OTUs belonging to 27 classes. The 10 most abundant classes comprised nearly 100% of the microbiota at all ages (Fig. 3). Stool samples from newborns and 4-month-old infants were significantly lower in alpha-diversity and significantly higher in beta-diversity than stool samples from 2-year-olds and their mothers (Fig. 4). At 1 year of age, both alpha- and beta-diversity estimates were significantly higher than that of 4 month-olds. There was a high dominance of Clostridia in stool samples from mothers, as well as from 1- and 2-year-olds. Five bacterial classes were relatively equal in abundance in neonatal stool samples collected soon after birth (3 days), whereas Actinobacteria became dominant thereafter (4–10 days) and remained so through at least first 4 months of age. By 1 year of age, the average profile of stool samples from children had started converging towards the adult profile. However, pronounced differences in the abundance of Actinobacteria and Bacteroidia were seen between adults and 2-year-old children, suggesting climax adult community was still not reached by 2 years of age.

Bacterial class composition of stool samples of infants (from 3 days to 2 years of age) and their mothers during early (1/2 trimester; EarlyPreg) and late (final trimester; LatePreg) pregnancy based on the deep sequencing of 16S rRNA gene amplicons.
s, Number of samples at each time period.

Diversity characteristics of stool samples of infants (from 3 days to 2 years of age) and their mothers during early (1/2 trimester; EarlyPreg) and late (final trimester; LatePreg) pregnancy based on the deep sequencing of 16S rRNA gene amplicons.
(a) Simpson’s reciprocal index of alpha-diversity. (b) Bray-Curtis dissimilarity index of beta-diversity. *p value < 0.05; and ***p value < 0.001.
Microbiota persistence and stability
The persistence of 599 most abundant OTUs in the dataset (with an abundance level ≥0.5% in at least one sample) were analyzed using PARAFAC. No significant associations of OTUs to age were identified when only considering the detected/non-detected information. When abundance levels were considered, two OTUs belonging to Bifidobacterium species (B. longum OTU594044 and B. breve OTU484303) and one assigned to Enterobacteriaceae family (OTU1109087), showed highest stability over time in the cohort (Fig. 5). Spearman correlation test identified the persistence of the B. longum-assigned OTU, which had a highest loading in PARAFAC, from 3–10 days to 4 months of age (correlation coefficient = 0.49; p = 0.007). The two other OTUs, however, did not show any significant correlations between the age groups.

Summary of PARAFAC analysis (multi-way decomposition which defines most influential OTUs over time) on the relative abundances of 599 most abundant bacterial OTUs.
(A) Most influential OTUs. (B) Time points associated with most influential OTUs.
Integron distribution and persistence
The distribution of integrons was analyzed by quantitative PCR of the int1 gene. All samples were included and amplification was controlled by 16S rRNA gene amplification. Out of initial 663 IMPACT samples, 16 failed to amplify PCR products using 16S rRNA gene-targeting primers and thus were excluded from the analysis. In total, 99 of the 647 samples analyzed showed the presence of integrons. The prevalence of the integron-positive samples was highest from 4-month-old children compared to any other age (Fig. 6a). The highest persistence patterns for integrons were seen in children between 3–10 days and 4 months and 4 months to 1 year (Fig. 6b). Persistence between some mother-child pairs was also detected. The int1 gene copy numbers of the positive samples, corrected for the estimated genome equivalents30, were significantly higher in samples from infants (3–10 days and 4 months) compared to both pregnant mothers and 2-year-old children (Fig. 6c).

Prevalence, persistence of integron-positive samples and relative quantity of integrons in the positive samples between time points.
(a) Relative prevalence of integron-positive samples in the dataset. *Binomial testing between the highest abundance (4 months) and the rest (p value = 0.005). (b) Persistence of integrons at each time point. The numbers represent the odds-ratio; the color gradient represents the percentage of persistence between time points. Significant p values by Fisher exact test are also indicated (*p value < 0.05; **p value < 0.01; ***p value < 0.001). (c) Relative integron quantification at each time point (log (int1 copies/genome equivalent1) for integron-positive samples. Error bars represent standard error of the mean (SEM). The significant difference between sample groups was calculated by Kruskal-Wallis test; p value < 0.05 is indicated by bracketing. Early_latePreg, samples collected from mothers during early (7–20 weeks) and late (32–40 weeks) pregnancy; 3–10 days, samples from 3- to 10-day-old infants; 4 months, 1 year and 2 years; samples from 4-month-old infants, 1-year-old and 2-year-old children, respectively. 116S rRNA copies of all samples from different age groups were normalized to reflect genome equivalents taking into account copy number information given by Vetrovsky et al.30.
For the children with persistent int1 genes, 17% (1 of 6 children with antibiotic usage information) received antibiotics during the first year of life. In addition, 31% (46 out of 147) of the children in the whole cohort had antibiotic usage information documented.
Correlation of int1 gene to 16S rRNA gene
Detection of int1 gene did not correlate to alpha-diversity (Simpson’s reciprocal index 1/D = 12.3 ± 1.74 [mean ± SEM] and 1/D = 13.7 ± 0.66 for int1-positive and int1-negative subgroups, respectively) or to beta-diversity (Bray-Curtis Dissimilarity index BC = 0.85 ± 0.03 and BC = 0.86 ± 0.04 for int1-positive and int1-negative subgroups, respectively). There was also no significant correlation detected between alpha-diversity and int1 gene relative abundance (correlation coefficient = −0.389, p = 0.45).
With respect to OTU quantity, the most persistent OTU (B. longum OTU594044) showed a positive correlation with the int1 gene (p = 0.03) at 3–10 days. No other significant correlations, however, were found (Supplementary Fig. S2). Additionally, it was investigated whether a change in OTU relative abundance could be associated to the change in int1 gene relative abundance over time, but there was not an OTU identified that was significantly associated to the int1 gene. Finally, the analyses concentrated on the OTUs that were detected in all samples for which int1 were detected (Supplementary Table S2); however, these OTUs did not show any quantitative correlations with int1 either.
There were additional attempts to find bacterial classes that might correlate to int1 detection or int1 gene abundance; however, no significant pairwise correlations between bacterial classes and int1 gene abundance were detected (Supplementary Fig. 3). Regression and classification decision trees were then built to test for the cumulative effects of bacterial classes, but these analyses also suggested weak correlations between 16S rRNA gene and int1 gene data (Supplementary Fig. S4 and Supplementary Fig. S5 for regression and classification, respectively).
Search for int1 gene in bacterial isolates
The detection of the int1 gene in the genomes of sequenced representatives of persistent/stable OTUs identified by PARAFAC was carried out by BLAST searching whole-genome sequencing data from 16 B. longum, 2 B. breve and 10 E. coli strains, isolated from previously published subset of the IMPACT dataset31,32. Despite high numbers of hits to 16S rRNA gene per isolate (451.5 ± 37.8), which was used as a proxy for the genome coverage, the analyses failed to identify reads that showed homology to the int1 gene sequence identified in our work.
Long-range PCR and amplicon sequencing
Six integron-positive samples were randomly selected for long-range PCR and sequencing. The reads were assembled into contigs and two contigs of lengths 1541 bp and 1019 bp showed BLAST hits to E. coli strain DK510 (GQ906578.1) containing dihydrofolate reductase (dfrA17) and aminoglycoside adenylyltransferase (aaDA5) genes (E-value = 0) with 100% identity (Fig. 7a) and E. coli strain A30 (KF921570.1) containing dihydrofolate reductase (dfrA12), hypothetical protein (orfF) and aminoglycoside adenylyltransferase (aadA2) gene cassettes (E-value = 0) with 100% identity (Fig. 7b), respectively.

Integrons detected by long-range PCR.
(a) 1.5 kb partially sequenced integron by long-range PCR product. (b) 1.1 kb partially sequenced integron by long-range PCR. Cylindrical boxes show individual genes that are size dependent, i.e. larger box is longer gene; dotted arrows indicate the direction of transcription; and gradient blue color the end of the acquired sequence. Gene and structural features: attI, primary recombination site; dfrA17 and dfrA12, dihydrofolate reductase; attC, recombination site; aadA5 and aadA2, aminoglycoside adenylyltransferase; and orfF, hypothetical protein.
Integron presence in shotgun metagenome data
Fifteen samples (late pregnancy, n = 1; 3–10 days, n = 6; 4 months, n = 5; and 2 years, n = 3) having microbiota profile information and highest relative abundance of int1 were selected for shotgun metagenome sequencing. On average, 837,048 reads with a size range from 35 bp to 301 bp were obtained for each sample. By NCBI BLAST searches 699 shotgun metagenomic reads from 12 samples were identified that showed high homology to the int1 gene (E-value < 10−5; average identity [range] 97.5% [85.1%; 100%]; average query coverage 99.7% [98.4%; 100.0%]).
Using the MG-RAST metagenome analyzer27, it was found that all the samples showed the presence of integrons and integron-related genes. The identity of the integron hits of the samples were obtained from PATRIC database (Supplementary Table S3).
Metagenome assembly and identification of complete integrons
The reads were extracted that showed int1 homology in only one direction of the paired-end reads (n = 71) to investigate the genetic background of their paired mates. By BLAST searching of these sequences against NCBI database, candidate plasmid pSH1148_107 (GenBank JN983049) was identified that was most prevalent among the hits (Supplementary Table S4). The metagenomic reads were then mapped onto the complete plasmid sequence and approximately 60% of the plasmid was encompassed by the metagenomic reads. Seventeen of the 25 conjugation proteins of the plasmid mapped to our reads, including the Inc1 conjugative transfer proteins, DNA primase and pilus biogene (Supplementary Fig. S6). The reads partially covered the origin of replication. There was one child who showed high prevalence of a plasmid related to pSH1148_107 (more than 1% of all reads) in stool samples from both 3–10 days and 4 months (20× and 34× mean coverage for 3–10 days and 4 months, respectively). The 3–10 days and 4 months reads mapped similarly to the plasmid. The de novo assembly of the reads mapped to a transposon containing integron with the sul1 gene and aadA gene cassette, which was similar to the resistance genes in pSH1148_107 and an additional dfrA17 gene cassette (Fig. 8). The gene cassettes encode resistance to sulphonamides, spectinomycin and streptomycin and trimethoprim respectively.

Graphical representation of a transposon-containing integron by de novo assembly.
The boxes illustrate the coding region of genes, dark blue represents genes of the transposon and the light blue indicates genes of the integron. Genetic features: dfrA17, trimethoprim resistance protein; aadA5, streptomycin and spectinomycin resistance protein; qacEdelta, quaternary ammonium compound resistance protein; sul1, sulphonamide resistance protein.
The long-range PCR amplicon contigs were also mapped to the integron assembled from our metagenome. The 1541 bp-long contig showed 97% coverage, suggesting both assemblies came from the same integron. The other contig of 1019 bp length had different gene cassettes and thus showed only partial coverage.
Taxonomic range of the integrons identified by long-range PCR
BLAST searching of the NCBI database with the int1-containing contigs identified by long-range PCR revealed high homology (100% pairwise identity with 100% query coverage) towards plasmids isolated from E. coli, Kluyvera georgiana, Salmonella enterica and Shigella flexneri (Supplementary Table S5), all belonging to Enterobacteriaceae family.
Search for integrons in other metagenomes
To search for the same integron in other publically available metagenomes, data was extracted from 60 metagenome samples from the cohort provided by Yatsunenko et al.33. The available cohort contained fecal samples from healthy children and adults in Malawi, United States and Venezuela; and 20 metagenomes from each of the respective countries was analyzed. Eleven (18.3%) of the metagenomes showed the presence of int1 gene. Seven of the int1-positive metagenome samples also contained reads mapping to the transposon flanking regions. However, the integron-associated gene cassettes were not similar to those detected in our dataset (Supplementary Table S6).
Discussion
Several studies have shown a high prevalence of AR genes in infants with the absence of antibiotic treatments3,34,35 which is in line with our findings. However, to our knowledge, this study is the first one to observe high int1 gene prevalence and persistence. A high prevalence of integrons was found at 3–10 days and 4 months of age. In the early periods of life the resistance against colonization by exogenous bacteria is low35, therefore opening for the possibility of establishment of bacteria from the environment. A plausible explanation for the high integron prevalence at early age could be the hospital environment, since children are first exposed to this atmosphere36. There was also persistence of int1 gene throughout the first two years of life and between mothers and their 2-year-old children, pointing towards maternal source as another potential route for transmission. Similar patterns have also been detected in transposon-associated genes in mother-infant pairs3,31. An alternative explanation, though, could be the colonization by various integrons at different ages. However, taking into account the increased likelihood of int1 detection at one time period given it was detected previously, the more probable explanation would be the persistence of the same integron rather than the detection of independent multiple colonization events.
The persistence of integrons in the gut microbiota indicates the versatility of MGEs to endure the drastic changes that occur during first years of life10,33. However, it is unlikely that antibiotic treatment influences the presence of multidrug resistance integrons since we did not find any alteration of persistence patterns in our dataset associated with antibiotic usage.
Diversity estimates of the cohort corresponded well with previously published observations of increase in alpha- and decrease in beta-diversity with age11,33. Interestingly, when int1 gene abundance was highest at early days of life, the microbial diversity was lowest, suggesting that int1 gene should be associated to those few bacteria that are established by then. However, there was no correlation between int1 gene and diversity estimates or bacterial classes. Moreover, despite numerous attempts, we could not associate int1 gene to any particular phylotype across individuals within our cohort. Hence, it is unlikely that the integrons have a strict phylotype association. In addition, when we tried to search for int1 gene in Bifidobacterium isolates that represent the most abundant bacterial group in infancy and that was the only bacterial genus correlating to int1 gene abundance at early infancy, we failed to find any indication of integron presence in its genomes. Lack of association between integrons and phylotypes across large phylogenetic distances has previously been observed37. Statistical inconsistencies have been reported when phylogenetic trees were obtained for int1 gene and molecular marker for phylogeny such as RNA polymerase subunit B (rpoB)37. Therefore, given the broad host range for integrons38, the most plausible explanation for the lack of phylotype association is high rates of HGT. In concordance with potentially high HGT rates, a possible transposon carrying an integron was identified in our samples, suggesting MGEs as the likely vehicle for mobility of integrons. The mobile nature of integrons-associated MGEs has been previously observed in pathogenic bacteria39,40, environmental samples41 and in hospital environments42. We also observed the persistence of a transposon-containing integron on a potential conjugative plasmid in one infant at two time periods. This integron contained genes associated with aminoglycosides and sulfonamide resistance similar to the conjugative plasmid pSH1148_107, along with and additional trimethoprim resistance gene.
We expanded our search for integrons from different samples in our dataset by involving long-range PCRs that could amplify the whole integron. Two class I integrons were identified with potential association to a mobile element having resistance genes to trimethoprim, streptomycin and spectinomycin. Interestingly, a study by Shahcheraghi et al. also found a similar integron containing resistance genes in enteropathogenic E. coli strains (JX442969.1) isolated from fecal samples of children less than 5 years of age43. These evidences give further support that integrons can be reservoirs for AR genes in infants, with the potential for transmission to pathogens4,35. Additionally, we also detected integrons with different gene cassettes in publicly available metagenomes, suggesting the diversity of integrons in global human populations.
Our observation of integron-containing elements regardless of antibiotics intake suggests that they can persist without outer selection pressure. A recent study on the gut microbiota of an isolated group of Yanomani Amerindian tribe showed a similar pattern of the carriage of a pool of mobilizable next-generation antibiotic resistance genes without any prior antibiotic pressure44. Moreover, Stern and colleagues found over 10,000 contigs containing potential mobile elements in the MetaHIT dataset45, which were likely to be quite common constituents of the gut microbiota since all were identified as targets for CRISPR elements. Interestingly, only around 10% of these contigs were of viral nature, leaving the rest to plasmids and MGEs, suggesting that the host actually counter selects these mobile elements. This finding supports the selfish parasitic-like spread of conjugative plasmids associated integrons in the gut.
The overall results of the study provide evidence for high prevalence of integrons in the fecal microbiota at early stages of life and further suggest that the commensal gut microbiota can serve as a reservoir for multidrug resistance, potentially contributing to its rapid spread.
Additional Information
How to cite this article: Ravi, A. et al. The commensal infant gut meta-mobilome as a potential reservoir for persistent multidrug resistance integrons. Sci. Rep. 5, 15317; doi: 10.1038/srep15317 (2015).
References
McKenna, M. Antibiotic resistance: the last resort. Nature 499, 394–396, 10.1038/499394a (2013).
Hu, Y. et al. Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nature communications 4, 2151, 10.1038/ncomms3151 (2013).
de Vries, L. E. et al. The Gut as Reservoir of Antibiotic Resistance: Microbial Diversity of Tetracycline Resistance in Mother and Infant. PloS one 6, e21644, 10.1371/journal.pone.0021644 (2011).
Sommer, M. O. A., Dantas, G. & Church, G. M. Functional Characterization of the Antibiotic Resistance Reservoir in the Human Microflora. Science 325, 1128–1131, 10.1126/science.1176950 (2009).
Ravi, A. et al. Integrons in the Intestinal Microbiota as Reservoirs for Transmission of Antibiotic Resistance Genes. Pathogens 3, 238–248 (2014).
Smillie, C. S. et al. Ecology drives a global network of gene exchange connecting the human microbiome. Nature 480, 241–244, 10.1038/nature10571 (2011).
Modi, S. R., Lee, H. H., Spina, C. S. & Collins, J. J. Antibiotic treatment expands the resistance reservoir and ecological network of the phage metagenome. Nature 499, 219–222, 10.1038/nature12212
Sekirov, I., Russell, S. L., Antunes, L. C. M. & Finlay, B. B. Gut Microbiota in Health and Disease. Physiol. Rev. 90, 859–904, 10.1152/physrev.00045.2009 (2010).
Jiménez, E. et al. Isolation of Commensal Bacteria from Umbilical Cord Blood of Healthy Neonates Born by Cesarean Section. Curr. Microbiol. 51, 270–274, 10.1007/s00284-005-0020-3 (2005).
Palmer, C., Bik, E. M., DiGiulio, D. B., Relman, D. A. & Brown, P. O. Development of the Human Infant Intestinal Microbiota. PLoS Biol. 5, e177, 10.1371/journal.pbio.0050177 (2007).
Avershina, E. et al. Major faecal microbiota shifts in composition and diversity with age in a geographically restricted cohort of mothers and their children. Fems Microbiol. Lett. n/a-n/a, 10.1111/1574-6941.12223 (2013).
Avershina, E. & Rudi, K. Is it who you are or what you do that is important in the human gut? Benef. Microbes. in press (2013).
Faith, J. J. et al. The Long-Term Stability of the Human Gut Microbiota. Science 341, 10.1126/science.1237439 (2013).
Kovalevskaya, N. P. Mobile Gene Cassettes and Integrons. Mol. Biol. 36, 196–201, 10.1023/A:1015361704475 (2002).
Fluit, A. C. & Schmitz, F. J. Resistance integrons and super-integrons. Clin. Microbiol. Infect. 10, 272–288, 10.1111/j.1198-743X.2004.00858.x (2004).
Mazel, D. Integrons: agents of bacterial evolution. Nat. Rev. Microbiol. 4, 608–620, 10.1038/nrmicro1462 (2006).
Gillings, M. R. et al. Using the class 1 integron-integrase gene as a proxy for anthropogenic pollution. ISME J. 10.1038/ismej.2014.226 (2014).
Gillings, M. R., Xuejun, D., Hardwick, S. A., Holley, M. P. & Stokes, H. W. Gene cassettes encoding resistance to quaternary ammonium compounds: a role in the origin of clinical class 1 integrons? ISME J. 3, 209–215, 10.1038/ismej.2008.98 (2009).
Rowe-Magnus, D. A. et al. The evolutionary history of chromosomal super-integrons provides an ancestry for multiresistant integrons. Proc. Natl. Acad. Sci. USA 98, 652–657, 10.1073/pnas.98.2.652 (2001).
Sorum, H., L’Abee-Lund, T. M., Solberg, A. & Wold, A. Integron-containing IncU R plasmids pRAS1 and pAr-32 from the fish pathogen Aeromonas salmonicida. Antimicrob. Agents Chemother. 47, 1285–1290 (2003).
Nadkarni, M. A., Martin, F. E., Jacques, N. A. & Hunter, N. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology 148, 257–266 (2002).
Yu, Y., Lee, C., Kim, J. & Hwang, S. Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol. Bioeng. 89, 670–679, 10.1002/bit.20347 (2005).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336, 10.1038/nmeth.f.303 (2010).
DeSantis, T. Z. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072, 10.1128/AEM.03006-05 (2006).
Bro, R. PARAFAC. Tutorial and applications. Chemometrics and Intelligent Laboratory Systems 38, 149–171, 10.1016/s0169-7439(97)00032-4 (1997).
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649, 10.1093/bioinformatics/bts199 (2012).
Meyer, F. et al. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9, 386, 10.1186/1471-2105-9-386 (2008).
Wattam, A. R. et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 42, D581–591, 10.1093/nar/gkt1099 (2014).
Sørensen, L. L., Coddington, J. A. & Scharff, N. Inventorying and Estimating Subcanopy Spider Diversity Using Semiquantitative Sampling Methods in an Afromontane Forest. Environ. Entomol. 31, 319–330, 10.1603/0046-225X-31.2.319 (2002).
Vetrovsky, T. & Baldrian, P. The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PloS one 8, e57923, 10.1371/journal.pone.0057923 (2013).
Avershina, E. et al. Bifidobacterial succession and correlation networks in a large unselected cohort of mothers and their children. Appl. Environ. Microbiol. 79, 497–507, 10.1128/AEM.02359-12 (2013).
de Muinck, E. J. et al. Diversity, transmission and persistence of Escherichia coli in a cohort of mothers and their infants. Environ. Microbiol. Rep. 3, 352–359, 10.1111/j.1758-2229.2010.00231.x (2011).
Yatsunenko, T. et al. Human gut microbiome viewed across age and geography. Nature 486, 222–227, 10.1038/nature11053 (2012).
Moore, A. M. et al. Pediatric fecal microbiota harbor diverse and novel antibiotic resistance genes. PloS one 8, e78822, 10.1371/journal.pone.0078822 (2013).
Sepp, E. et al. The occurrence of antimicrobial resistance and class 1 integrons among commensal Escherichia coli isolates from infants and elderly persons. Ann. Clin. Microbiol. Antimicrob. 8, 34 (2009).
Davies, J. & Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 74, 417–433, 10.1128/MMBR.00016-10 (2010).
Boucher, Y., Labbate, M., Koenig, J. E. & Stokes, H. W. Integrons: mobilizable platforms that promote genetic diversity in bacteria. Trends Microbiol. 15, 301-309, 10.1016/j.tim.2007.05.004 (2007).
Nandi, S., Maurer, J. J., Hofacre, C. & Summers, A. O. Gram-positive bacteria are a major reservoir of Class 1 antibiotic resistance integrons in poultry litter. Proc. Natl. Acad. Sci. USA 101, 7118–7122, 10.1073/pnas.0306466101 (2004).
Meng, H. et al. Characterization and horizontal transfer of class 1 integrons in Salmonella strains isolated from food products of animal origin. Int. J. Food Microbiol. 149, 274–277, 10.1016/j.ijfoodmicro.2011.07.006 (2011).
Dalsgaard, A., Forslund, A., Serichantalergs, O. & Sandvang, D. Distribution and content of class 1 integrons in different Vibrio cholerae O-serotype strains isolated in Thailand. Antimicrob. Agents Chemother 44, 1315–1321 (2000).
Vaisvila, R., Morgan, R. D., Posfai, J. & Raleigh, E. A. Discovery and distribution of super-integrons among pseudomonads. Mol. Microbiol. 42, 587–601 (2001).
Gonzalez, G. et al. Presence of integrons in isolates of different biotypes of Acinetobacter baumannii from Chilean hospitals. FEMS Microbiol. Lett. 161, 125–128 (1998).
Shahcheraghi, F. et al. Identification and characterization of class 1 integrons among atypical enteropathogenic Escherichia coli isolated from children under 5 years of age. Iran. J. Microbiol. 6, 156–162 (2014).
Yeagle, P. Microbiome of uncontacted Amerindians. Science 348, 298, 10.1126/science.348.6232.298-a (2015).
Stern, A., Mick, E., Tirosh, I., Sagy, O. & Sorek, R. CRISPR targeting reveals a reservoir of common phages associated with the human gut microbiome. Genome Res. 22, 1985–1994, 10.1101/gr.138297.112 (2012).
Acknowledgements
This work was supported by Quota scholarship and funding from Norwegian University of Life Sciences, Ås, Norway and Norwegian University of Science and Technology, Trondheim, Norway.
Author information
Authors and Affiliations
Contributions
A.R.- main author of the article. E.A.-main author of the article. S.F.- Provided plasmids and integron-related experiments. J.L.- Culturing of fecal samples. O.S.- Sample collection for IMPACT study. T.Ø.- Sample collection for IMPACT study. R.J.- Sample collection for IMPACT study. A.L.M.- Isolation of Bifidobacterium. T.M.L.- review of scientific content in the article. KR- main lead of the article
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ravi, A., Avershina, E., Foley, S. et al. The commensal infant gut meta-mobilome as a potential reservoir for persistent multidrug resistance integrons. Sci Rep 5, 15317 (2015). https://doi.org/10.1038/srep15317
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep15317
This article is cited by
-
Antibiotic resistome from the One-Health perspective: understanding and controlling antimicrobial resistance transmission
Experimental & Molecular Medicine (2021)
-
Experimental support for multidrug resistance transfer potential in the preterm infant gut microbiota
Pediatric Research (2020)
-
Human Milk Microbiota: Transferring the Antibiotic Resistome to Infants
Indian Journal of Microbiology (2019)
-
Maternal gut and breast milk microbiota affect infant gut antibiotic resistome and mobile genetic elements
Nature Communications (2018)
-
Early microbiota, antibiotics and health
Cellular and Molecular Life Sciences (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
