
Abstract
Ultralow stability of gold clusters prohibits the understanding of their intrinsic reactivity (that is vital for revealing the origin of gold’s catalytic properties). Using density functional theory including many-body dispersion method, we aim to ascertain effective ways in exploiting gold clusters’ intrinsic reactivity on carbon nanotubes (CNTs). We find that the many body van der Waals interactions are essential for gold clusters’ reactivity on CNTs and even for O2 activation on these supported clusters. Furthermore, curvature and dopant of CNTs are found to qualitatively change the balance between physisorption and chemisorption for gold clusters on CNTs, determining the clusters’ morphology, charge states, stability and reactivity, which rationalize the experimental findings. Remarkably, N doped small curvature CNTs, which effectively stabilize gold clusters and retain their inherent geometric/electronic structures, can be promising candidates for exploiting gold clusters’ intrinsic reactivity.
Similar content being viewed by others

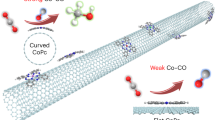

Introduction
As catalysts, gold nanoparticles have attracted great interest over past decades, due to its unexpected activity and high selectivity towards many reactions1,2,3,4. In particular, gold clusters with a few atoms Aun exhibit unusual intrinsic reactivity because of their geometric and electronic structures5,6,7,8,9,10. The understanding of Aun clusters’ intrinsic reactivity is of great importance, both for revealing the origin of gold’s catalytic properties and for applying gold cluster catalysts5,6,7. Gold clusters thus have been extensively studied in gas phase experimentally and theoretically5,6,9,10,11,12,13,14,15,16,17,18,19, showing that Aun clusters adopt planar structures up to Au1218,19, while their reactivity is sensitive to size, shape and charge states. However, the nature of catalytic properties of gold clusters is still ambiguous, since gold clusters suffer issue of ultralow stability, which leads to their short lifetimes in free states that hampers the understanding of their intrinsic reactivity practically. Many attempts have been made to stabilize metal nanoparticles by protecting them with coordinating ligands or depositing them on active substrates20,21,22,23,24,25,26,27. Although ligand-protection and active substrates support yielded some stable and active gold cluster catalysts22,23,24,25,26,27, these catalysts are entangled with the gold-ligand/gold-substrate covalent bonding, the saturation of clusters’ low-coordinate atoms and the substantial change of clusters geometry, which blur the intrinsic reactivity of gold clusters. Therefore, it is essential to achieve the long-term stable gold clusters that retain their inherent geometric and electronic structures.
Carbon materials as inert substrates are advantageous over active substrates and have also been widely used for growing metal nanoparticles28,29,30,31. The studies by Corma et al. have synthesized Au5–10 on functionalized carbon nanotubes (CNTs) that are highly active for the aerobic oxidation of thiophenol with O2 but are unfortunately passivated rapidly by forming larger and inactive nanoparticles, meanwhile the Au4 clusters are found to be inactive on CNTs8. In addition, the morphology and catalytic mechanism of the supported Au4–10 are still unclear because of the limit of experimental detection, substantially prohibiting the attempt to improve the stability of these catalysts. Density functional approximations (DFA) with (semi-)local functionals, which miss long-range van der Waals (vdW) interactions for nonhomogeneous electron gas, cannot be applied to nanomaterials either. The main challenge for theoretically accurate determination of adsorption properties of nanomaterials originates from their nonlocal anisotropic polarization that is coupled with a pronounced contribution of many-body electronic correlations32. This contribution is missed in the pairwise approximation for dispersion forces too.
To reveal the properties of Aun clusters on CNTs, we employ DFA augmented with accurate description of nonlocal many-body dispersion interactions (DFA+MBD) to study the configurations of Aun32,33, which should be affected by both chemisorption and physisorption. The possible contributions consist of covalent bonding, electrostatic interactions, Pauli repulsions and vdW interactions. Therefore, we focus on the influence of CNTs’ curvature on Aun, which are accompanied by different chemical reactivity and electrodynamic response effects depending on the diameters of CNTs34, corresponding to distinct chemisorption and physisorption. Indeed, the curvature effect is a rather general phenomenon: Graphene often experiences curl or bending in reality due to tensions, defects and so on35, intrinsically exhibiting part of CNTs.
Next, we attempt to improve the stability of Aun clusters by doping CNTs (with different substitution dopants). We propose a design basis that the optimal dopant should enlarge the electrostatic attractions (but avoid forming covalent bonding) between gold clusters and substrates. Furthermore, to retain the intrinsic reactivity of gold clusters on CNTs, the candidate dopant should remain the clusters’ geometric and electronic properties unchanged compared to the isolated cases’. By investigating the influences of different dopants, we find that nitrogen dopant combined with curvature control, which perfectly meets the above requirements, can effectively accelerate the exploitation of gold clusters’ intrinsic reactivity on CNTs. In addition, our DFA + MBD calculations also provide fundamental insights into catalytic mechanism of gold clusters on CNTs.
Results and Discussions
We first study the influence of curvature on the morphology of the gold clusters, showing the optimal configurations and adsorption energies in Fig. 1 (more results are referred to Figure S1 and Table S1 in Supplementary Information). To simulate CNTs and curved-graphene, we use the bending graphenes, with six kinds of curvature denoted as Dm (m = 10, 20, 30, 40, 60 and 80), corresponding to the different diameters of CNTs 10 Å, 20 Å, 30 Å, 40 Å, 60 Å and 80 Å. In addition, we choose the Aun clusters with 4 ≤ n ≤ 7 that are in the range of experimental Au3–108. To better understand the adsorption properties of Aun, we separate the contribution of vdW interactions (Ead,MBD) from the total adsorption energy by Perdew-Burke-Ernzerhof36 combined with MBD method (PBE+MBD). The rest is PBE contribution (Ead,PBE).

Adsorption properties of the Au5–6clusters on D10 ~ D80.
(a) Configurations of the optimal Au5–6 clusters on D10 ~ D80. (b) Adsorption energies of the standing and lying modes for the Au5–6 clusters on D10 ~ D80. The lines guide the eye. With decreasing of the curvature (from D10 to D80), Au5–6 gradually transform from the standing mode to the lying mode, which are determined by the competition of chemisorption and physisorption.
We find that the Au4–7 clusters on the substrates resemble the geometry of the isolated Aun clusters, with planar structures more stable than three-dimensional ones5,6,7,8,9,10,18,19. These clusters experience two interesting adsorption modes — lying and standing — depending on the curvature of substrates (Fig. 1 and S1). We expect the further experimental studies with IR spectroscopy to identify these two modes. The Au4 cluster always stands on D10~D80 regardless of the curvature, while Au5–7 gradually transform from the standing mode to the lying mode on D10 ~ D80 with decreasing of the curvature (Fig. 1 and S1), which are determined by the competition of chemisorption and physisorption (mainly the competition of covalent bonding and vdW interactions. see Table S1). The standing mode of gold clusters forms stronger chemical bonds with substrates than the lying mode does, by optimizing the overlap of gold clusters’ the highest occupied molecular orbtials (HOMO) and substrates’ the lowest unoccupied molecular orbtials (LUMO). In contrast, the lying mode of gold clusters is conducive to maximize vdW interactions with substrates by minimizing the distance between the gold atoms and the substrates. With decreasing of the curvature, chemical reactivity of substrates gradually decreases, while vdW interactions play an increasingly important role in the adsorption of the Au4–7 clusters, forcing the transformation of the Au4–7 clusters from the standing mode to the lying mode. As the Aun size increases, vdW interactions grow with 0.13 ~ 0.20 eV/atom and favor the lying mode. In particular, the lying mode Au5–7 experience pure physisorption on CNTs, where these clusters are anchored by vdW interactions, electrostatic interactions and Pauli repulsions.
To elucidate the reactivity of the Aun clusters, we study the O2 activation that is crucial for plenty of reactions7,8,9,10,12,13,14,15,16,17,37. Our calculations demonstrate that the HOMOs of the lying mode Au5–7 have lobes exclusively localized on the low-coordinated Au atoms (Fig. 2 and S2), which are all accessible to the LUMO of O2 (LUMOsO2), since no covalent bonding is formed between the clusters and the substrates. As O2 adsorbs on these clusters, the charge states of O atoms and the lengths of O–O bond (L) are similar with those on the isolated Aun clusters (Fig. 2 and S2 and Table S2), showing that the substrates greatly preserve the reactivity of the lying mode Au5–7. In contrast, the standing modes Au4 (and the standing modes Au5 on D10 ~ D30) bind to the substrates with covalent bonding, which saturates the low-coordinated Au atoms, making their HOMOs be partially inaccessible for LUMOsO2 and limiting the adsorption of other reactants (Fig. 1, S1 and S2). These results reveal the importance of taking the lying mode for the supported Aun clusters to exhibit intrinsic reactivity.

Electronic and geometric properties of the Au5 clusters.
(a,b) Atomic distribution of the HOMOs of the isolated Au5 clusters and those supported on D80, with the optimal configurations of the adsorption of O2 on these clusters. (c) HOMOs of the Au5 clusters on D80N, D80O and D80B where the subscript N, O, B denote the doping element. (d) The electron density difference of the Au5 clusters on these doped D80 (both lying and standing modes for D80N). The different colours of HOMOs represent opposite signs of wavefunctions. LUMOs of O2 are not shown for simplification. For the electron density difference, blue-green (red) indicates the deletion (accumulation) of electron density. The numbers in bold are the bond lengths of adsorbed O2, while those in italic are the charges on O2 Q (e) with Hirshfeld definition.
On the lying modes, O2 activation follows the same manner as on the isolated gold clusters: O2 is likely activated by gold clusters with an odd (not even) number of electrons (Figure S2)7,8,9,10,15,16. In addition, the electron density, which is transferred to the π*OO molecular orbital of O2, is originally from the Aun clusters, while the substrates act as an electron reservoir that exchanges electrons with adsorbates during catalytic process (Tables 1 and S2). To mitigate the impact of DFT electron self-interaction error on the description of electronic properties, we adopted hybrid functional Heyd-Scuseria-Ernzerhof (HSE)38 to study the electron transfer among the substrates, Aun clusters and O2. We find that the HSE results are compatible with the PBE ones (Table 1), confirming the independence of our results on exchange-correlation functional and further supporting our conclusions. It is noteworthy that the contribution of vdW interactions to O2 adsorption increases from 0.06 eV on Au4/Dm to 0.44 eV on Au7/Dm with increasing of the Aun size. The maximum contribution is up to 58% (0.34 ~ 0.44 eV) on Au7/D10 ~ D80 for the lying mode gold clusters, although it is negligible to that on the isolated gold clusters (≤0.06 eV) and the bare D10 ~ D80 (≤0.1 eV). The synergy effects of gold clusters and CNTs make vdW interactions be essential even for activating small molecules like O2.
The stability of the supported Aun clusters is critical to the application of these clusters as catalysts, which can be evaluated from their diffusion barriers. Herein, we compute the diffusion barriers using the adsorption energy difference of the Aun clusters at different adsorption sites, which are consistently less than 0.2 eV on all considered substrates (0.05 ~ 0.1 eV for the lying modes), indicating a quick migration of the supported Aun clusters even at low temperature. The tiny diffusion barriers are due to the dominant role of vdW interactions in the adsorption of the Aun clusters.
Overall, our results explain the experimental findings on the CNTs (the sizes correspond to D40) that the Au5–7 clusters are highly active for O2 activation but quickly aggregate into larger and inactive nanoparticles, while the Au4 clusters present negligible reactivity8. This strongly confirms the promise of our bending-graphene models in studying the properties of CNTs.
To improve the catalytic performance of the Aun clusters on CNTs, we consider the role of doping in stabilizing these clusters. Elements N, O, B, P, Li, Be, Cr and Ag are implanted into substrates respectively by replacing one C atom with one impurity atom (the concentration of 1/90). Taking Au5/D80 as an example, none but D80 doped with N (D80N) surprisingly remain the lying mode of the Au5 clusters (D80N-Au5-l in Fig. 2 and S2). N dopant, which is sp2 hybridization (see electron density difference of D80N-Au5-s in Fig. 2d), perfectly saturates the dangling bonds as one C atom is replaced. Therefore, the lying mode Au5 cluster does not form any covalent bonding with D80N, with the distance between D80N and Au atoms around 3.36 Å. Nevertheless, the amount of electron-density 0.32e is transferred from D80N to the Au5 cluster towards N doping (Table S2), significantly increasing electrostatic attractions (monopole-monopole and multipole-multipole interactions) between D80N and Au5. The resulting adsorption energies for the Au5 cluster are Ead,PBE = 0.19 eV and Ead,MBD = 0.81 eV, being larger than those on D80 (Ead,PBE = −0.05 eV and Ead,MBD = 0.78 eV). Clearly, the N dopant stabilizes the lying mode Au5 on D80 by 0.27 eV without forming any covalent bonding, which remarkably meets the design basis we proposed.
In D80N, each N dopant contributes two p electrons to the π system and leaves the p orbital of N dopant unoccupied, while these two electrons are partially localized in the p orbital of the N dopant’s neighboring C atoms. Therefore, the standing mode Au5, which needs the electron-density donation from binding atoms, binds with the N dopant’s neighboring C atoms rather than the N dopant itself. In this case, electron-density is also transferred from the Au5 cluster to the unoccupied p orbital of the N dopant as shown in the D80N-Au5-s of Fig. 2d. The N dopant can hardly modify the nature of sp2 hybridization for its neighboring C atoms. Thus, the overlap of Au-5d and C-2p orbitals yields weak covalent bonding between Au5 and D80N for the standing mode, having Ead,PBE = 0.27 eV (Ead,MBD = 0.64 eV) and LC-Au = 2.36 Å. As results, the Au5 cluster lies on D80N.
In the case of the O doped D80 (D80O), O dopant is sp3 hybridization and has two unpaired electrons that cannot fully saturate its neighboring C atoms. Thus these C atoms undergo sp3 hybridization, leading to the overlap of Au-5d and C-sp3 orbitals that makes the Au5 cluster strongly bind with D80O (Ead,PBE = 1.32 eV). Regarding the B, Li and Be dopants, dangling C atoms cannot be fully saturated either, which form strong covalent bonding with the Au5 clusters (Ead,PBE = 1.38 ~ 2.43 eV). The sizes of P, Cr and Ag atoms are much larger than that of C atom, making the dopants locate outside CNTs by altering bond angles of C-dopant-C rather than shortening bond lengths of C-dopant, which consequently enable forming covalent P-Au, Cr-Au and Ag-Au bonds with Ead,PBE of 1.14 ~ 2.20 eV.
More importantly, the optimal Au5 cluster on D80N exhibits the identical atomic distribution of the HOMOs compared to the isolated case (Fig. 2 and S2), implying that N doped substrates retain not only the geometry but also the electronic structure of the supported clusters, which is critical for the application of gold clusters’ intrinsic reactivity. We thus adopt N as the dopant to stabilize gold clusters on CNTs.
As N is implanted into D10 ~ D80, the geometries of the supported Au4–7 are not changed. Importantly, the adsorption energies of Au4–7 are overall increased at doped sites (Fig. 3), especially, those of the lying modes Au5 and Au7 are increased by 0.27 ~ 0.53 eV. The resulting diffusion barriers of the Au5 and Au7 clusters are in the range of 0.35 ~ 0.63 eV on (D10 ~ D80)N [2.5 ~ 10 times larger than those on (D10 ~ D80)], indicating a significantly improved stability of the Au5 and Au7 clusters even at elevated temperature of 200 ~ 350 K. The increase of adsorption energy exhibits an odd-even alteration depending on the number of gold atoms, which reflects the redistribution of electron density towards N doping. The open-shell Au5 and Au7, which can readily accept more electrons from the substrates than the close-shell Au4 and Au6 do, effectively stabilize Au5/(D10 ~ D80)N and Au7/(D10 ~ D80)N. This stabilization can hardly change the vdW interactions between Au4–7 and substrates (ΔEad,MBD ≈ 0.05 eV in Table S1). If the N concentration is increased to 1/45, the diffusion barrier of the lying mode Au5 is further increased to 0.76 eV on D80N (0.35 eV at concentration of 1/90).

The comparison of adsorption energy for the Au4–7 clusters on the substrates of D10 ~ D80 and (D10 ~ D80)N.
Adsorption energies of the Au4–7 clusters are overall increased towards Nitrogen doping.
The Au4–7/(D10 ~ D80)N are more active in activating O2 compared to Au4–7/D10 ~ D80: more electrons (0.02 ~ 0.12 e) are transferred to O2 and the bond length of adsorbed O2 are elongated further by 0.01 ~ 0.05 Å towards N doping (Fig. 2 and S2). Namely, nitrogen dopants effectively improve the reactivity of the supported Aun clusters by making the substrates donate more electrons for activating O2. Furthermore, our HSE calculations predict the same trend as PBE calculations for the electron transfer among O2, Au5/Au6 and D40N (Table 1). In addition, HSE functional also identifies a comparable diffusion barrier for the Au5 cluster on D40N as PBE functional (0.42 eV vs 0.47 eV at concentration of 1/90). Clearly, our results are independent on exchange-correlation functionals, explicitly confirming that N dopant is able to promote both reactivity and stability of the gold clusters on CNTs. In addition, the dispersed N dopant can also enhance the dispersion of the gold clusters. Recalling that the experiments of the aerobic oxidation of thiophenol with O2 were carried out at 300 K8, we also performed ab inito molecular dynamics simulations for Au5/D40N and Au7/D40N at 300 K, finding slight change of the adsorption position of the Au5 and Au7 clusters during 10 ps runs. Therefore, we conclude that the N doped CNTs largely mitigate the problem of low stability of the gold clusters on CNTs8 and serve as promising substrates for the application of the gold clusters’ intrinsic reactivity.
It is gratifying that CNTs and graphene exhibit sufficient flexibility towards doping functionalization39,40, while the N doped CNTs and graphene have been synthesized experimentally and exhibit remarkable reactivity for variant reactions41,42,43. In particular, negatively charged 10 nm gold nanoparticles were successfully anchored to the N doped CNTs43, while the Au3–10 clusters have been synthesized on CNTs8. These encouraging results show robust prospects for growing gold clusters on the N doped CNTs and graphene.
By comparing the MBD results to those by the pairwise approximation of Tkatchenko-Scheffler (TS)44 method (Table S1), we find that many body effects are essential for the lying mode Aun (20 ~ 35% contribution) not for the standing mode, reflecting the highly anisotropic polarization of gold clusters and CNTs. In the case of O2 adsorption on these gold clusters, many body effects can critically affect O2 activation with up to 41% contribution on Au7/D80 (Table S3). In particular, these effects first reduce and then enhance the adsorption energy with decreasing of the curvature, indicating the significant influence of anisotropic polarization of nanomaterials on O2 activation. Recalling the dominant role of MBD force in anchoring gold clusters on CNTs and activating O2 on Aun/CNTs, DFA+MBD methods are thus essential for accurate prediction of properties of gold clusters on nanomaterials as well as catalysis on these catalysts.
Conclusions
In conclusions, our DFA+MBD results reveal the fundamental mechanism of Au4–7 in activating O2 on CNTs, where Au4–7 catalytic properties are determined by the balance of chemisorption and physisorption. We find that curvature and dopant of CNTs combined with the clusters size qualitatively change this balance, determining clusters’ morphology, charge states, stability and reactivity. Remarkably, N doped small curvature CNTs, which effectively promote gold clusters’ stability by enlarging electrostatic attractions (without forming covalent bonding), retain gold clusters’ inherent geometric and electronic structures. These results enable us to explain the experimental findings of gold clusters on CNTs8 and to predict N doped CNTs as promising substrates for exploiting gold clusters’ intrinsic reactivity. The methodologies we employed, including the DFA+MBD method and the principles of tuning substrates for gold clusters, can serve as a tool to engineer other clusters (i.e. Ptn) supported on nanomaterials where strong covalent bonding ought to be avoided.
Methods
vdW interactions were calculated in the scheme of non-local MBD method on top of DFA (DFA+MBD)32,33, using the FHI-aims all electron code with “tight” settings45. The DFA+MBD approaches add the vdW energy given as a sum of C6R−6 terms to the DFA total energy. The DFA+MBD methods compute the long-range correlation energy through the coupled harmonic oscillator model Hamiltonian32,33,46,47,48, which is an effective random phase approximation-like treatment of many body effect, going beyond the pairwise vdW approaches. In addition, the MBD approach, which avoids the explicit use of single-electron orbitals, allows for a favorable N3 scaling (N is the number of atoms) and a negligible computational cost relative to a self-consistent DFA calculation. More importantly, The MBD methods has been found to be highly accurate for many molecular and solid-state systems32,33,46,47,48.
All geometries were obtained using CASTEP49 code with Vanderbilt-type ultrasoft pseudopotentials50 and the PBE + TS method36,44. Forces and stresses for TS calculations were calculated numerically and used to obtain fully consistent TS geometries for all calculations44: the normal self-consistency cycle is first completed for PBE; second, the resulting self-consistent electron density is used to create the vdW energy; After adding this vdW energy to PBE total energy, one can effectively compute the forces of PBE+TS. The PBE+TS method has been found to yield the interlayer distance of 3.34 Å for graphite51, in perfect agreement with the experimental value 3.34 Å. The careful convergence tests allow us to adopt a cutoff energy of 400 eV and a k-point mesh of 2 × 2 × 1 for 90 atoms supercell of graphene (5 × 9). All calculations are spin unrestricted. The DFA + MBD calculations are shown to converge MBD energy to a meV/atom level. To explicitly elucidate the interplay between substrates and gold clusters, we also compute the electronic structures of considered systems using hybrid HSE functional38.
To obtain the most stable structures of gold clusters on substrates, we defined adsorption energy of gold clusters (Ead) as:

where Etotal is the energy of the bound Aun/substrates, Esub is the energy of the substrates and Egold is the energy of the isolated, fully relaxed Aun clusters. Since the vdW energy is added as an additional term to the PBE total energy, it is thus convenient to separate the vdW contribution (by MBD) from the total adsorption energy.
Additional Information
How to cite this article: Gao, W. et al. Design Principles of Inert Substrates for Exploiting Gold Clusters’ Intrinsic Catalytic Reactivity. Sci. Rep. 5, 15095; doi: 10.1038/srep15095 (2015).
References
Hughes, M. D. et al. Tunable gold catalysts for selective hydrocarbon oxidation under mild conditions. Nature 437, 1132–1135 (2005).
Corma, A. & Garcia, H. Supported gold nanoparticles as catalysts for organic reactions. Chem. Soc. Rev. 37, 2096–2126 (2008).
Corma, A. & Serna, P. Chemo selective hydrogenation of nitro compounds with supported gold catalysts. Science 313, 332–334 (2006).
Grirrane, A., Corma, A. & García, H. Gold-catalyzed synthesis of aromatic azo compounds from anilines and nitroaromatics. Science 322, 1661–1664 (2008).
Wang, L.-M. & Wang, L.-S. Probing the electronic properties and structural evolution of anionic gold clusters in the gas phase. Nanoscale 4, 4038–4053 (2012).
Gruene, P. et al. Structures of neutral Au7, Au19 and Au20 clusters in the gas phase. Science 321, 674–676 (2008).
Boronat, M., Leyva-Pérez, A. & Corma, A. Theoretical and experimental insights into the origin of the catalytic activity of subnanometric gold clusters: Attempts to predict reactivity with clusters and nanoparticles of gold. Acc. Chem. Res. 47, 834–844 (2014).
Corma, A. et al. Exceptional oxidation activity with size-controlled supported gold clusters of low atomicity. Nat. Chem. 5, 775–781 (2013).
Yoon, B., Häkkinen, H. & Landman, U. Interaction of O2 with gold clusters: Molecular and dissociative adsorption. J. Phys. Chem. A 107, 4066–4071 (2003).
Huang, W., Zhai, H.-J. & Wang, L.-S. Probing the interactions of O2 with small gold cluster anions (Aun−, n=1–7): Chemisorption vs physisorption. J. Am. Chem. Soc. 132, 4344–4351 (2010).
Li, J., Li, X., Zhai, H.-J. & Wang, L.-S. Au20: A tetrahedral cluster. Science 299, 864–867 (2003).
Pal, R., Wang, L.-M., Pei, Y., Wang, L.-S. & Zeng, X. C. Unraveling the mechanisms of O2 activation by size-selected gold clusters: Transition from superoxo to peroxo chemisorption. J. Am. Chem. Soc. 134, 9438–9445 (2012).
Zhai, H.-J., Kiran, B., Dai, B., Li, J. & Wang, L.-S. Unique CO chemisorption properties of gold hexamer: Au6(CO)n− (n = 0–3). J. Am. Chem. Soc. 127, 12098–12106 (2005).
Fielicke, A. et al. Gold cluster carbonyls: Saturated adsorption of CO on gold cluster cations, vibrational spectroscopy and implications for their structures. J. Am. Chem. Soc. 127, 8416–8423 (2005).
Woodham, A. P., Meijer, G. & Fielicke, A. Charge separation promoted activation of molecular oxygen by neutral gold clusters. J. Am. Chem. Soc. 135, 1727–1730 (2013).
Woodham, A. P., Meijer, G. & Fielicke, A. Activation of molecular oxygen by anionic gold clusters. Angew. Chem. Int. Ed. 51, 4444–4447 (2012).
Woodham, A. P. & Fielicke, A. Superoxide formation on isolated cationic gold clusters. Angew. Chem. Int. Ed. 53, 6554–6557 (2014).
Furche, F. et al. The structures of small gold cluster anions as determined by a combination of ion mobility measurements and density functional calculations. J. Chem. Phys. 117, 6982–6990 (2002).
Häkkinen, H. et al. On the electronic and atomic structures of small AuN− (N=4–14) clusters: A photoelectron spectroscopy and density-functional study. J. Phys. Chem. A 107, 6168–6175 (2003).
Häkkinen, H. The gold-sulfur interface at the nanoscale. Nat. Chem. 4, 443–455 (2012).
Desireddy, A. et al. Ultrastable silver nanoparticles. Nature 501, 399–402 (2013).
Farmer, J. A. & Campbell, C. T. Ceria Maintains smaller metal catalyst particles by strong metal-support bonding. Science 329, 933–936 (2010).
Lopez-Acevedo, O., Kacprzak, K. A., Akola, J. & Häkkinen, H. Quantum size effects in ambient CO oxidation catalysed by ligand-protected gold clusters. Nat. Chem. 2, 329–334 (2009).
Yang, H. et al. All-thiol-stabilized Ag44 and Au12Ag32 nanoparticles with single-crystal structures. Nat. Comm. 4, 2422 (2013).
Yoon, B. et al. Charging effects on bonding and catalyzed oxidation of CO on Au8 clusters on MgO. Science 307, 403–407 (2005).
Herzing, A. A., Kiely, C. J., Carley, A. F., Landon, P. & Hutchings, G. J. Identification of Active Gold nanoclusters on iron oxide supports for CO oxidation. Science 321, 1331–1335 (2008).
Lu, J., Aydin, C., Browning, N. D. & Gates, B. C. Imaging isolated gold atom catalytic sites in zeolite NaY. Angew. Chem. Int. Ed. 51, 5842–5846 (2012).
Joo, S. H. et al. Ordered nanoporous arrays of carbon supporting high dispersions of platinum nanoparticles. Nature 412, 169–172 (2001).
Gao, W., Mueller, J. E., Anton, J., Jiang, Q. & Jacob, T. Ni cluster growth on defect sites of graphene: A computational study. Angew. Chem. Int. Ed. 52, 14237–14241 (2013).
Rinaldi, A. et al. Dissolved carbon controls the initial stages of nanocarbon growth. Angew. Chem. Int. Ed. 50, 3313–3317 (2011).
Sanz-Navarro, C. F. et al. Molecular dynamics simulations of carbon-supported Ni clusters using the reax reactive force field. J. Phys. Chem. C 112, 12663–12668 (2008).
Tkatchenko, A., DiStasio, R. A., Jr., Car, R. & Scheffler, M. Accurate and efficient method for many-body van der Waals interactions. Phys. Rev. Lett. 108, 236402 (2012).
Ambrosetti, A., Reilly, A. M., DiStasio, R. A., Jr. & Tkatchenko, A. Long-range correlation energy calculated from coupled atomic response functions. J. Chem. Phys. 140, 18A508 (2014).
Gobre, V. V. & Tkatchenko, A. Scaling laws for van der Waals interactions in nanostructured materials. Nat. Comm. 4, 2341 (2013).
Thompson-Flagg, R. C., Moura, M. J. B. & Marder, M. Rippling of graphene. EPL 85, 46002 (2009).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Turner, M. et al. Selective oxidation with dioxygen by gold nanoparticle catalysts derived from 55-atom clusters. Nature 454, 981–984 (2008).
Heyd, J., Scuseria, G. E. & Ernzerhof, M. Erratum: “Hybrid functionals based on a screened Coulomb potential” [J. Chem. Phys. 118, 8207 (2003)]. J. Chem. Phys. 124, 219906 (2006).
Zhao, Y.-L. & Stoddart, J. F. Noncovalent functionalization of single-walled carbon nanotubes. Acc. Chem. Res. 42, 1161–1171 (2009).
Dreyer, D. R., Todd, A. D. & Bielawski, C. W. Harnessing the chemistry of graphene oxide. Chem. Soc. Rev. 43, 5288–5301 (2014).
Gong, K., Du, F., Xia, Z., Durstock, M. & Dai, L. Nitrogen-doped carbon nanotube arrays with high electrocatalytic activity for oxygen reduction. Science 323, 760–764 (2009).
Qu, L., Liu, Y., Baek, J.-B. & Dai, L. Nitrogen-doped graphene as efficient metal-free electrocatalyst for oxygen reduction in fuel cells. ACS Nano 4, 1321–1326 (2010).
Jiang, K., Eitan, A., Schadler, L. S., Ajayan, P. M. & Siegel, R. W. Selective attachment of gold nanoparticles to nitrogen-doped carbon nanotubes. Nano Lett. 3, 275–277 (2003).
Tkatchenko, A. & Scheffler, M. Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 102, 073005 (2009).
Blum, V. et al. Ab initio molecular simulations with numeric atom-centered orbitals. Comput. Phys. Commun. 180, 2175–2196 (2009).
Ambrosetti, A., Alfè, D., DiStasio, R. A., Jr. & Tkatchenko, A. Supporting information for “Hard numbers for large molecules: Toward exact energetics for supramolecular systems”. J. Phys. Chem. Lett. 5, 849–855 (2014).
Reilly, A. M. & Tkatchenko, A. Role of dispersion interactions in the polymorphism and entropic stabilization of the aspirin crystal. Phys. Rev. Lett. 113, 055701 (2014).
Tkatchenko, A. Current understanding of van der Waals effects in realistic materials. Adv. Funct. Mater. 25, 2054–2061 (2015).
Segall, M. D. et al. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys.: Condens. Matter 14, 2717–2744 (2002).
Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 41, 7892–7895 (1990).
Gao, W. & Tkatchenko, A. Sliding mechanisms in multilayered hexagonal boron nitride and graphene: The effects of directionality, thickness and sliding constraints. Phys. Rev. Lett. 114, 096101 (2015).
Acknowledgements
This work was supported by the Program for New Century Excellent Talents in University (No. NCET-13-0255), the National Natural Science Foundation of China (No. 51201069, 51422103) and the Key Grant Project of Chinese Ministry of Education (No. 313026) and by the computing resources of High Performance Computing Center of Jilin University and National Supercomputing Center in Jinan, China.
Author information
Authors and Affiliations
Contributions
W.G. and Q.J. conceived and designed the research. W.G. and T.T.C. perforemed calculations. W.G., T.T.C. and Q.J. wrote the paper, Y.F.Z., Z.W., M.Z. and J.C.L. discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Gao, W., Ting Cui, T., Fu Zhu, Y. et al. Design Principles of Inert Substrates for Exploiting Gold Clusters’ Intrinsic Catalytic Reactivity. Sci Rep 5, 15095 (2015). https://doi.org/10.1038/srep15095
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep15095
This article is cited by
-
Probing the structural evolution and electronic properties of divalent metal Be2Mgn clusters from small to medium-size
Scientific Reports (2020)
-
Aromatic molecules on low-index coinage metal surfaces: Many-body dispersion effects
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
