
Abstract
Activating transcription factor 3 (ATF3) is induced by inflammatory responses, cell death, cytokines and oxidative stress conditions. ATF3 is a negative regulator in the Toll-like receptor 4 signalling pathway. The principal molecule in this pathway is nuclear factor κB (NF-κB) that translocates into the nucleus to initiate the transcription of inflammatory mediators. However, scarce data are available regarding the interaction of ATF3 and p65, a part of the NF-κB dimer. Therefore, we studied the mechanism of regulation of p65 by ATF3 in RAW 264.7 cells. First, LPS-mediated NF-κB activation was confirmed and then the direct interaction of ATF3 and p65 was observed through immunoprecipitation (IP). The presence of histone deacetylase 1 (HDAC1) was also detected in the complex. In ATF3 deficient cells, NF-κB activity was up-regulated and HDAC1 was not detected by IP. These observations suggest that p65 is attenuated by ATF3 such that ATF3 recruits HDAC1 to the ATF3/p65 complex and facilitates the deacetylation of p65. Likewise, inflammatory response genes were induced by translocated NF-κB in ATF3-deficient cells. Cumulatively, we uncovered a novel mechanism for the negative regulation of NF-κB by ATF3 via direct interaction with p65.
Similar content being viewed by others


Introduction
Toll-like receptors (TLRs) are categorized as pattern recognition receptors usually expressed in sentinel cells including antigen-presenting cells. Activation of TLR signalling pathways that converge on nuclear factor κB (NF-κB) trigger the production of pro-inflammatory cytokines and initiate antigen presentation functions1,2,3. Because, TLRs can stimulate the immune system, their ligands are useful in treating a variety of diseases, including cancers4,5,6.
NF-κB plays significant roles in biological responses such as immunity, cell proliferation, inflammation, differentiation and apoptosis7. The mammalian NF-κB family is composed of RelA (p65), c-Rel, RelB, p50 and p52, which form homo- or heterodimers at the κB sites of the promoters of target genes. Whereas RelA/c-Rel, RelA/p50, RelB/p50 and RelB/p52 heterodimers are transcriptional activators, homodimers such as p50/p50 and p52/p52 generally behave as transcriptional repressors8,9,10. For normal physiology, regulated NF-κB-induced inflammation is desirable and is achieved through diverse mechanisms of post-translational modification, ubiquitination of various components, deacetylation of promoter sites and the translocation of NF-κB into cytosol. The dysregulation of NF-κB-induced inflammation induces human disorders including inflammatory and neurodegenerative diseases11 and various cancers12,13,14.
NF-κB is normally held in an inactive form in the cytosol by inhibitor of κB (IκB) proteins15,16. Various members of the IκB family target different NF-κB dimers, e.g., IκB-α and IκB-β preferentially interact with RelA (p65)/p50 and c-Rel/p50 heterodimers, respectively, IκB-∊ binds only with p65 and c-Rel hetero- and homodimers17,18,19 and Bcl-3 associates only with p50/p50 or p52/p52 homodimers20. Although various IκB proteins interact with the same NF-κB homo- or heterodimers, the transcription factors are regulated differentially owing to functional differences among the proteins. For example, IκB-α is rapidly degraded in stimulated cells, resulting in immediate release of NF-κB, but IκB-β degrades over a longer time, causing sustained activation of transcription factors21,22.
Activating transcription factor 3 (ATF3) is a member of the ATF/cyclic adenosine monophosphate response element–binding (CREB) protein family of bZip transcription factors that binds to the consensus cyclic adenosine monophosphate response element site23,24. ATF3 functions as a transcriptional repressor by forming a homodimer25. In macrophages, ATF3 is increased by lipopolysaccharide (LPS; a TLR4 ligand), bacillus Calmette-Guérin26,27 and interferon in human peripheral blood mononuclear cells. Moreover, ATF3 is also up-regulated by TLR4 signalling and is part of the negative feedback loop that regulates the LPS-stimulated inflammatory response22.
Formerly, ATF3 was thought to deacetylate κB sites in the promoter of target genes by recruiting histone deacetylase 1 (HDAC1) and attenuating cytokine release28. However, it was unknown whether ATF3 directly interacts with p65 to regulate its activity. Therefore, this study was designed to explore the underlying mechanism of the negative regulation of p65 in TLR4 signalling. This study revealed the novel perspective that ATF3 not only attenuates the promoter binding of NF-κB but also inhibits NF-κB activity through physical association, which may help modulate the TLR4 signalling pathway in some diseases.
Results
LPS-induced inflammatory responses in RAW 264.7 cells
First, we treated RAW 264.7 cells with LPS and observed the translocation of various proteins into the nucleus using Western blot (Fig. 1a). ATF3 expression was rapidly up-regulated after 2 h, whereas the amount of HDAC1 in the nucleus was unaffected by LPS treatment. Previous reports have shown that ATF3 is a negative regulator that binds to the promoter region of target genes and recruits HDAC1 to form a complex to modulate inflammation28. The p65 protein rapidly accumulated in the nucleus within 30 min, induced the expression of target genes for 2 h and then vanished. The level of ATF3 corresponded inversely with the level of p65 protein in the nucleus, likely because the accumulating ATF3 decreased the nuclear concentration of p65 (Fig. 1b). Moreover, the results from confocal microscopy showed that LPS treatment for 2 h increased acetylated p65 (ac-p65) and induced the translocation of ATF3 into the nucleus (Fig. 1c,d).

Sensitization of cells to lipopolysaccharide (LPS)-induced inflammatory responses.
(a) LPS (1 μg/mL)-mediated induction of p65, IκBζ, ATF3, β-actin, HDAC1 and topoisomerase II-α protein expression in cytoplasm and nucleus. RAW 264.7 cells were treated with 1 μg/mL LPS for the indicated times. Cell lysates were processed with NE-PER lysate and the protein expression levels were measured with western blot. β-actin was used as a loading control in cytoplasm. HDAC1 and topoisomerase II-α were used as loading controls in nucleus. (b) Intensities of ATF3, p65 and topoisomerase II-α for the indicated times, calculated using ImageJ software. Data represent three independent experiments (n = 3; *P < 0.05, **P < 0.01). (c,d) Levels of acetylated p65 (ac-p65), ATF3 and HDAC1 proteins in nucleus were measured with confocal microscopy. Nuclei were stained with Hoechst stain (blue color). Scale bar: 20 μm.
Direct binding and regulation of p65 by ATF3
Using previous observations, we hypothesized that ATF3 is associated with p65 activation. Therefore, we treated cells with LPS in a time-dependent manner and performed immunoprecipitation (IP) to decipher the interaction of ATF3 with p65. In several independent experiments, antibodies specific to either ATF3 or p65 were agarose bead-labelled and used to extract the cognate protein bound to the labelled protein. In anti-ATF3 antibody beads, we observed p65 bound to ATF3 in as quickly as 30 min (Fig. 2a). HDAC1 associated with ATF3 could bind at the target promoter region and inhibit target gene expression by deacetylating the promoter and was bound with ATF3 at 30 min. Moreover, when anti-p65 antibody was attached to the agarose beads and the complex was extracted, we observed that the ATF3/p65 complex formed with the same binding kinetics (Fig. 2b). These results supported our hypothesis that ATF3 physically interacts with p65. Furthermore, these results could be corroborated the hypothesis that the reduction of p65 protein in the nucleus after 30 min was correlated with ATF3.

ATF3 interacts with the p65 subunit of nuclear factor κB (NF-κB).
(a) Cells were treated with 1 μg/mL LPS for the indicated times. Beads were labelled with ATF3 antibody and the interacting proteins, which were bound to ATF3, were precipitated. A total of 20 μg protein was separated with electrophoresis. The levels of these proteins were determined with Western blot using whole-cell lysates. (b) Cells were treated with 1 μg/mL LPS for the indicated times. Beads were labelled with p65 antibody and the proteins bound to p65 were precipitated. A total of 20 μg protein was separated with electrophoresis. Protein levels were determined with Western blot using whole-cell lysates. Data show a representative set among the results of three independent experiments (n = 3).
Inhibition of LPS-induced nuclear localization of p65 by ATF3
From analyses of previous data, we hypothesized that the translocation of p65 to the nucleus is closely related to ATF3 expression. Therefore, we monitored the activation of p65 in the absence of ATF3 using ATF3 small interfering RNA (siRNA) in TLR4 signalling. In ATF3-deficient RAW 264.7 cells, the up-regulated expression of p65 was maintained from 30 min to 2 h compared with p65 expression in cells treated with scrambled siRNA (Fig. 3a). In addition, we observed ATF3 and p65 in the nucleus after 2 h of LPS treatment using confocal microscopy (Fig. 3b). After 2 h of LPS treatment, ATF3 was detected in the nucleus in scrambled siRNA-treated cells, but p65 was not. However, in ATF3-deficient RAW 264.7 cells, p65 protein was significantly detected in the nucleus with confocal microscopy even after 2 h of LPS treatment, as has been seen in the Western blot results (Fig. 3a). These observations confirmed that the translocation of p65 is associated with ATF3 protein expression.

Activation of NF-κB via ATF3 down-regulation.
(a) Cells were transfected with 100 nM ATF3 small interfering RNA (siRNA) and reagent alone for 48 h and then treated with 1 μg/mL LPS for the indicated times. The protein levels of ATF3, p65, α-tubulin and topoisomerase II-α were detected with Western blot using the respective antibodies. α-tubulin and topoisomerase II-α were used as a loading control for cytoplasm and nucleus, respectively. (b) p65 and ATF3 were assessed with confocal microscopy. Cells were transfected with 100 nM ATF3 siRNA and reagent for 48 h and then treated with 1 μg/mL LPS for 2 h. (c) RAW 264.7 cells (left panel) were transfected with ATF3 siRNA and reseeded from a 10-cm dish to a 96-well plate to quantify NF-κB activity using enzyme-linked immunosorbent assay (ELISA). Human embryonic kidney 293 (HEK293) cells (right panel) were also transfected with ATF3 siRNA and NF-κB activity was detected with SEAP assay using ELISA. Data represent three independent experiments (n = 3; *P < 0.05, **P < 0.01). (d) Cells were treated with 1 μg/mL LPS for the indicated times. Beads were bound with p65 antibody and p65 and HDAC1 were precipitated. A total of 20 μg protein was separated with electrophoresis. Proteins levels were determined with Western blot using whole-cell lysates. (e) Cell lysates were processed with NE-PER lysate and the protein expression levels of ATF3, ac-p65, HDAC1 and β-actin were measured by Western blot. β-actin and HDAC1 were used as a loading control for cytoplasm and nucleus, respectively. (f,g) Human PBMCs were transfected with 100 nM ATF3 siRNA and reagent alone for 48 h and then treated with 1 μg/mL LPS for the indicated times. (f) The protein levels of ATF3 and β-actin were detected by Western blot; β-actin was used as a loading control. (g) The protein expression levels of ATF3, ac-p65, topoisomerase II-α and β-actin were measured with Western blot. β-actin and topoisomerase II-α were used as a loading control for cytoplasm and nucleus, respectively. Western data show a representative set among the results of three independent experiments (n = 3).
Recruitment of HDAC1 by ATF3 to regulate NF-κB activation
We confirmed that the expression of ATF3 protein is associated with the translocation of p65. We also determined whether ATF3 affects the activation of p65. To confirm the down-regulation of NF-κB activity via binding with ATF3, we performed NF-κB activity assays using enzyme-linked immunosorbent assay (ELISA) and secreted embryonic alkaline phosphatase (SEAP). ELISA is a powerful detection method that measures the activity of NF-κB in nuclear extracts. Activated p65 binds to the κB site in DNA and additional horseradish peroxidase-conjugated secondary antibody reacts with the substrate. When the expression of ATF3 was inhibited using siRNA in RAW 264.7 cells, NF-κB activity was up-regulated threefold to that of the control in response to LPS (Fig. 3c, top). SEAP assay detects NF-κB activity using the κB site of immune response target genes29. The deacetylated-p65, which was ATF3-assisted deacetylated, was eliminated in the nucleus and could not bind with the κB site of the target genes, thereby resulting in the down-regulation of NF-κB target genes. Using this assay, we found that NF-κB activity was up-regulated 1.5-fold by LPS treatment, whereas ATF3 expression was inhibited by siRNA in human embryonic kidney 293 (HEK293)-Blue TLR4 reporting cells (Fig. 3c, bottom).
In addition, we performed IP under ATF3-deficient conditions (Fig. 3d). After 2 h of LPS treatment, p65 bound with HDAC1 in scrambled siRNA but failed to bind under ATF3-deficient conditions. Moreover, treatment of ATF3-deficient cells with LPS resulted in the increased expression of ac-p65 in the nucleus compared with scrambled siRNA cells at 8 h (Fig. 3e). In human peripheral blood mononuclear cells (hPBMCs), LPS induced the expression of ATF3 at 2 and 4 h that was inhibited by ATF3 siRNA (Fig. 3f). Furthermore, ATF3-deficient cells also showed an increase in the expression of ac-p65 in the nucleus following its treatment with LPS compared to scrambled siRNA cells at 4 h (Fig. 3g). These results showed that ATF3 has critical importance in mediating interaction between p65 and HDAC1, as well as in the acetylation of p65. The reduced acetylation of p65 in ATF3 presence implied an indirect role of ATF3 in governing the acetylation status of p65 that in turn mediates the transcription of inflammatory genes (Fig. 3g).
ATF3 regulates NF-κB-dependent gene expression
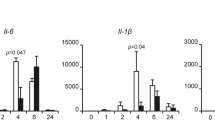
Interleukin (IL)-6 and nitric oxide (NO) are representative inflammatory response molecules30,31. NO is produced by inducible NO synthase induced by NF-κB signalling. IL-6 is also induced by NF-κB signalling via binding to the κB site in the IL-6 promoter30. Therefore, we confirmed IL-6 and NO expression using ELISA (Fig. 4a) and observed that both were up-regulated under ATF3-deficient conditions. Moreover, a plethora of inflammatory mediators were expressed, including IκBζ, IL-1β, IL-6, IL-12β and inducible NO synthase when NF-κB was activated. It has been reported that ATF3 can bind to the IL-6 and IL-12β promoter regions and inhibit transcription via deacetylation of the κB site28.

Inflammatory response genes in NF-κB signalling were up-regulated under ATF3-deficient conditions.
(a) Cells were transfected with 100 nM ATF3 siRNA for 48 h and then treated with 1 μg/mL LPS for 24 h. The secretion of interleukin (IL)-6 and nitric oxide (NO) were detected with ELISA. (b) RAW 264.7 cells were transfected with scrambled siRNA or ATF3 siRNA for 48 h and then treated with 1 μg/mL LPS for 2 h. Messenger RNA expression was measured using conventional reverse transcription-polymerase chain reaction. (c) RAW 264.7 cells were transfected with scrambled siRNA or ATF3 siRNA for 48 h and then treated with 1 μg/mL LPS for 2 and 4 h. Messenger RNA expression of IL-6 was measured using real-time polymerase chain reaction. Expression of IL-6 was normalized to β-actin and compared to control cells. Data represent three independent experiments (n = 3; *P < 0.05, **P < 0.01).
Furthermore, we evaluated messenger RNA expression of the target genes of well-known primary inflammatory mediators using reverse transcription-polymerase chain reaction (RT-PCR) after treating cells with LPS for 2 h (Fig. 4b). As the results indicated, these genes were up-regulated compared with those in the control. Of particular interest, the expression level of IL-6 was significantly altered in ATF3-deficient cells compared with scrambled siRNA cells in a time-dependent manner (Fig. 4c). Cumulatively, the results of this study showed that ATF3 binds to NF-κB (p65) directly and creates a docking site for HDAC1 to deacetylate p65 to inhibit inflammatory response cytokines induced by NF-κB signalling. Our findings present a mechanism other than the canonical one stating that ATF3 binds directly with the cytokine promoter region, not the NF-κB subunit.
Discussion
The stress-induced transcription factor ATF3 is a negative regulator of cytokines such as IL-6, IL-12β and tumour necrosis factor-α in LPS-induced TLR4 signalling22,28,32. NF-κB is the principal mediator of TLR4 signalling that, upon activation, translocates into the nucleus and induces inflammation. ATF3 is also a target gene of NF-κB and is induced when NF-κB is activated33. The canonical mechanism of ATF3-negative regulation proposes that ATF3 recruits HDAC1 to the transcriptionally active complex and assists in deacetylating the promoter region of inflammatory cytokines such as IL-6, thereby impeding p65 binding to the target promoters and in turn attenuating their transcription22,28. In this study, we demonstrated that NF-κB activity is attenuated via direct interaction of ATF3 and the p65 subunit of NF-κB rather than through an indirect mechanism.
The detection of an immune-activating ligand such as LPS culminated in the activation of the immune system; as a result, inflammatory mediators came into play to combat the invading pathogen. This host–pathogen interaction is tightly regulated so that neither the pathogen harms the organism nor an exuberant immune response yields unwanted consequences34. This tight regulation is orchestrated by multiple proteins that execute the response in a coordinated fashion. Similarly, when TLR4 senses LPS, it generates many cytokines (Fig. 1a). This activation also triggers the mechanisms of negative regulation, such as ATF3 translocation into the nucleus. The time frames of p65 degradation and ATF3 accumulation coincide, pointing to the negative influence of ATF3 on p65 (Fig. 1b).
The results of recent studies have suggested that ATF3 recruits HDAC1 to the p65-harbouring transcriptional complex and deacetylates the promoters, resulting in p65 dissociation28,35,36,37. Furthermore, these studies have implied that an alternative spliced form of ATF3, ATF3Δzip2, but not ATF3, binds to p65 and modulates the response35. In this study, we demonstrated through IP analysis that not only ATF3Δzip2 but also ATF3 can bind to mp65 (Fig. 2a,b). Moreover, p65 binds to HDAC1 (Fig. 2b), but under ATF3-deficient conditions (Fig. 3d), p65 is unable to bind to HDAC1. This intriguing behaviour could be attributed to ATF3 interaction with p65 that creates a docking site for HDAC1. The docked HDAC1 then plausibly deacetylates p65, facilitating its export from the nucleus. Therefore, we now believe that ATF3 is a key molecule in mediating the interaction between p65 and HDAC1 and that it attenuates p65 activity.
To unravel the requirement of ATF3 for the binding of HDAC1 in these interactions, we conducted experiments under ATF3-deficient conditions. We observed that under these conditions, p65 was maintained in the nucleus for a longer time in response to LPS treatment compared to that in the control (Fig. 3a). Additional data showed that NF-κB was significantly up-regulated in cells lacking ATF3 (Fig. 3b,c). The formation of ternary complex can be deduced by a partial direct contribution from each molecule. In the presence of ATF3, HDAC1 bound to p65 was detected (Fig. 2b), whereas in the absence of ATF3, this association was impaired (Fig. 3d), clearly implying the necessary role of ATF3 in mediating this ternary complex. In contrast to those of previous studies, our results revealed a lack of interaction between p65 and HDAC1 in the absence of ATF3, which precluded the deacetylation of p65 and increased p65 activity compared with that in the control (Fig. 3e). The presence of ATF3 suppressed the level of ac-p65, whereas the ATF3-siRNA enhanced the ac-p65 level, implying the ATF3 role in acetylation of p65 and the plausible mechanism is the recruitment of HDAC1 on various complexes that carried out deacetylation of p65. We observed the similar results when the experiments were repeated in hPBMCs, indicating that this might be a canonical mechanism that is followed in various cells (Fig. 3f,g). Therefore, we concluded that ATF3 facilitates the interaction of HDAC1 and p65, leading to reduced p65 activity. When cells are lacking ATF3, p65 has ample time to induce its target genes11, in particular, those of inflammation-related cytokines that were also observed (Fig. 4).
ATF3 is a key molecule in the regulation of innate immune response and it is associated with a variety of immune and inflammatory responses. Most of these responses are triggered by NF-κB activation. Further experiments are needed to unravel the details of this regulatory checkpoint and this study provides a basis for those studies. Moreover, the results of this study furnish a novel mechanism of innate immune regulation by ATF3 that will be helpful in designing therapeutic interventions for inflammation-related abnormalities.
Materials and Methods
Cell cultures
RAW 264.7 (Korean Cell Line Bank, Seoul, Korea) and HEK-Blue™-hTLR4 (Invivogen, San Diego, CA, USA) cell lines were cultured in Dulbecco’s modified Eagle medium (DMEM) with low glucose and DMEM growth medium containing 10% FBS and 1% penicillin/streptomycin (Thermo Scientific, Waltham, MA, USA), respectively. Human PBMCs were purchased from PromoCell (Heidelberg, Germany) and cultured in RPMI1640 medium containing 10% FBS, 1% penicillin/streptomycin (Thermo Scientific) and 1% L-glutamine (Gibco, Grand Island, NY, USA). These cells were incubated in an atmosphere of 5% CO2 at 37 °C (Thermo Scientific).
Confocal microscopy analysis
Cells were fixed by 3.7% formaldehyde (Sigma-Aldrich, St. Louis, MO, USA) for 15 min and permeabilized by 0.2% Triton X-100 for 15 min. Cells were blocked by 5% FBS (Thermo Scientific) in PBS (AMRESCO, Solon, OH, USA) for 1 h and then incubated with primary antibodies (1:1000, 1 h) targeting p65, ATF3 (Santa Cruz Biotechnology, Dallas, TX, USA), ac-p65 (Abcam, Cambridge, MA, USA) or HDAC1 (Millipore, Billerica, MA, USA). Later, cells were incubated with secondary antibodies (1:1000, 1 h) conjugated with Alexa Fluor 488 or 546 (Invitrogen, Carlsbad, CA, USA). Nuclei were stained with Hoechst33258 (5 μM; Sigma-Aldrich) for 15 min. Stained cells were visualized using confocal microscopy (LSM-700, Carl Zeiss Microimaging, Germany) and analyzed by Zen 2009 software.
RNA isolation and RT- and qRT-PCR
Total RNA was isolated using an RNeasy mini kit (QIAGEN, Valencia, CA, USA) and the total RNA concentration was detected using a Micro UV-Vis fluorescence spectrophotometer (Malcom, Tokyo, Japan). Total RNA (500 ng) was reverse-transcribed to complementary DNA with Oligo dT using an Accupower RT PreMix complementary DNA kit (Bioneer, Daejeon, Korea). PCR was performed using GoTaq® Green Master Mix (Promega, Madison, WI, USA). The RT-PCR primers were as follows: IL-1β F; 5′-GCA ACT GTT CCT GAA CTC AAC T-3′ and R; 5′-ATC TTT TGG GGT CCG TCA ACT-3′; IL-6, F; 5′-TGG GGC TCT TCA AAA GCT CC-3′ and R; 5′-AGG AAC TAT CAC CGG ATC TTC AA-3′; IL-12β, F; 5′-TGG TTT GCC ATC GTT TTG CTG-3′ and R; 5′-ACA GGT GAG GTT CAC TGT TTC T-3′; iNOS, F; 5′-CAG GGA GAA CAG TAC ATG AAC AC-3′ and R; 5′-TTG GAT ACA CTG CTA CAG GGA-3′ and the primers for ATF3, TNF-α, IκBζ and β-actin are as described previously24. Thermal cycling was carried out with an initial denaturation phase of 10 min at 95 °C, followed by 30 cycles each of 30 sec of denaturation at 95 °C, annealing for 30 sec at 57 °C and extension for 1 min at 72 °C. Amplification was carried out in a Veriti™ thermal cycler (Applied Biosystems, Carlsbad, CA, USA) and the amplified product was subjected to electrophoresis on 1.5% agarose gel. Gel images were captured by UV transilluminator system (TFX-20M; Vilber-Lourmat, Marne-la-Vallée, France). In qRT-PCR analysis, the expression level of IL-6 was measured using a Maxima SYBR Green/ROX qPCR master mix (Thermo Scientific) and examined using a qRT-PCR (Bio-Rad Laboratories, Hercules, CA, USA) with primers including IL-6, as described previously38. The two-step PCR protocol was as follows: 10 min at 95 °C, followed by 30 cycles of denaturing (5 s at 95 °C) and annealing (10 s at 57 °C). Relative IL-6 level was calculated using the ΔΔCT method and IL-6 level was normalized to β-actin level.
Western blot analysis
After treatment, protein was isolated by resuspending cells in M-PER® Mammalian Protein Extraction Reagent (Thermo Scientific) and NE-PER lysate (Thermo Scientific) for immunoprecipitation with protease & phosphatase inhibitor cocktails (Thermo Scientific), incubated for 10 min at 4 °C and centrifuged at 16,000 × g for 10 min to remove cell debris. Extracts were loaded onto 10% polyacrylamide gels and transferred to Hybond-ECL membranes (Amersham, Pollards Wood, UK). Membranes were blocked with 5% skim milk in phosphate-buffered saline containing 0.05% Tween 20 (PBST). The membranes were then incubated with specific primary antibodies diluted with PBST at 4 °C with gentle shaking overnight: p65, ATF3, topoisomerase II-α, IκBζ, α-tubulin and β-actin (Santa Cruz Biotechnology); ac-p65 (Abcam) and HDAC1 (Millipore). After several washings, the membranes were incubated with anti-mouse or anti-rabbit immunoglobulin G peroxidase-conjugated antibody (Thermo Scientific) diluted in PBST (1:1,000) for 2 h. After the membranes were washed several times with PBST, detection was carried out using Pierce ECL Western blotting substrate (Thermo Scientific) and exposure of the blots to a LAS-1000 system (Fuji, Tokyo, Japan). The intensities of protein bands were measured using the ImageJ software (http://rsb.info.nih.gov/ij/)39.
IP analysis
RAW 264.7 cells were seeded at an approximate density of 1 × 106 cells/10-cm dish. After treatment, IP with antibodies specific to ATF3 and p65 (Santa Cruz Biotechnology) was performed. Protein was isolated following the method used for Western blot analysis.
Quantification of IL-6 and p65 with ELISA
The concentrations of IL-6 and p65 in the supernatant of the culture media were determined using commercially available ELISA kits (eBioscience, San Diego, CA, USA). First, respective cells (5 × 105/well) were dispensed into a 24-well plate and stimulated with LPS (1 μg/ml) for IL-6 at 24 h or p65 at 2 h. Then supernatant was collected and added to the antibody-coated microplate to capture IL-6 and p65. The plates were then maintained at room temperature for 2 h. Next, the plates were washed five times, biotin-conjugated detecting antibody was added to each well and the plates were further incubated at room temperature for 1 h. After incubation, the plates were washed five times and incubated with avidin–horseradish peroxidase for 30 min. Finally, detection was carried out with 3,3′,5,5′-tetramethylbenzidine solution by measuring absorbance at 450 nm with an ELISA reader and analysed using SoftMax® Pro 5.3 software (Molecular Devices, Sunnyvale, CA, USA).
SEAP reporter assay
SEAP was detected using QUANTI-Blue™ (Invivogen) according to the manufacturer’s instructions. Briefly, 10 μL of supernatant was collected by centrifuging 1 mL of culture medium at 11,200 × g. This supernatant was then added to a 96-well plate and heated to 65 °C for 10 min to deactivate endogenous phosphatases. To each well, 50 μL of 1 × dilution buffer, 100 μL of 1 × assay buffer, 20 μL of 100 mM l-homoarginine and 20 μL H2O were added and incubated at 37 °C for 10 min. Finally, 20 μL of staining solution was added and optical density was measured at 405 nm. LPS stimulation induced the HEK-Blue™-hTLR4 cells to secret alkaline phosphatase as an indication of TLR4 activity. SEAP activity was quantified using QUANTI-Blue™ by measuring optical density at 620–655 nm with an ELISA reader (Molecular Devices).
NO assay
RAW 264.7 cells were plated at a density of 1 × 106 cells/mL into 96-well plates, incubated for 12 h and treated with the LPS for 24 h. Supernatant (100 μL) from the cultured cells was transferred to new 96-well plates and quantified using an NO detection kit according to the manufacturer’s protocol (iNtRON Biotech, Seongnam, Korea). Values were calculated by measuring the absorbance at 540 nm using an ELISA reader (Molecular Devices).
siRNA design and transfection
siRNA for ATF3 was designed and purchased from Bioneer (Daejeon, Korea) as follows: 5-CTG TGA GAT AAG CGG GAC TCA G-340,41. Lipofectamine 2000 transfection reagent (Life Technologies, Grand Island, NY, USA) was used for siRNA delivery. RAW 264.7 cells were seeded at 5 × 105 cells in a 6-cm dish containing 2 mL DMEM with low glucose for 12 h. hPBMCs were seeded at 2 × 106 cells in a 6-cm dish containing 2 mL of RPMI1640 medium for 12 h. Opti-MEM Reduced serum medium (2 mL; Life Technologies) containing 100 nM of siRNA and 20 μL of Lipofectamine 2000 reagent was incubated at room temperature for 15 min and added to each well. After transfection with ATF3 siRNA for 48 h, the medium was replaced with normal medium for further experiments.
Statistical analysis
The statistical analysis was done by the one-way ANOVA using the SigmaPlot software version 12.0 (Systat Software Inc., San Jose, CA, USA). The data shown are the results of at least three independent experiments and the statistical significance was defined as a P-value of *P < 0.05 or **P < 0.01.
Additional Information
How to cite this article: Kwon, J.-W. et al. Activating transcription factor 3 represses inflammatory responses by binding to the p65 subunit of NF-κB. Sci. Rep. 5, 14470; doi: 10.1038/srep14470 (2015).
References
Kaisho, T. & Akira, S. Toll-like receptor function and signaling. J Allergy Clin Immunol 117, 979–987, quiz 988 (2006).
Krishnan, J., Selvarajoo, K., Tsuchiya, M., Lee, G. & Choi, S. Toll-like receptor signal transduction. Exp Mol Med 39, 421–438 (2007).
Basith, S., Manavalan, B., Lee, G., Kim, S. G. & Choi, S. Toll-like receptor modulators: a patent review (2006–2010). Expert Opin Ther Pat 21, 927–944 (2011).
Prins, R. M. et al. The TLR-7 agonist, imiquimod, enhances dendritic cell survival and promotes tumor antigen-specific T cell priming: relation to central nervous system antitumor immunity. J Immunol 176, 157–164 (2006).
Whitmore, M. M., Li, S., Falo, L., Jr. & Huang, L. Systemic administration of LPD prepared with CpG oligonucleotides inhibits the growth of established pulmonary metastases by stimulating innate and acquired antitumor immune responses. Cancer Immunol Immunother 50, 503–514 (2001).
Krishnan, J., Lee, G. & Choi, S. Drugs targeting Toll-like receptors. Arch Pharm Res 32, 1485–1502 (2009).
Ghosh, S. & Karin, M. Missing pieces in the NF-kappaB puzzle. Cell 109 Suppl, S81–96 (2002).
Siebenlist, U., Franzoso, G. & Brown, K. Structure, regulation and function of NF-kappaB. Annu Rev Cell Biol 10, 405–455 (1994).
Verma, I. M., Stevenson, J. K., Schwarz, E. M., Van Antwerp, D. & Miyamoto, S. Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes Dev 9, 2723–2735 (1995).
Hayden, M. S. & Ghosh, S. Signaling to NF-kappaB. Genes Dev 18, 2195–2224 (2004).
Tak, P. P. & Firestein, G. S. NF-kappaB: a key role in inflammatory diseases. J Clin Invest 107, 7–11 (2001).
Rayet, B. & Gelinas, C. Aberrant rel/nfkb genes and activity in human cancer. Oncogene 18, 6938–6947 (1999).
Gilmore, T. D., Koedood, M., Piffat, K. A. & White, D. W. Rel/NF-kappaB/IkappaB proteins and cancer. Oncogene 13, 1367–1378 (1996).
Basith, S., Manavalan, B., Yoo, T. H., Kim, S. G. & Choi, S. Roles of toll-like receptors in cancer: a double-edged sword for defense and offense. Arch Pharm Res 35, 1297–1316 (2012).
Gilmore, T. D. The Rel/NF-kappaB signal transduction pathway: introduction. Oncogene 18, 6842–6844 (1999).
Basith, S., Manavalan, B., Gosu, V. & Choi, S. Evolutionary, structural and functional interplay of the IkappaB family members. PloS one 8, e54178 (2013).
Thompson, J. E., Phillips, R. J., Erdjument-Bromage, H., Tempst, P. & Ghosh, S. I kappa B-beta regulates the persistent response in a biphasic activation of NF-kappaB. Cell 80, 573–582 (1995).
Whiteside, S. T., Epinat, J. C., Rice, N. R. & Israel, A. I kappa B epsilon, a novel member of the I kappa B family, controls RelA and cRel NF-kappa B activity. EMBO J 16, 1413–1426 (1997).
Manavalan, B., Basith, S., Choi, Y. M., Lee, G. & Choi, S. Structure-function relationship of cytoplasmic and nuclear IkappaB proteins: an in silico analysis. PloS one 5, e15782 (2010).
Nolan, G. P. et al. The bcl-3 proto-oncogene encodes a nuclear I kappa B-like molecule that preferentially interacts with NF-kappa B p50 and p52 in a phosphorylation-dependent manner. Mol Cell Biol 13, 3557–3566 (1993).
Baldwin, A. S., Jr. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol 14, 649–683 (1996).
Whitmore, M. M. et al. Negative regulation of TLR-signaling pathways by activating transcription factor-3. J Immunol 179, 3622–3630 (2007).
Hai, T. & Hartman, M. G. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene 273, 1–11 (2001).
Kim, E. Y. et al. ATF3 plays a key role in Kdo2-lipid A-induced TLR4-dependent gene expression via NF-kappaB activation. PloS one 5, e14181 (2010).
Chen, B. P., Liang, G., Whelan, J. & Hai, T. ATF3 and ATF3 delta Zip. Transcriptional repression versus activation by alternatively spliced isoforms. J Biol Chem 269, 15819–15826 (1994).
Drysdale, B. E., Howard, D. L. & Johnson, R. J. Identification of a lipopolysaccharide inducible transcription factor in murine macrophages. Mol Immunol 33, 989–998 (1996).
Farber, J. M. A collection of mRNA species that are inducible in the RAW 264.7 mouse macrophage cell line by gamma interferon and other agents. Mol Cell Biol 12, 1535–1545 (1992).
Gilchrist, M. et al. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature 441, 173–178 (2006).
Moon, K. Y., Hahn, B. S., Lee, J. & Kim, Y. S. A cell-based assay system for monitoring NF-kappaB activity in human HaCat transfectant cells. Anal Biochem 292, 17–21 (2001).
Libermann, T. A. & Baltimore, D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol 10, 2327–2334 (1990).
Xie, Q. W., Kashiwabara, Y. & Nathan, C. Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J Biol Chem 269, 4705–4708 (1994).
Anwar, M. A., Panneerselvam, S., Shah, M. & Choi, S. Insights into the species-specific TLR4 signaling mechanism in response to Rhodobacter sphaeroides lipid A detection. Sci Rep 5, 7657 (2015).
Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010).
Anwar, M. A., Basith, S. & Choi, S. Negative regulatory approaches to the attenuation of Toll-like receptor signaling. Exp Mol Med 45, e11 (2013).
Hua, B. et al. A splice variant of stress response gene ATF3 counteracts NF-kappaB-dependent anti-apoptosis through inhibiting recruitment of CREB-binding protein/p300 coactivator. J Biol Chem 281, 1620–1629 (2006).
Li, H. F., Cheng, C. F., Liao, W. J., Lin, H. & Yang, R. B. ATF3-mediated epigenetic regulation protects against acute kidney injury. Journal of the American Society of Nephrology: JASN 21, 1003–1013 (2010).
Kiernan, R. et al. Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J Biol Chem 278, 2758–2766 (2003).
Kawane, K., Tanaka, H., Kitahara, Y., Shimaoka, S. & Nagata, S. Cytokine-dependent but acquired immunity-independent arthritis caused by DNA escaped from degradation. Proceedings of the National Academy of Sciences of the United States of America 107, 19432–19437 (2010).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Yoshida, T. et al. ATF3 protects against renal ischemia-reperfusion injury. Journal of the American Society of Nephrology: JASN 19, 217–224 (2008).
Park, E. J., Kwon, H. K., Choi, Y. M., Shin, H. J. & Choi, S. Doxorubicin induces cytotoxicity through upregulation of pERK-dependent ATF3. PloS one 7, e44990 (2012).
Acknowledgements
This work was supported by the Mid-Career Researcher Program through the National Research Foundation of Korea, funded by the Ministry of Education, Science and Technology (2012R1A2A2A02016803) and by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (HI14C1992). This work was also partially supported by a grant from the Priority Research Centers Program (NRF 2012-0006687).
Author information
Authors and Affiliations
Contributions
J.K. and S.C. conceived and designed the research. J.K., H.K., H.S. and Y.C. performed the experiments. J.K., M.A.A. and S.C. analysed the data and prepared the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kwon, JW., Kwon, HK., Shin, HJ. et al. Activating transcription factor 3 represses inflammatory responses by binding to the p65 subunit of NF-κB. Sci Rep 5, 14470 (2015). https://doi.org/10.1038/srep14470
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep14470
This article is cited by
-
ATF3-induced activation of NF-κB pathway results in acquired PARP inhibitor resistance in pancreatic adenocarcinoma
Cellular Oncology (2023)
-
Role of metformin in inflammation
Molecular Biology Reports (2023)
-
Activating transcription factor 3 inhibits NF‑κB p65 signaling pathway and mediates apoptosis and cell cycle arrest in cervical cancer cells
Infectious Agents and Cancer (2022)
-
Sex differences in the inflammatory response of the mouse DRG and its connection to pain in experimental autoimmune encephalomyelitis
Scientific Reports (2022)
-
Research on the correlation between activating transcription factor 3 expression in the human coronary artery and atherosclerotic plaque stability
BMC Cardiovascular Disorders (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
